1. Introduction
Iridium (Ir) exhibits a high melting point (2443 °C), excellent high-temperature chemical inertness [
1], and good compatibility with PuO
2 fuel and external graphitic components at about 1400 °C. Consequently, Ir alloys provide an optimal series of cladding materials for radioisotope batteries [
2]. The nuclear fuel cladding of radioisotope thermoelectric generators (RTGs) is also expected to have good strength and toughness properties. This is to prevent nuclear fuel from leaking in the case of an impact with the Earth in the event of a launch failure [
3]. However, Ir, unlike other face-centered cubic (FCC) metals (e.g., Cu, Ag, Au, etc.), exhibits inherent brittleness at room temperature (RT) [
4]. It is reported that brittle transgranular fracture is the sole fracture mode of high-purity Ir at RT [
4]. Research on the inherent brittleness of Ir and strategies to improve its toughness is a longstanding issue.
Doping and alloying efforts have been carried out to improve its mechanical strength and toughness [
5,
6,
7,
8]. It is reported that trace doping of Th and Ce (<30 ppm) significantly enhances the fracture toughness of Ir alloys [
9]. This fact is closely related to the nanoscale ThIr
5 and CeIr
5 precipitates at grain boundaries. As early as the 1980s, the Oak Ridge National Laboratory in the United States had already developed DOP-26 (Ir-0.3W-0.006Th-0.005Al, wt%) with excellent mechanical properties and ductility [
9,
10]. DOP-26 has since become the cladding material of choice for NASA [
11].
Theoretical calculations [
12,
13] suggested that the solid solutions of some elements (3.125 at %), such as Th, La, Ce, and Y, can significantly improve the toughness of Ir. However, the solubility of Th, La, Ce, and Y in Ir is extremely low. For example, the solubility of Th in Ir is about 30 ppm [
10]. Though supersaturated solid solutions can be obtained with the rapid quenching technique, it is nearly impossible to improve their solubility in FCC Ir with traditional casting methods. Density functional theory (DFT) calculations suggest that W and Pt may improve the brittleness of Ir [
12,
13]. The early literature reported that the ductility of Ir alloys was slightly improved by alloying with 0.3% W [
10]. Several experimental studies [
14,
15,
16] on Ir-W-Al alloys suggested that alloying with W and Al can improve the high-temperature mechanical properties of iridium alloys. W is likely to be a preferred alloying element to improve the toughness of Ir. Improving the ductility of refractory high-entropy alloys (HEAs) is also a research hotspot because of their high strength and obvious brittleness [
17]. It is reported that solid solution alloying [
18], second-phase dissolution [
19], compositional gradient design [
20], valence electron concentration [
21], and microstructure design can improve the toughness of HEAs. The primary purpose of this work is to investigate the effect of W solid solution alloying on the mechanical toughness of Ir-based alloys through DFT calculations. Hu et al. [
22] predicted that the addition of V shortens the pseudo-energy gap and enhances the interaction force between Mo and W atoms in a NbMoTaW HEA by first-principles calculation. The addition of V improves the mechanical properties of the NbMoTaW HEA but does not improve the toughness.
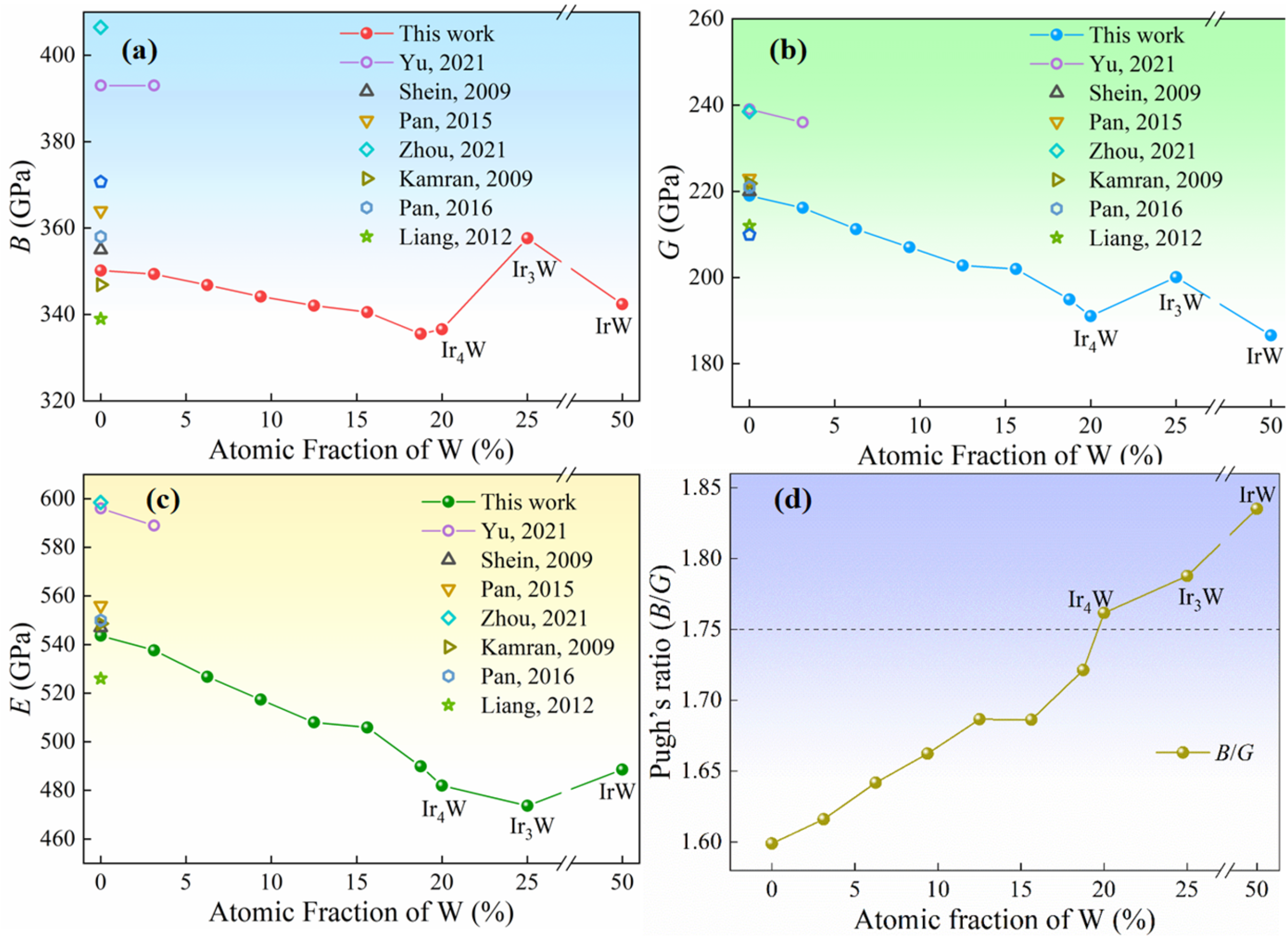
The reported Pugh’s ratio (
B/
G) and Poisson’s ratio (ν) of pure Ir, with inherent brittleness, are about 1.61 and 0.24, respectively [
23]. Elastic properties are closely related to the toughness of metal materials. When Pugh’s ratio is greater than 1.75 or Poisson’s ratio is greater than 0.26, the material is considered tough [
24]. If they are below these values, the material is considered brittle.
The generalized stacking fault energy (GSFE), originally proposed by Vitek [
25], is closely related to the slip and dislocation behavior. FCC metals generally exhibit high ductility, attributed to their close-packed atomic arrangement and 12 slip systems. The intrinsic stacking fault energy (γ
isf) of Ir (about 420 mJ/m
2 [
26]) is much higher than that of Cu (about 51 mJ/m
2), Ag (about 21 mJ/m
2 [
27], and Au (about 33 mJ/m
2 [
26]), which have good ductility. These values are closely related to the deformation and plane sliding nature of those metals. Pugh’s ratio (
B/
G) and stacking fault energy (SFE) are always employed to predict or estimate the toughness evolution of a material after alloying [
28,
29,
30]. Shang et al. [
28] reported that alloying elements in diluted Ni-based alloys and increasing the temperature induce a decrease in the SFE, which is linked to easier shear deformation. Tian et al. [
29] reported that alloying elements such as Cr and W in FCC Co-based binary alloys can tune the SFE to modulate dislocation behavior. A lower SFE facilitates the dissociation of full dislocations into Shockley partials. This dissociation hinders cross-slips, reduces creep rates, and thus contributes to enhanced ductility.
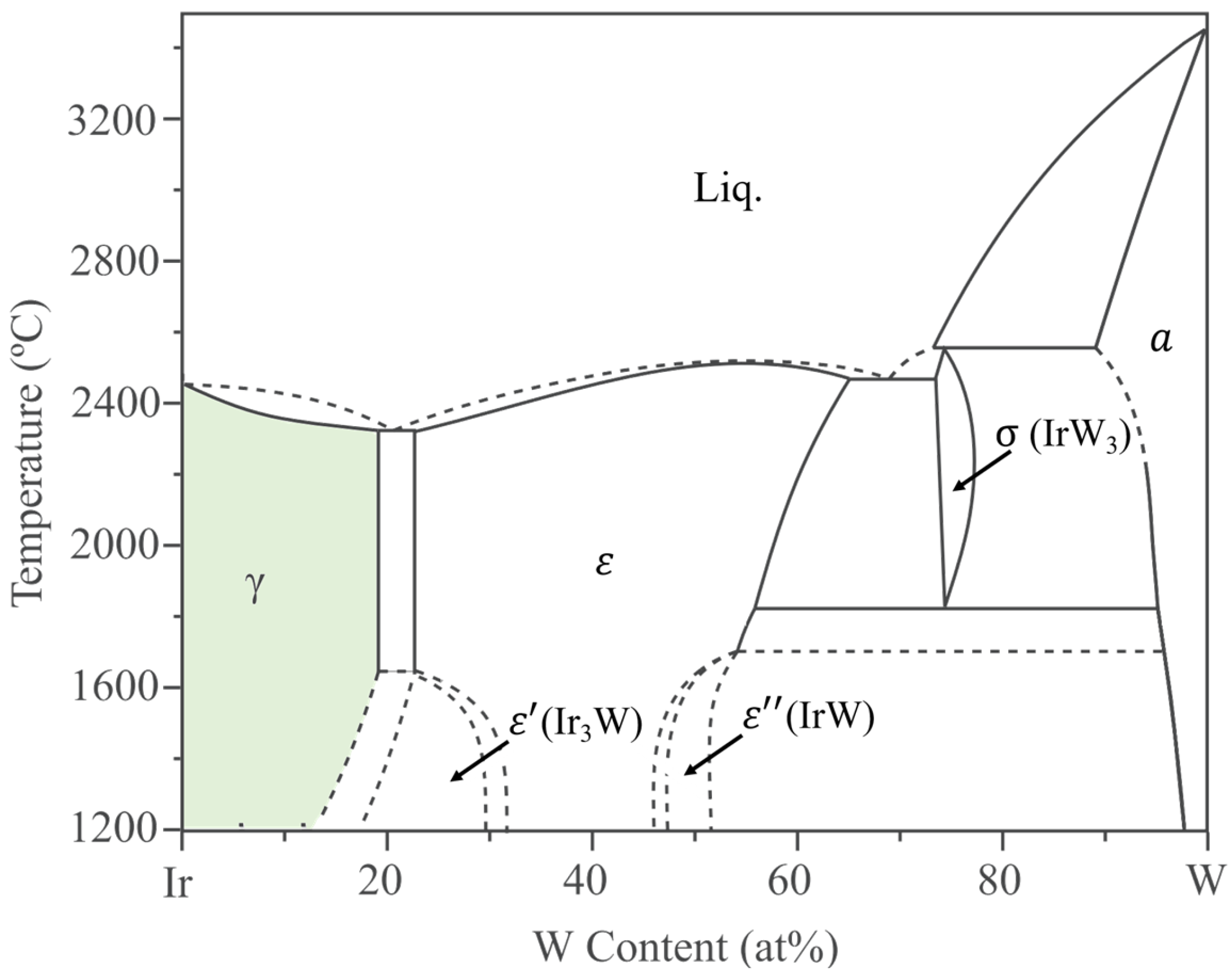
The Ir-W equilibrium phase diagram shows that W exhibits high solubility in Ir (~19 at.% above 1700 °C, 17.8 at.% at 1400 °C) [
31]. Alloying with W may significantly improve the toughness of Ir without reducing the alloy strength. However, the effects of W on the elastic properties, stacking fault energy, and the bonding characteristics are still not exclusively reported. In the present work, Ir-W solid solutions (Ir
31W
1, Ir
30W
2, Ir
29W
3, Ir
28W
4, Ir
27W
5, and Ir
26W
6) cubic atomic models and eight-layer ABCABCAB stacking models were constructed. Their equilibrium lattice constants, formation energies, elastic properties, electronic structures, and GSFE were calculated using first-principles calculations. For the Ir-W solid solutions, the effects of W on lattice distortion and electronic charge distribution within the Ir matrix were analyzed. The elastic properties of three intermetallic compounds (Ir
4W, Ir
3W, and IrW) were also investigated for comparison. The evolution of Cauchy pressure (
C12−
C44), Pugh’s ratio (
B/
G), GSFE, and bonding characteristics was discussed to predict the evolution of toughness. These findings will improve the understanding of why W enhances the toughness of Ir-based alloys.
2. Methodology
First-principles calculations were performed using the Vienna ab initio simulation package [
32,
33] (VASP) based on density functional theory [
34] (DFT). The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof [
35] (PBE) functional was employed to describe the electron–ion interaction. The electron configurations for Ir and W atoms are [Xe] 4f
145d
76s
2 and [Xe] 4f
145d
46s
2, respectively. The maximum plane-wave basis cutoff energy was 550 eV. The convergence criteria for the electronic and ionic steps were set to 1
10
−6 eV/atom and 0.005 eV/Å. The k-point mesh was divided into 8
8
8 by the Monkhorst–Pack method for pure Ir and Ir-W solid solutions. The electronic density of states (DOS) was calculated using the linear tetrahedron method with Blöchl correction. All the calculations were carried out assuming a non-magnetic state. The crystal orbital Hamilton population (COHP) analysis was performed using the Local Orbital Basis Suite Towards Electronic-Structure Reconstruction (LOBSTER) procedure [
36]. COHP partitions the electron density into bonding, non-bonding, and antibonding sections. Negative COHP values indicate bonding states, while positive COHP values indicate antibonding states [
37,
38,
39,
40].
The present research focuses on polycrystalline Ir-W alloys by casting. According to the Ir-W binary phase diagram [
31] (
Figure 1), the γ-Ir solid solution (with space group Fm
m) has a solubility limit of approximately19 at.% W above 1700 °C.
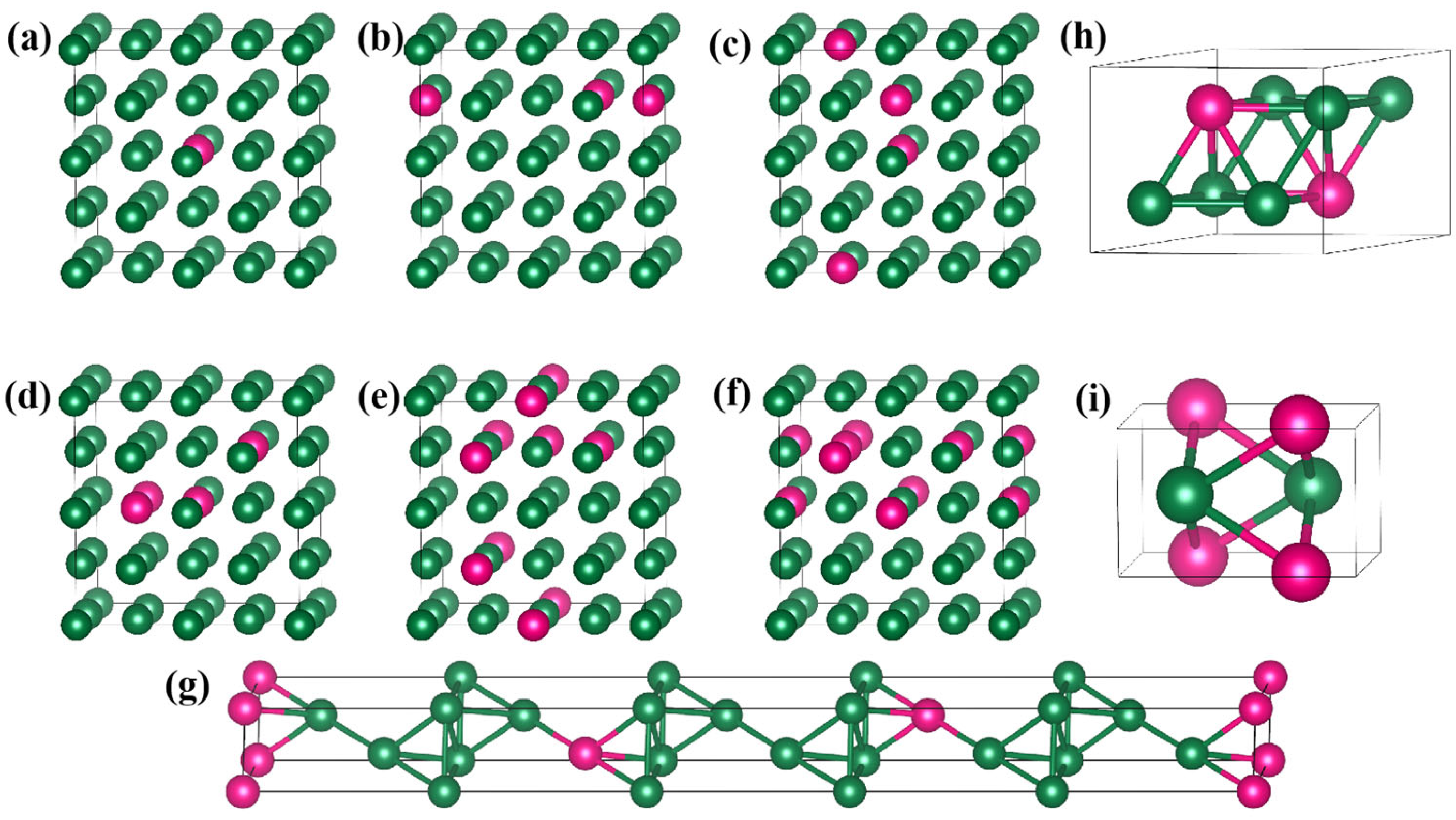
Pure Ir and six Ir-W solid solutions (Ir
31W
1, Ir
30W
2, Ir
29W
3, Ir
28W
4, Ir
27W
5, and Ir
26W
6) with 2
2
2 supercell models containing 32 atoms were constructed in this work. Within those solid solution supercells, Ir atoms were randomly replaced by W atoms. The special quasi-random structure (SQS) was established using the Alloy Theory Automation Toolkit [
41,
42,
43] (ATAT) code. SQS simulates the random state of solute alloys by constructing supercells with special atomic positions that minimize the difference between their atomic correlation functions and that of the absolute random crystal cells. It is possible that the W is not uniformly distributed in a real alloy. There may be W-rich regions or W-regions in a real Ir-W alloy. The non-uniform distribution of W atoms in a real solid solution alloy was not addressed in the present research.
According to the Ir-W binary phase diagram [
31] (
Figure 1), there are high temperature phases (ε) with W content of 22~66 at.%, stable-phase Ir
3W (ε′, with space group P6
3/mmc) and IrW (ε′′, with space group Pmma) on the Ir-rich side, and high-temperature-phase IrW
3 (σ, with space group P4
2/mnm) on the W-rich side. Diffusion coupling experiments with EPMA confirmed there are γ, ε′, ε, and ε′′ phases on the Ir-rich side with an increase in W content at 1500 °C [
15]. Above 1650 °C, there are γ phases and ε phases on the Ir-rich side, with a two-phase region (γ + ε) located at approximately 19~22 at % W. The structure of Ir
4W (with space group R
m) was reported by the Materials Project database (mp-1223666) [
44]; however, experimental studies on Ir
4W are not reported. The
supercells of Ir
4W, Ir
3W, and IrW used in this work were derived from the Materials Project database [
44]. For Ir
4W, Ir
3W, and IrW phases, the Brillouin zone was sampled with k-point meshes of 24
24
1, 12
11
11, and 11
18
10, respectively, generated by the Monkhorst–Pack scheme for geometry optimization and mechanical properties calculations. The high-temperature phases ε and σ (IrW
3) were not considered in this work. The details of the Ir-W alloy supercells are illustrated in
Figure 2. The key DFT calculation parameters used in the modeling are compared in
Table 1.
2.1. Calculation of Lattice Constant and Supercell Length
The Birch–Murnaghan equation of state (EOS) [
45] was employed to fit the pressure–volume data points for pure Ir and Ir-W solid solutions to determine their equilibrium volumes. The EOS is expressed as follows:
where
,
,
are the external pressure, volume, equilibrium volume, bulk modulus, and first derivative of bulk modulus to pressure, respectively. The zero-pressure volume
V0 is defined as the equilibrium volume. The supercell length is calculated from
. The lattice constant of pure Ir is half its supercell length. The lattice constants of Ir
4W, Ir
3W, and IrW were obtained by the structural relaxation method in VASP with the GGA-PBE functional.
2.2. Calculation of Formation Energy
The formation energy of Ir-W alloy,
, was calculated by the following equation [
46]:
where
is total energy of the Ir-W alloy supercell.
and
are the energy of individual atoms of the FCC structure Ir and BCC structure W, respectively.
x and
y are the number of Ir and W atoms, respectively.
2.3. Calculation of Elastic Properties
The elastic properties were calculated by the stress–strain method [
47]. The bulk modulus (
B) and shear modulus (
G) were obtained by the Voigt–Reuss–Hill approach [
48]. For FCC structure alloys,
where
,
,
, and
are the bulk and shear modulus from Voigt and Reuss models, respectively.
For the FCC system, the elastic constants follow the relationship
C11 =
C22 =
C33,
C44 =
C55 =
C66,
C12 =
C23 =
C13, exhibiting three independent components, namely
C11,
C12, and
C44. However, the addition of W, for example, in Ir
31W
1, Ir
30W
2, Ir
29W
3, Ir
28W
4, Ir
27W
5, and Ir
26W
6, slightly distorts the FCC symmetry of the supercell, resulting in minor differences between these parameters, e.g.,
C11,
C22, and
C33. The elastic constants were averaged according to Formulas (6)–(8), and the elastic moduli were subsequently calculated using Formulas (3)–(5).
Formulas for calculating the elastic modulus of trigonal (Ir
4W), hexagonal (Ir
3W), and orthorhombic (IrW) structured alloys can be found in [
49]. The bulk modulus (
B) and shear modulus (
G) were calculated using the Hill approximation, which is represented by the following formulas [
48].
The Young’s modulus (E) and Poisson’s ratio (
) were derived from
B and
G.
2.4. Calculation of Generalized Stacking Fault Energy
The specific formula for calculating GSFE is as follows [
50]:
where
is the energy of the crystal after deformation with a slip displacement
d,
E0 is the energy of the stacked configuration without slipping, and
A is the area of the face defect region.
The close-packed (111) plane was employed as the slipping plane in this work. Supercell models with an 8-layer ABCABCAB stacking sequence, containing 128 atoms, were constructed by cutting along the [
] and [11
] directions, with a 15 Å vacuum layer along the [111] direction. The supercell top view is shown in
Figure 3a. In the FCC Ir structure, full dislocations b
1 dissociate into partial dislocations (b
2, b
3). The slip path for the GSFE calculation was along the [11
] direction, denoted as b
2. The [
] direction view of the supercell without slipping is shown in
Figure 3b.
Pure Ir and six Ir-W solid solutions (Ir31W1, Ir30W2, Ir29W3, Ir28W4, Ir27W5, and Ir26W6) with 2 2 2 supercell models containing 32 atoms were constructed in this work. For these solid solution supercells, Ir atoms were randomly replaced by W atoms. SQS simulates the random state of solute alloys by constructing supercells with special atomic positions, minimizing the difference between their atomic correlation functions and those of absolutely random crystal cells. It is possible that the W is not uniformly distributed in a real alloy. There may be W-rich regions or W-regions in a real Ir-W alloy. The non-uniform distribution of W atoms in a real solid solution alloy was not considered in the present research.
For the GSFE calculations, the total slip displacement (b = a/6 <11>) along the (111) plane was divided into ten equal increments of 0.1b each. The crystal energies at various slip displacements (0.2b, 0.4b, 0.5b, 0.6b, 0.8b, 1.0b, 1.2b, 1.4b, 1.5b, 1.6b, 1.8b, and 2.0b) were calculated, and the generalized stacking fault energies of the supercell model in different configurations were derived using Formula (13). All atoms were allowed to relax only in the direction perpendicular to the slip surface for structural optimization after each slip. The maximum plane-wave basis cutoff energy was set to 450 eV for calculating GSFE. The convergence criteria for electronic and ionic steps were set to 1 × 10−5 eV/atom and 0.01 eV/Å, respectively. The k-point mesh was divided into 441 using the Monkhorst–Pack method.
4. Discussion
Ir, unlike other metallic elements of the FCC structure, exhibits inherent brittleness at RT. Kontsevoi et al. [
71] suggested the brittleness of Ir is a result of the pseudo-covalent bond features of Ir-Ir bonds. Cawkwell et al. [
72] suggested that there are two core screw dislocations in iridium, a glissile planar core and a metastable non-planar core. The athermal transformation between the two core structures leads to exceptionally high rates of cross-slip during plastic deformation, associated with an exponential increase in the dislocation density. Cawkwell et al. [
72] reported that the mechanism of thermal cross-slip is because of the mixed metallic and covalent interatomic bonding. Kontsevoi et al. [
71] suggested the effective way to increase plasticity of Ir-based alloy is to decrease the covalent contribution to chemical bonding by alloying methods.
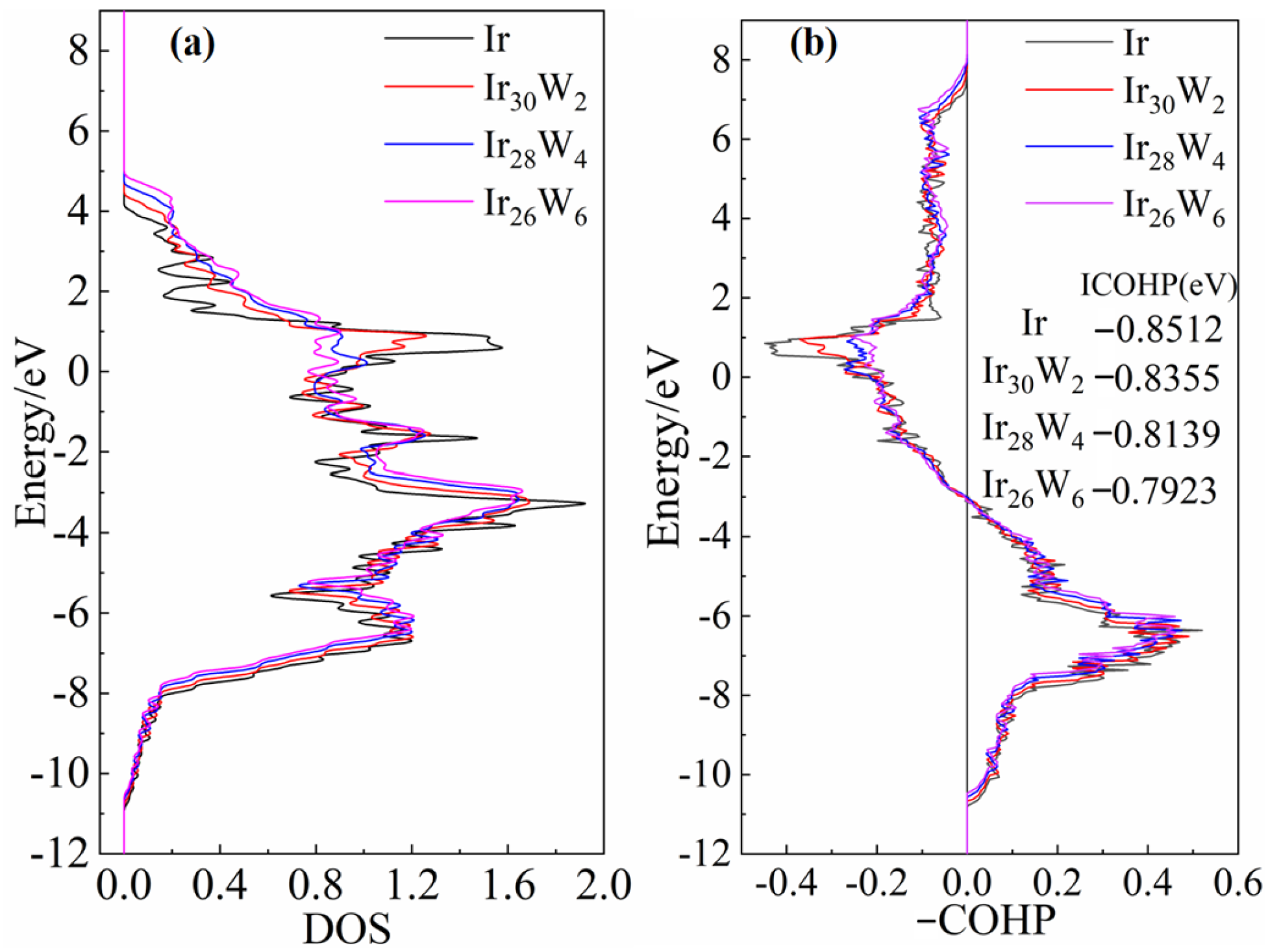
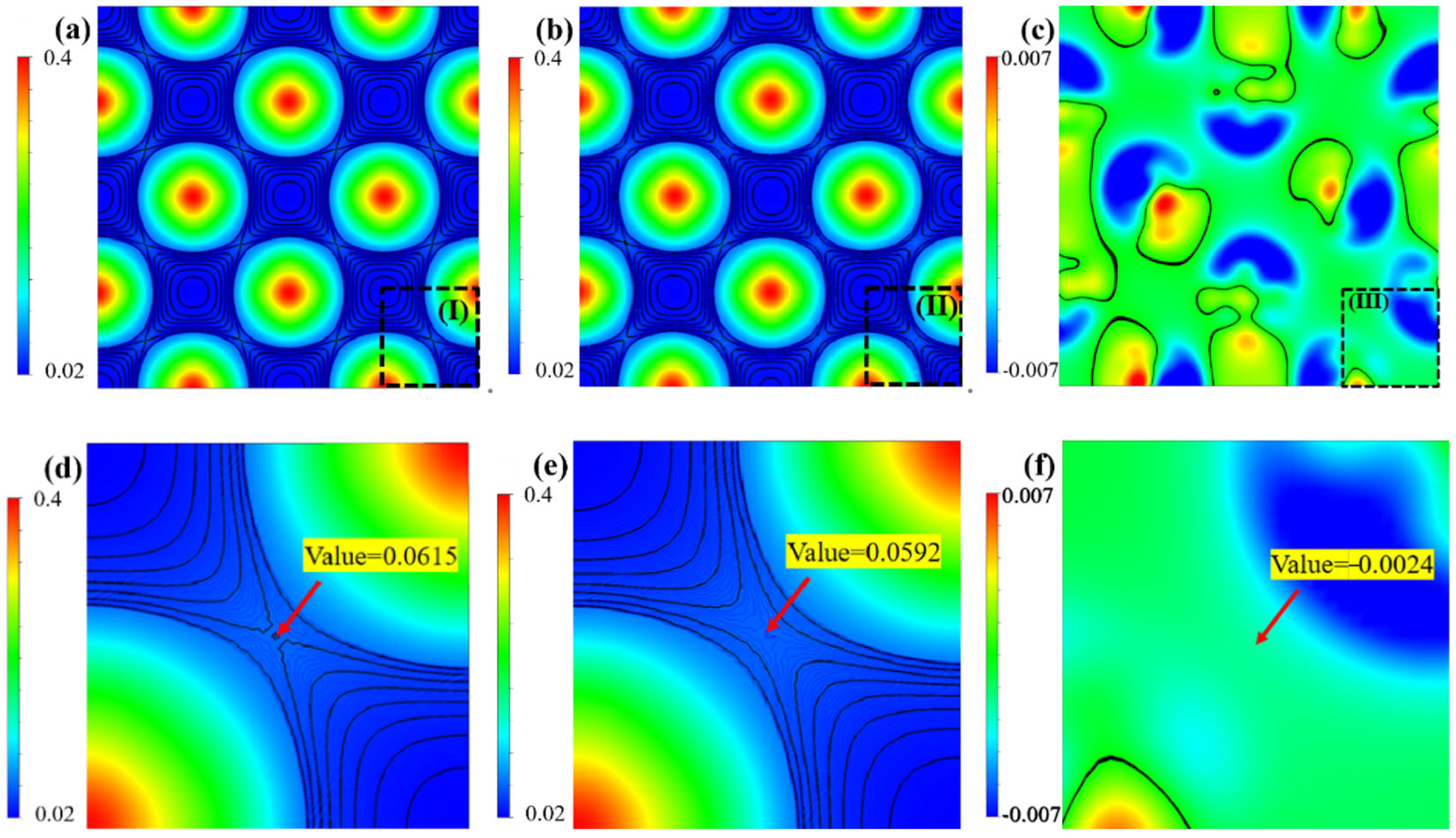
The above DOS and COHP analyses evidence that the chemical bond of Ir-Ir is a combining of metallic and covalent natures. The charge density analyses evidence that the density of the shared electrons by neighboring Ir-Ir atoms decreases with increasing W concentration. The covalent nature of Ir-Ir bonds is gradually weakened. The reduction of ICOHP values with increasing W content also suggests that the Ir-Ir bond strength is weakened. This approach of studying alloying effects through electronic structure (e.g., charge density, bond characteristics) and mechanical property correlations is consistent with DFT studies on other refractory systems. For instance, Hu et al. [
22] employed first-principles calculations to reveal how V addition modulates pseudo-energy gaps in NbMoTaW-based high-entropy alloys. It also shows how V addition affects interatomic interactions and mechanical properties.
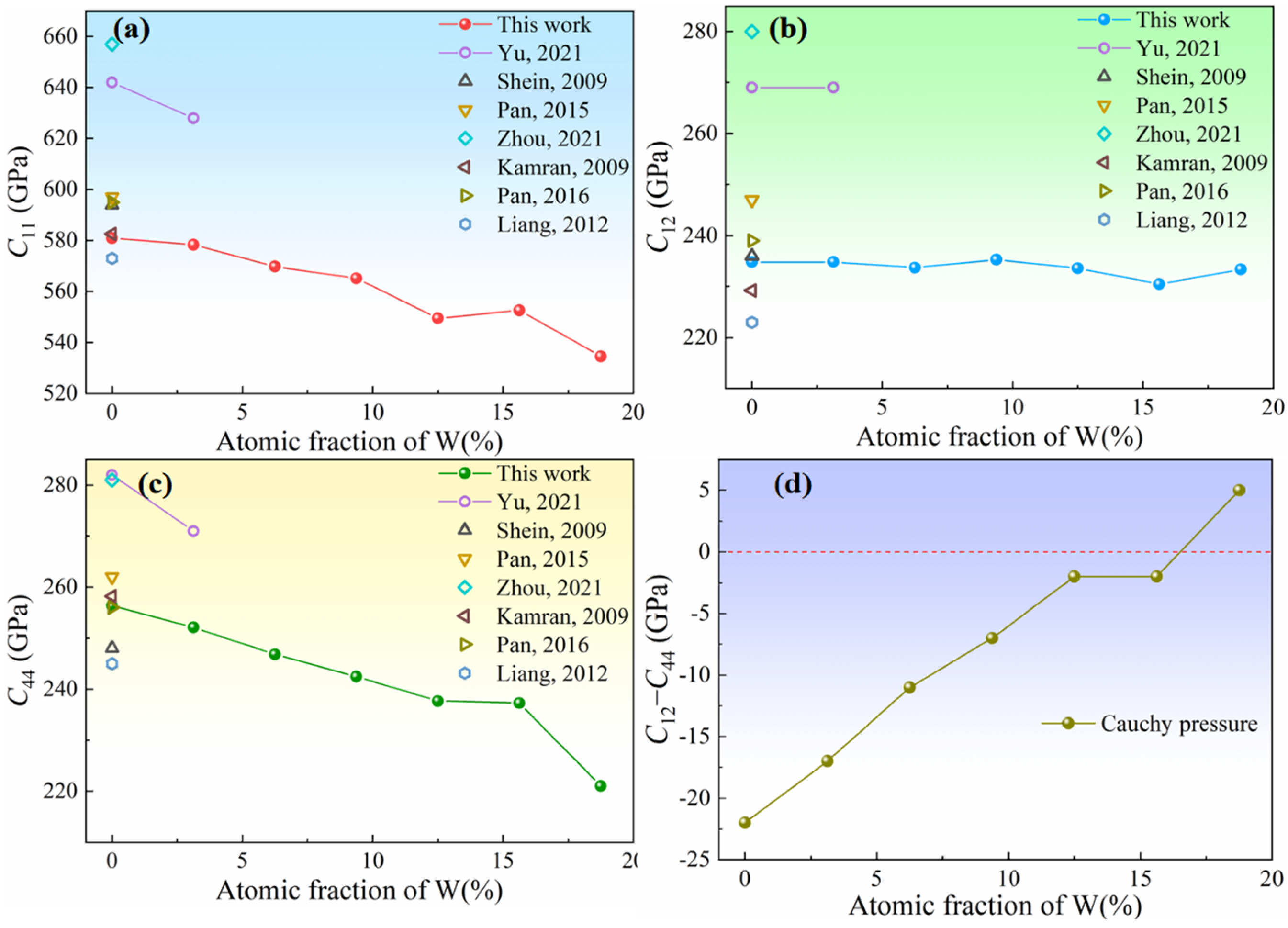
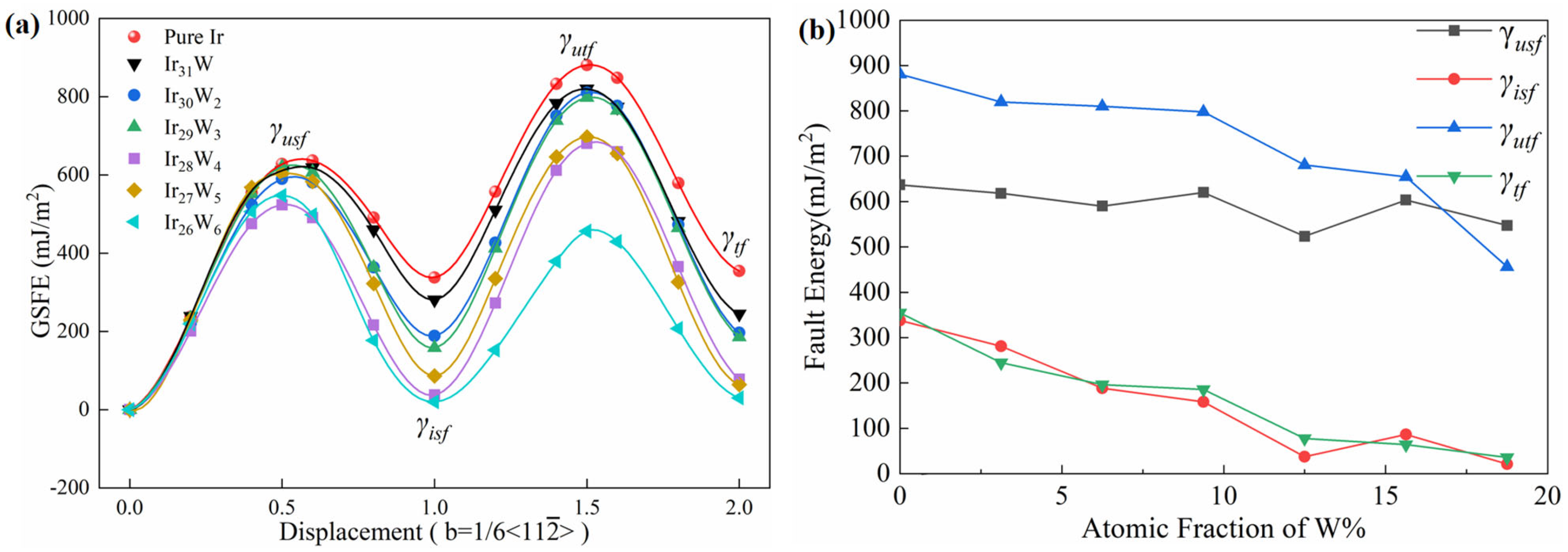
Table 7 summarizes the evolution trends with increasing W concentration from 0 to 18.75 at % for Cauchy pressure, Pugh’s ratio, Poisson’s ratio, stacking fault energy, twinning propensity parameter, and ICOHP values. The above findings, the increasing of Cauchy pressure (
C12−
C44), bulk Pugh’s ratio (
B/
G), and Poisson’s ratio with increasing W concentration, suggest that the toughness of Ir-W solid solution alloys is improved. The addition of W results in a reduction of intrinsic SFE and unstable SFE of Ir. This evolution trend may promote the full dislocations to extended dislocations. This trend is similar to the findings in Co-based alloys reported by Tian et al. [
29]. Alloying of Cr and W leads to a reduction of SFE and facilitates the dissociation of full dislocations into Shockley partials. These behaviors enhance ductility by hindering cross-slip and reducing creep rates. The reduction of unstable SFE also indicates that the energy barrier of slipping decreases [
69]. With increasing the concentration of W, both the TFE (
γtf) and unstable TFE (
γutf) of Ir-W solid solutions decrease, resulting in increasing the twinning propensity parameters (τ
a,
T, and
η), thus improving the twinning propensity of Ir.
Efforts to balance the strength and ductility of a material have been ongoing for centuries. Liu et al. [
73] designed Ti–Zr–Nb–Ta and Ti–Zr–Nb–Mo refractory HEAs balancing strength and ductility through tailoring valence electron concentration by changing the alloying composition. Chen et al. [
74] investigated a CoCuFeNiPd HEA, which undergoes short-range ordering (SRO). The SRO leads to a pseudo-composite microstructure, which surprisingly enhances both the ultimate strength and ductility. Face-center-cubic-preferred clusters enhance strength, while body-center-cubic-preferred clusters improve ductility. Qi et al. [
75] developed single-phase ordered B
2 aluminum-enriched refractory HEAs demonstrating high strength and ductility. It is reported that valence electron count (VEC) defines domains for alloy ductility and brittleness. Strategically avoiding the VEC valley is advised in designing future ductile alloys.
Pei et al. [
76] designed HEAs with enhanced strength and ductility. They highlighted that negative SFE, combined with a
k parameter (ratio of short-ranged interactions between closed-packed planes) with |
k| close to 1/2, promotes the formation of various nanoscale close-packed structures (such as twins, stacking faults, and nano-sized HCP domains). These structures balance strength and ductility by tailoring SFE and atomic interactions. Ren et al. [
77] reported that the alternating FCC and BCC nanolamellae in AlCoCrFeNi eutectic HEAs synergistically enhanced strength and ductility. The present research established that it is theoretically practicable to improve the toughness of Ir by alloying methods with tailoring valence electron concentration to decrease the covalent contribution to chemical bonding. Alloying Ir with W results in the weakening of Ir-Ir covalent properties and the reduction of SFE. The toughening mechanism is consistent with these advanced alloy design concepts.
Shang et al. [
28] stated that SFE in Ni-based alloys decreases with increasing temperature, which may further modulate deformation mechanisms under service conditions. Yang et al. [
78] reported that the ductile–brittle transition temperature of Ir is in the range of 700–800 °C. They attributed low-temperature brittleness (25–700 °C) to high SFE. High SFE raises the activation barrier for dislocation slip, which hinders screw dislocation migration and causes dense dislocation tangles. The 0 K simulation results elucidate the intrinsic properties for the enhancement of Ir toughness by W alloying (SFE and electronic properties). Thermal effects at finite temperatures may improve the toughness of Ir alloys. These effects include lattice parameter changes caused by thermal expansion and elastic modulus softening due to phonons. The evolution of toughness as a function of temperature may also be affected by W. For example, W may influence toughness and the brittle transition temperature. The evolution of elastic properties and SFE of Ir and Ir-W solid solution alloys as a function of temperature will be investigated in future work.
5. Conclusions
In the present research, first-principles calculations were carried out, focusing on the effect of W on the elastic properties, GSFE, electronic structures, and Ir-Ir chemical bonding properties of Ir-W solid solution alloys. The calculated lattice constants, elastic constants, and elastic modulus of Ir, Ir3W (ε′), and IrW (ε′′) are in good agreement with the reported theory and experimental values. The calculated GSFEs of Ir are in good agreement with the reported values. The evolution of lattice constants, formation energies, elastic constants, elastic modulus, GSFE, and electronic structures of Ir-W solid solutions alloys as a function of W concentration was addressed and compared. The main conclusions are summarized as follows.
- (1)
Cauchy pressure (C12−C44) increases from −22 GPa to 5 GPa, and Pugh ratio (B/G) increases from 1.60 to 1.72 with increasing W content from 0 to 18.75 at %. These facts suggest that the toughness of Ir-W alloys is improved.
- (2)
The intrinsic SFE and unstable SFE of Ir alloy decrease with increasing W concentration. It is reasonable to predict that the reduction of SFE accompanies the lowering of the slipping barrier, which is responsible for the improvement of the material’s plastic deformation capability.
- (3)
The Ir-Ir bonds exhibit both metallic and covalent natures. The covalent nature of Ir-Ir bonds is weakened with the increasing of W concentration, evidenced by the growing ICOHP values from −0.8512 eV to −0.7923 eV and the reduction of common shared electrons between neighboring Ir-Ir atoms.
The weakening covalent character of Ir-Ir bonds through W alloying results in a decrease in stacking fault energy and lowering the energy barrier for sliding. It is a feasible approach to improve the toughness of Ir and mitigate its inherent brittleness by alloying methods. Further research, including both theoretical calculations and experimental studies, is strongly desired to design and develop high-temperature Ir-based alloys with higher toughness and strength.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}