

Derivatization of PVA into Polyols Suitable for Fabrication of Rigid Polyurethane Foams—Preliminary Studies and Perspectives

Abstract

1. Introduction
2. Experimental Section
2.1. Materials
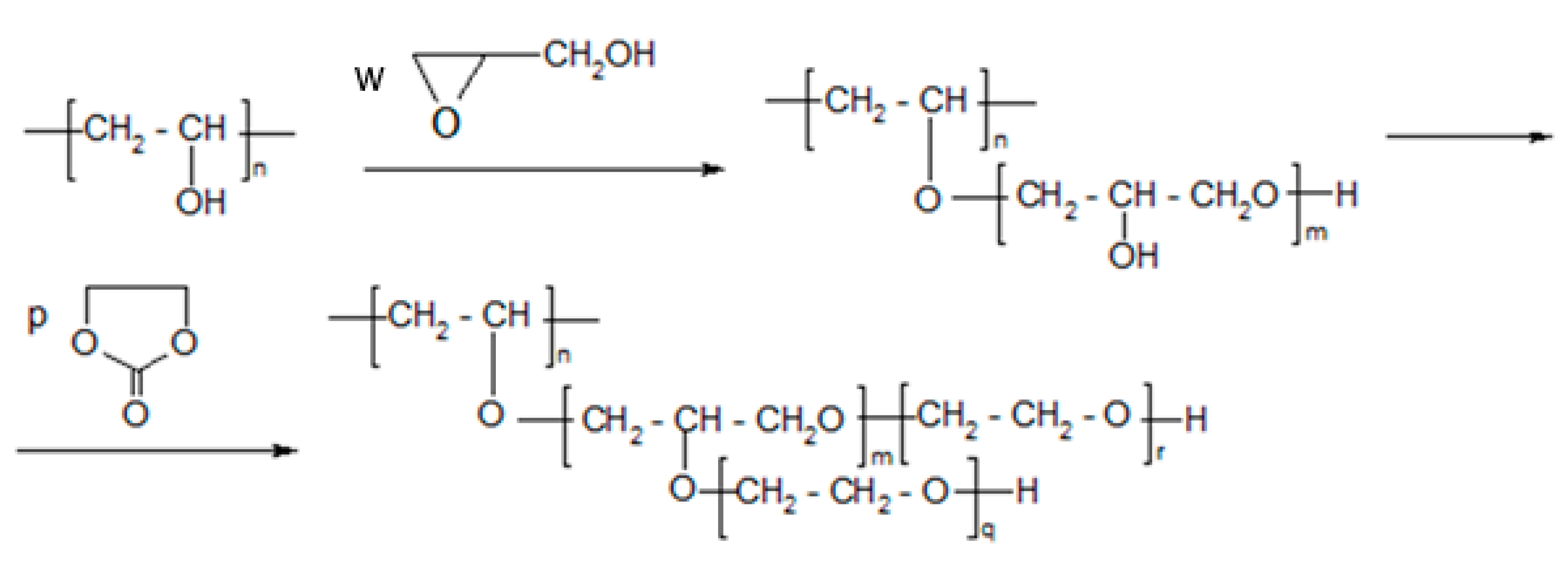
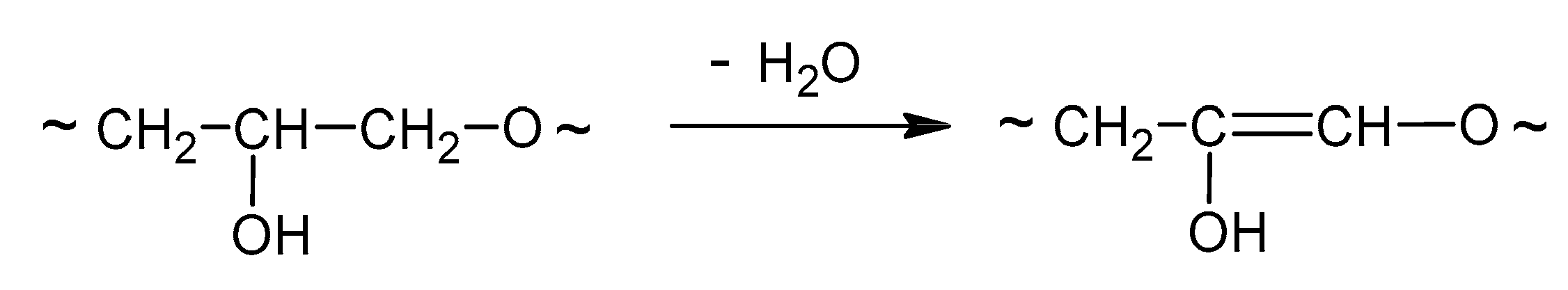
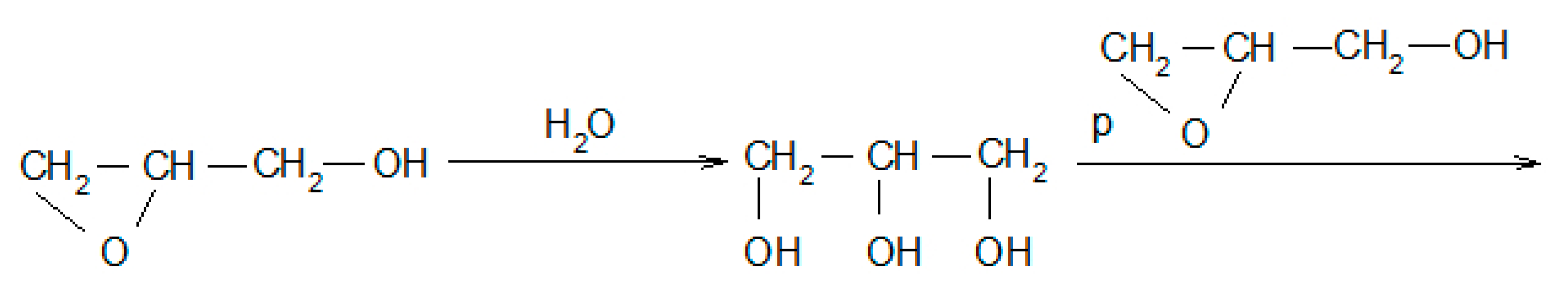
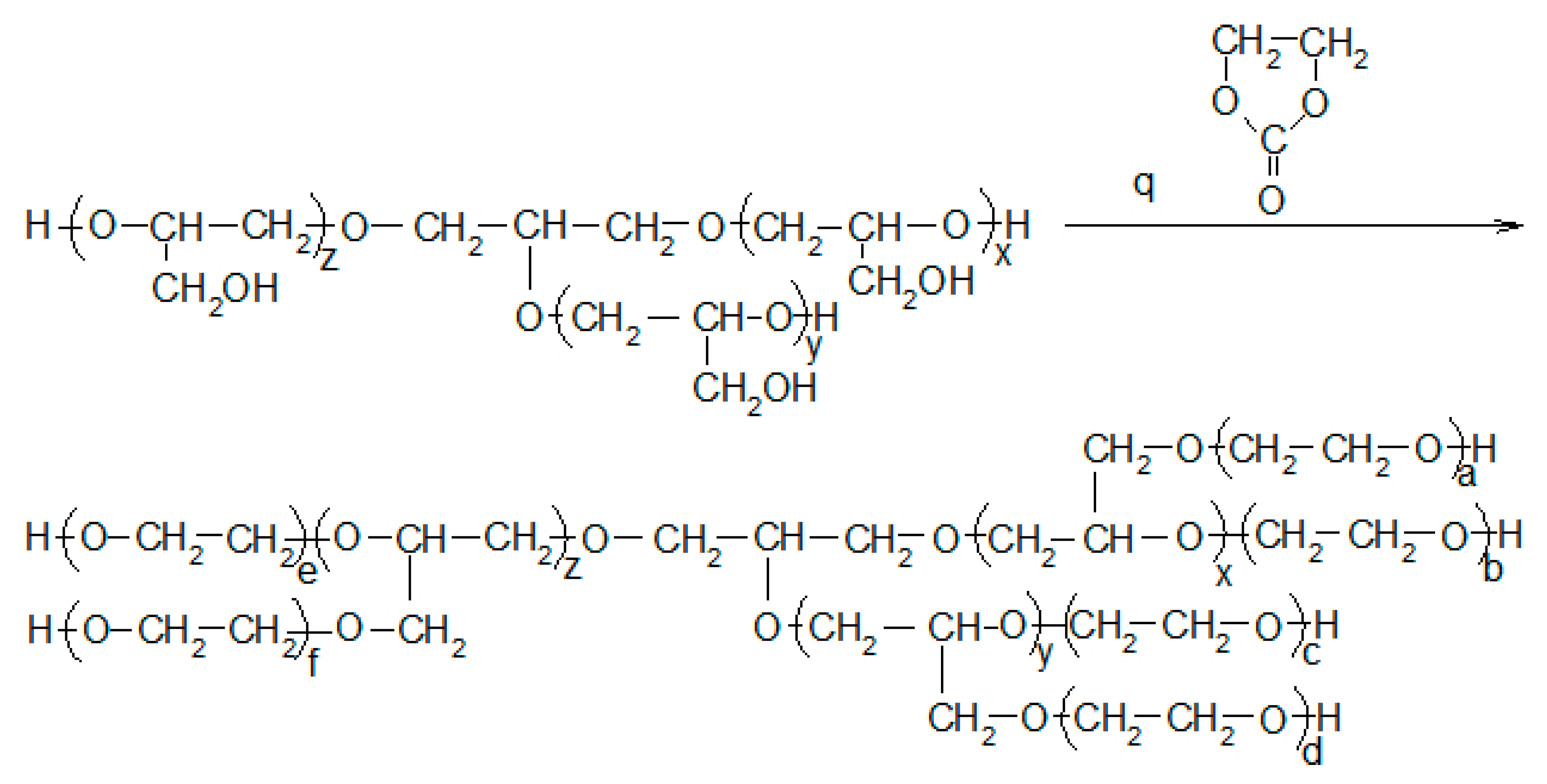
2.2. Synthesis of Polyols
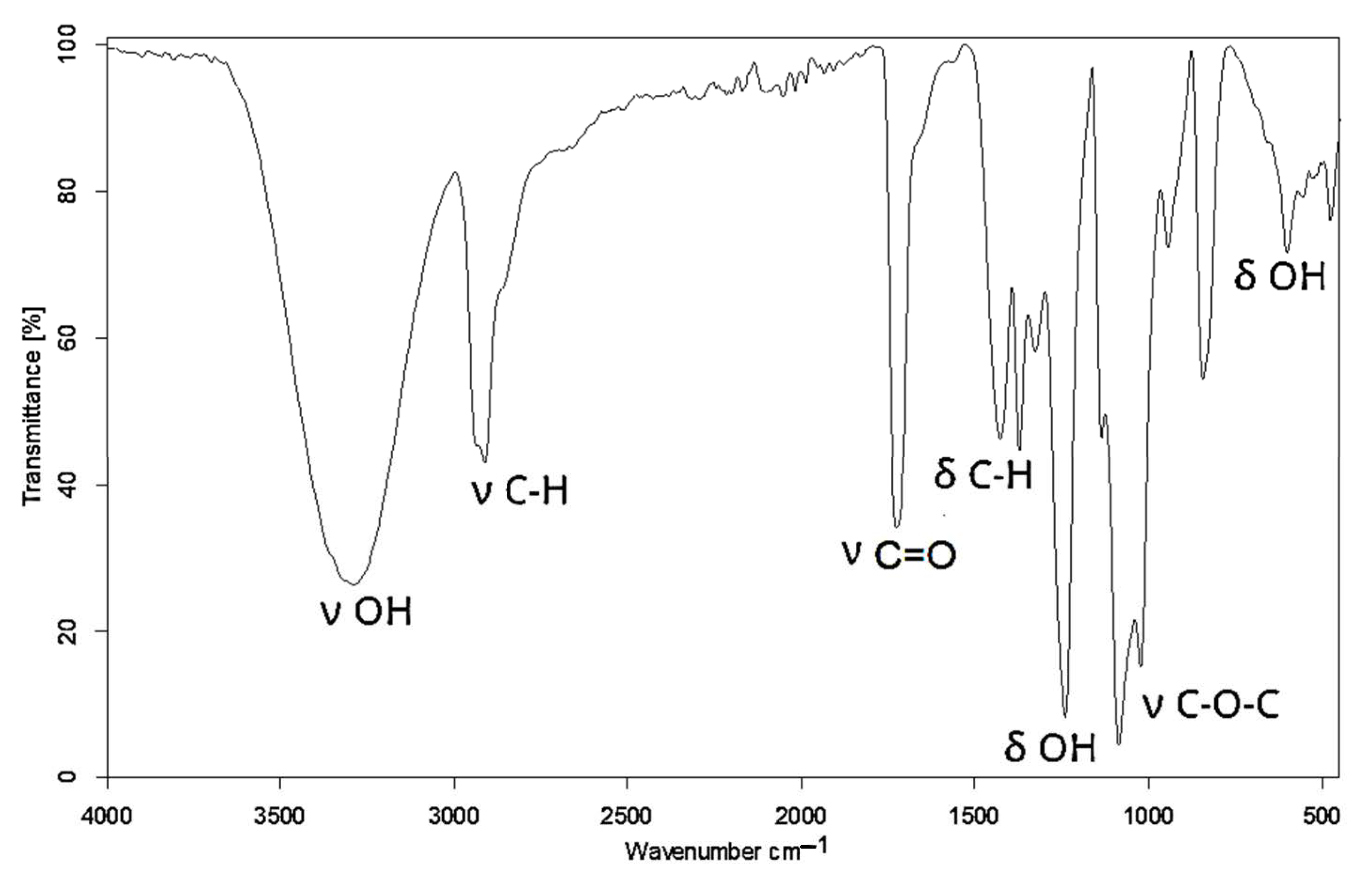
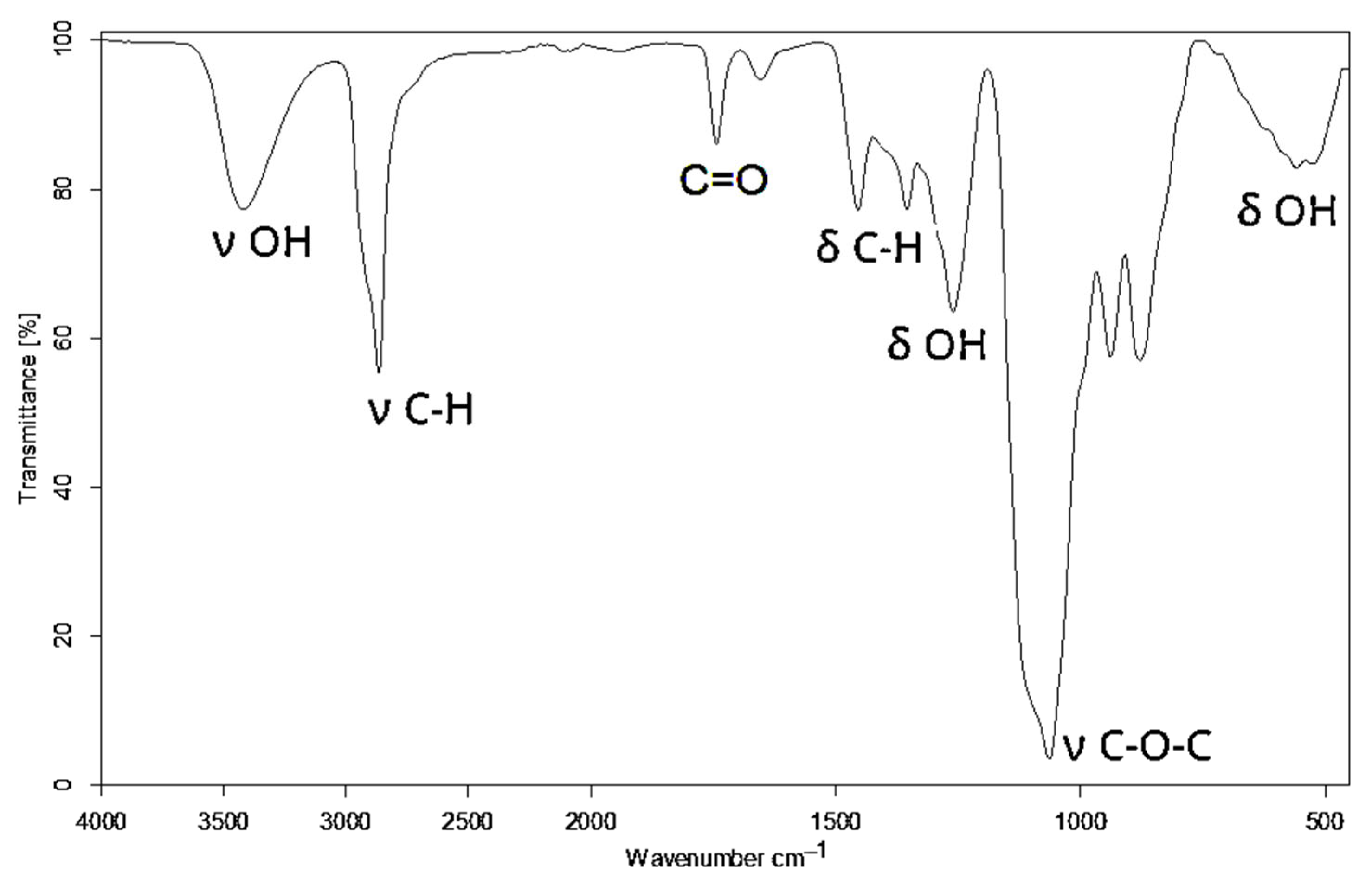
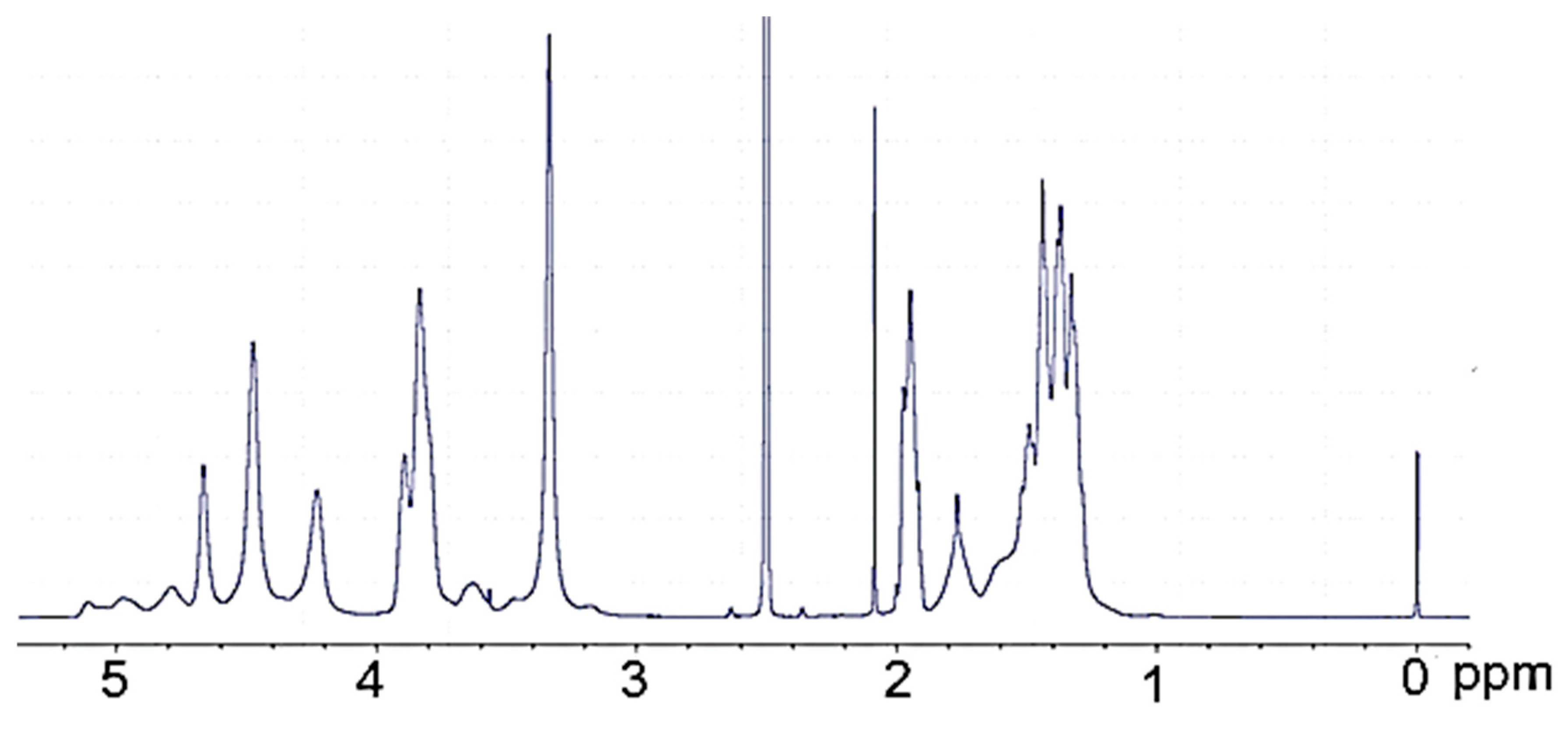
2.3. Analytical Methods
2.4. Physical Properties of Polyols
2.5. Obtaining the Polyurethane Foams
2.6. Properties of Foams
2.7. Biodegradation of Foams
3. Results and Discussion
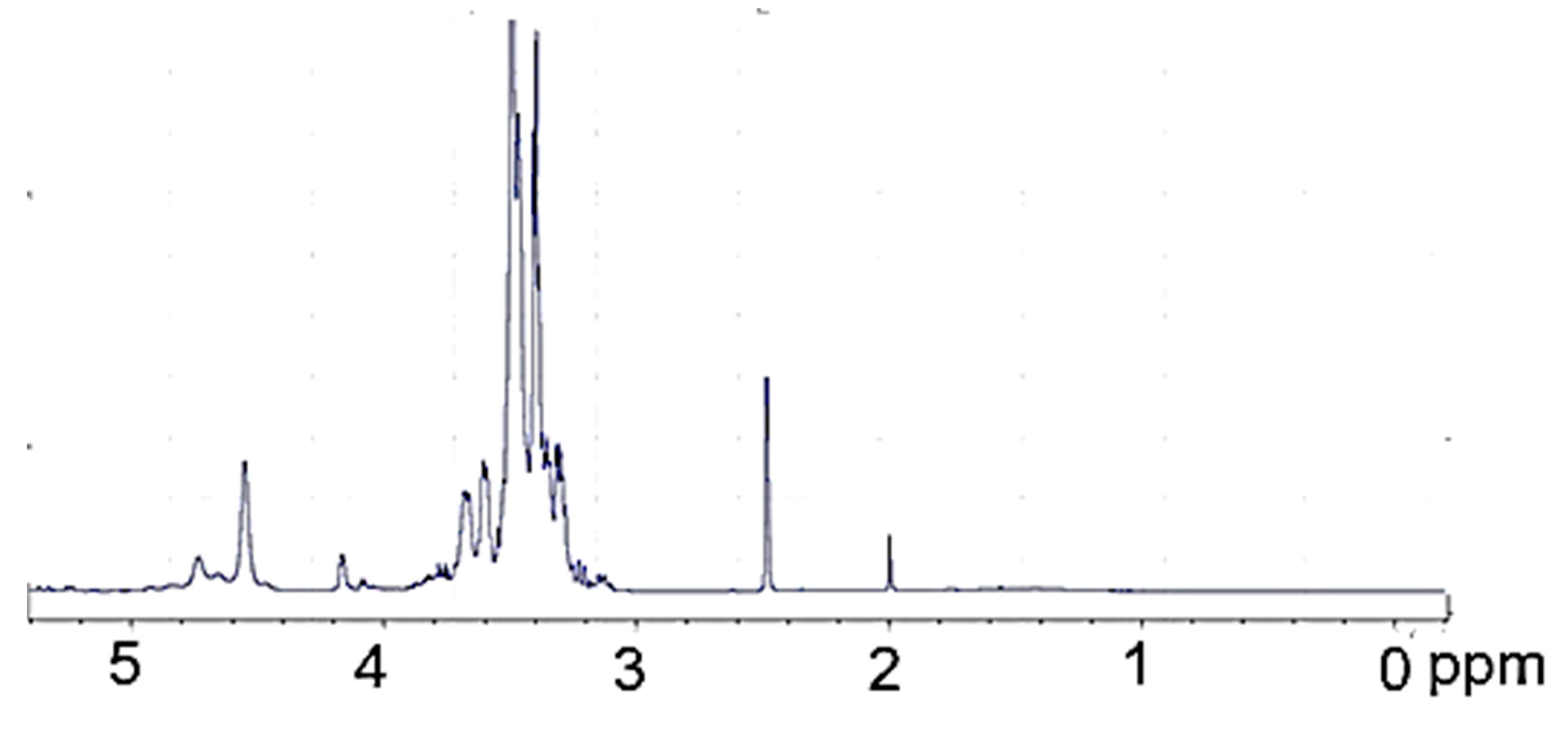
3.1. Synthesis and Properties of Polyol
3.2. Polyurethane Foam
4. Summary and Conclusions
- A new method for the synthesis of polyol from PVA, which is suitable for obtaining rigid polyurethane foams, was elaborated.
- The density, viscosity, and surface tension of the obtained polyol were determined. The average chemical structure of the polyol was postulated together with the well-defined admixture of low-molecular-weight products of the oxyalkylation of glycidol with ethylene carbonate and oligomers of glycidol.
- The foams obtained from PVA-derived polyol exhibited properties typical of rigid polyurethane foams, but their thermal resistance was enhanced. The obtained PUFs can thus be exploited for a long time at a temperature of 150 °C.
- The obtained PUFs are ecologically friendly. Powdered foam degrades completely, while a cube of PUF is degraded by up to 54.6% following 28 days of exposure to soil, based upon biological oxygen demand measurements.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pizzi, A.; Mittal, K.L. Handbook of Adhesive Technology, Revised and Expanded, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Kim, N.; Sudol, E.D.; Dimonie, V.L.; Aasser, M.S.E. Poly (vinyl alcohol) Stabilization of Acrylic Emulsion Polymers Using the Miniemulsion Approach. Macromolecules 2003, 36, 5573–5579. [Google Scholar] [CrossRef]
- Baker, M.I.; Walsh, S.P.; Schwartz, Z.; Boyan, B.D. A review of polyvinyl alcohol and its uses in cartilage and orthopedic applications. J. Biomed. Mater. Res. B Appl. Biomater. 2012, 100, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Paradossi, G.; Cavalieri, F.; Chiessi, E. Poly (vinyl alcohol) as versatile biomaterial for potential biomedical applications. J. Mater. Sci. Mater. Med. 2003, 14, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Ebnesajjad, S.; Modjarrad, K. Handbook of Polymer Applications in Medicine and Medical Devices (Plastics Design Library), 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Goodship, V.; Jacobs, D.K. Polyvinyl Alcohol: Materials, Processing and Applications; Rapra review reports; Smithers Rapra Technology: Shrewsbury, UK, 2009. [Google Scholar]
- Rabek, J.F. Polymers, Preparation, Research Methods, Application; Wydawnictwo Naukowe PWN: Warszawa, Poland, 2013. [Google Scholar]
- Cohen, S.G.; Haas, H.C.; Slotnick, H. Studies on Hydroxyethylpolyvinyl alcohol. J. Polymer Sci. 1953, 11, 193–2019. [Google Scholar] [CrossRef]
- Inskip, H.K.; Klabunde, W. Hydroxyethylation of Polyvinyl Alcohol. U.S. Patent 2990398, 27 June 1961. [Google Scholar]
- Scardiglia, F.; Heights, A.; Ranky, W.O.; Knaggs, E.A. Process for Etoxylating Polyvinyl Alkohol. U.S. Patent 3099646, 30 July 1963. [Google Scholar]
- Milne, J.N.; Bois, C. Reaction Product of an Alkylene Oxide-Polyvinyl Alkohol Mixture with Untreated Polyvinyl Alkohol. U.S. Patent 3106543, 1963. [Google Scholar]
- Halpern, B.D.; Krueger, B.O. Alkoxylated Polyvinyl Alkohol. U.S. Patent 3052652, 1962. [Google Scholar]
- Arno, H.; Perplies, E. Verfahren zur Herstellung von quellfaehigen, vernetzen Ethern des Polyvinylalkohol und deren Verwendung. European Patent 0021131, 1980. [Google Scholar]
- Shui, T.; Chae, M.; Bressler, D.C. Cross-Linking of Thermally Hydrolyzed Specified Risk Materials with Epoxidized Poly (Vinyl Alcohol) for Tackifier Applications. Coating 2020, 10, 630. [Google Scholar] [CrossRef]
- Carlotti, S.J.; Giani-Beaune, O.; Schue, F. Water-Soluble Poly (vinyl alcohol) Grafted with Propylene Oxide and Epichlorohydrin: Characterization, Mechanical Properties, and Model Reactions. J. Appl. Polymer Sci. 2001, 81, 2868–2874. [Google Scholar] [CrossRef]
- Clemens, J.H. Reactive Applications of Cyclic Alkylene Carbonates. Ind. Eng. Chem. Res. 2003, 42, 663–674. [Google Scholar] [CrossRef]
- Wang, X.L.; Du, F.G.; Meng, Y.Z.; Li, R.K.Y. Novel in situ crosslinking reaction of ethylene-vinyl alcohol copolymers by propylene carbonate. Mater. Lett. 2006, 60, 509–513. [Google Scholar] [CrossRef]
- Brojer, Z.; Hertz, Z.; Penczek, P. Epoxy Resins; WNT: Warsaw, Poland, 1972. [Google Scholar]
- Kijowska, D.; Wołowiec, S.; Lubczak, J. Kinetics and mechanism of initial steps of synthesis of polyetherols from melamine and ethylene carbonate. J. Appl. Polym. Sci. 2004, 93, 294–300. [Google Scholar] [CrossRef]
- PN-93/C-89052.03; Polyethers for Polyurethanes. Test Methods. Determination of the Hydroxyl Number. Polish Committee for Standardization: Warsaw, Poland, 1993.
- Misiorek, M.; Sekuła, J.; Ruman, T. Mass Spectrometry Imaging of low Molecular Weight Compounds in Garlic (Allium sativum L.) with Gold Nanoparticle Enhanced Target. Phytochem. Anal. 2017, 28, 479–486. [Google Scholar] [CrossRef]
- PN-EN ISO 845-2000; Cellular Plastics and Rubbers. Determination of Apparent (Bulk) Density. Polish Committee for Standardization: Warsaw, Poland, 2000.
- PN-EN ISO 2896-1986; Cellular Plastics, Rigid. Determination of Water Absorption. Polish Committee for Standardization: Warsaw, Poland, 1986.
- PN-EN ISO 2796-1986; Cellular Plastics, Rigid. Test of Dimensional Stability. Polish Committee for Standardization: Warsaw, Poland, 1986.
- PN-EN ISO 844-1978; Cellular Plastics, Compression Test for Rigid Materials. Polish Committee for Standardization: Warsaw, Poland, 1978.
- ISO 17556-2019; Plastics—Determination of the Ultimate Aerobic Biodegradability of Plastic Materials in Soil by Measuring the Oxygen Demand in a Respirometer or the Amount of Carbon Dioxide Evolved. International Organization for Standardization: Geneva, Switzerland, 2019.
- ISO 11274-2019; Soil Quality—Determination of the Water-Retention Characteristic—Laboratory Methods. International Organization for Standardization: Geneva, Switzerland, 2019.
- ISO 10390-2005; Soil Quality—Determination of pH. International Organization for Standardization: Geneva, Switzerland, 2025.
- Lubczak, J.; Lubczak, R. Method of Producing Polyols. PL Patent 448709, 29 May 2024. [Google Scholar]
- Czupryński, B. Topics in the Chemistry and Technology of Polyurethanes; Bydgoszcz Academy Publishing House: Bydgoszcz, Poland, 2004. [Google Scholar]
- Wirpsza, Z. Polyurethanes: Chemistry, Technology, Application; WNT: Warsaw, Poland, 1991. [Google Scholar]
- Szpiłyk, M.; Lubczak, R.; Lubczak, J. Cellulose-Based Polyurethane Foams of Low Flammability. Polymers 2024, 16, 1438. [Google Scholar] [CrossRef]
- Szpiłyk, M.; Lubczak, R.; Lubczak, J. The biodegradable cellulose-derived polyol and polyurethane foam. Polymer Test. 2021, 100, 107250. [Google Scholar] [CrossRef]
- Szpiłyk, M.; Lubczak, R.; Walczak, M.; Lubczak, J. Polyol and polyurethane foam from cellulo hydrolysate. J. Chem. Technol. Biotechnol. 2021, 96, 881–889. [Google Scholar] [CrossRef]
- Chmiel, E.; Lubczak, J. Synthesis of oligoetherols from mixtures of melamine and boric acid and polyurethane foams formed from these oligoetherols. Polymer Bull. 2019, 76, 2253–2275. [Google Scholar] [CrossRef]
- Strzałka, A.; Debska, B.; Lubczak, J. Polyols and polyurethane foams based on chitosans of various molecular weights. J. Appl. Polym. Sci. 2024, 141, e55393. [Google Scholar] [CrossRef]
- McHenry, E.; Piper, E. High-Strength Carbon Foams. U.S. Patent 3387940, 1968. [Google Scholar]
- Lubczak, J.; Chmiel-Szukiewicz, E.; Duliban, J.; Głowacz Czerwonka, D.; Lubczak, R.; Łuksiewicz, B.; Zarzyka, I.; Łodyga, A.; Tynski, P.; Minda-Data, D.; et al. Polyurethane foams with 1,3,5-triazine ring of improved thermal stability. Przem. Chem. 2014, 10, 1690–1697. [Google Scholar]
- Jiao, L.; Xiao, H.; Wang, Q.; Sun, J. Thermal degradation characteristics of rigid polyure-thane foam and the volatile products analysis with TG-FTIR-MS. Polym. Degrad. Stab. 2013, 98, 2687–2696. [Google Scholar] [CrossRef]
- Ketata, N.; Sanglar, C.; Waton, H.; Alamercery, S.; Delolme, F.; Raffin, G.; Grenier-Loustalot, M.F. Thermal Degradation of Polyurethane Bicomponent Systems in Controlled Atmospheres. Polym. Polym. Compos. 2005, 13, 1–26. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Signal Position m/z | Relative Intensity of Signal [%] | Molecular Ion Structure | Calc. Molecular Weight [g/mol] |
|---|---|---|---|---|
| 1 | 113.133 | 8.79 | GL + K+ | 113.000 |
| 2 | 196.994 | 15.93 | H2O + 2GL + CH3OH | 198.110 |
| 3 | 212.897 | 20.61 | H2O + 2GL + OE + H+ | 211.118 |
| 4 | 236.921 | 5.39 | 2GL + 2OE + H+ | 237.134 |
| 5 | 274.928 | 9.78 | 2GL + 2OE + K+ | 275.090 |
| 6 | 291.107 | 3.56 | H2O + GL + 4OE + Na+ | 291.142 |
| 7 | 307.085 | 8.63 | H2O + 3GL + OE + Na+ | 307.137 |
| 8 | 335.134 | 26.05 | 3GL + 2OE + Na+ | 333.153 |
| 9 | 351.096 | 80.39 | H2O + 3GL + 2OE + Na+ | 351.163 |
| 10 | 363.097 | 19.87 | 4GL + OE + Na+ | 363.163 |
| 11 | 379.163 | 23.67 | 4G +OE + K+ | 379.137 |
| 12 | 393.928 | 89.51 | 2GL + 6OE-H2O | 394.220 |
| 13 | 395.126 | 90.69 | 4 GL + OE + CH3OH + Na+ | 395.189 |
| 14 | 407.130 | 18.62 | 2GL + 5OE + K+ | 407.168 |
| 15 | 409.153 | 16.40 | 4GL + 3OE-H2O + H+ | 411.223 |
| 16 | 421.143 | 10.75 | GL + 7OE + K+ | 421.184 |
| 17 | 423.165 | 10.30 | 4GL + 2OE + K+ | 423.520 |
| 18 | 425.119 | 18.54 | 5 GL + CH3OH + Na+ | 425.200 |
| 19 | 437.121 | 20.19 | 3GL + 4OE + K+ | 437.179 |
| 20 | 439.144 | 40.74 | 4 GL + 2OE + CH3OH + Na+ | 439.179 |
| 21 | 451.165 | 5.36 | 2GL + 6OE + K+ | 451.195 |
| 22 | 465.158 | 5.65 | GL + 8OE + K | 465.598 |
| 23 | 469.147 | 17.00 | 5 GL + OE + CH3OH + Na+ | 469.226 |
| 24 | 481.149 | 11.43 | 3GL + 5OE + K+ | 481.205 |
| 25 | 495.170 | 3.40 | 2GL + 7OE + K+ | 495.221 |
| 26 | 497.197 | 4.58 | 6 GL + CH3OH + Na+ | 499.237 |
| 27 | 590.901 | 100 | H2O + 6GL + 2OE + K+ 7GL + 2OE-H2O + H+ | 589.247589.307 |
| Composition [g/100 g of Polyol] | Isocyanate Index | Foaming Process | Product Characterization | |||||
|---|---|---|---|---|---|---|---|---|
| pMDI | Water | Silicon L-6900 | TEA | Cream Time [s] | Rise Time [s] | Tack-Free Time [s] | ||
| 100 | 2 | 2.9 | 2.0 | 0.8 | 10 | 5 | 10 | Low foaming of foam |
| 115 | 2 | 2.9 | 2.0 | 1.0 | 10 | 6 | 8 | Viscous surface of foam |
| 125 | 2 | 2.9 | 2.0 | 1.1 | 10 | 7 | Immediate | Rigid foam, irregular pores |
| 135 | 2 | 2.9 | 2.0 | 1.2 | 13 | 8 | 5 | Rigid foam, irregular pores |
| 125 | 2 | 2.9 | 1.1 | 1.1 | 20 | 10 | Immediate | Rigid foam, regular pores |
| 125 | 2 | 2.9 | 0.9 | 1.1 | 21 | 12 | Immediate | Rigid foam, regular pores |
| 125 | 2 | 2,9 | 0.8 | 1.1 | 25 | 12 | Immediate | Rigid foam, regular pores |
| 125 | 2 | 2.9 | 0.7 | 1.1 | 25 | 12 | 1 | Rigid foam, regular pores |
| 125 | 2 | 2.9 | 0.4 | 1.1 | 30 | 15 | 2 | Rigid foam, regular pores |
| 120 | 3 | 2.9 | 2.0 | 0.9 | 8 | 7 | Immediate | Viscous surface of foam |
| 135 | 3 | 2.9 | 2.0 | 1.1 | 10 | 8 | Immediate | Rigid foam, regular pores |
| 145 | 3 | 2.9 | 2.0 | 1.2 | 12 | 10 | 2 | Rigid foam, irregular pores |
| 170 | 3 | 2.9 | 2.0 | 1.4 | 13 | 11 | 6 | Viscous surface of foam |
| 135 | 3 | 2.9 | 1.0 | 1.1 | 20 | 5 | Immediate | Rigid foam, regular pores |
| 135 | 3 | 2.9 | 0.4 | 1.1 | 25 | 25 | 1 | Rigid foam, regular pores |
| 135 | 3 | 2.9 | 0.3 | 1.1 | 30 | 28 | 1 | Rigid foam, regular pores |
| 135 | 3 | 2.9 | 0.1 | 1.1 | 35 | 35 | 1 | Semi-rigid foam |
| Amount of Water/100 g of Polyol | Density [kg/m3] | Absorption of Water [wt%] | Heat Conductance Coefficient [W/m·K] | Mass Loss [wt%] After One Month Exposure at Temperature | Compressive Strength [MPa] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 min | 3 h | 24 h | 150 °C | 175 °C | Before Exposure | After Exposure at Temperature | ||||
| 150 °C | 175 °C | |||||||||
| 2 | 76.0 | 0.36 | 1.09 | 1.99 | 0.0328 ± 0.0006 | 15.75 | 35.85 | 0.361 | 0.431 | 0.510 |
| 3 | 56.1 | 0.50 | 1.59 | 2.15 | 0.0388 ± 0.0009 | 18.62 | 38.72 | 0.259 | 0.330 | 0.554 |
| Amount of Water/100 g of Polyol | Length Change | Width Change | Height Change | |||
|---|---|---|---|---|---|---|
| 20 h | 40 h | 20 h | 40 h | 20 h | 40 h | |
| 2% | −1.44 | −2.05 | −1.36 | −1.86 | −1.24 | −1.80 |
| 3% | −2.34 | −3.52 | −3.23 | −3.35 | −1.98 | −2.92 |
| Amount of Water/100 g of Polyol | Larger Diameter [µm] | Smaller Diameter [µm] | Thickness of Cell Wall |
|---|---|---|---|
| 2 | 290 ± 38 | 228 ± 34 | 18 ± 3 |
| 3 | 197 ± 47 | 169 ± 23 | 9 ± 2 |
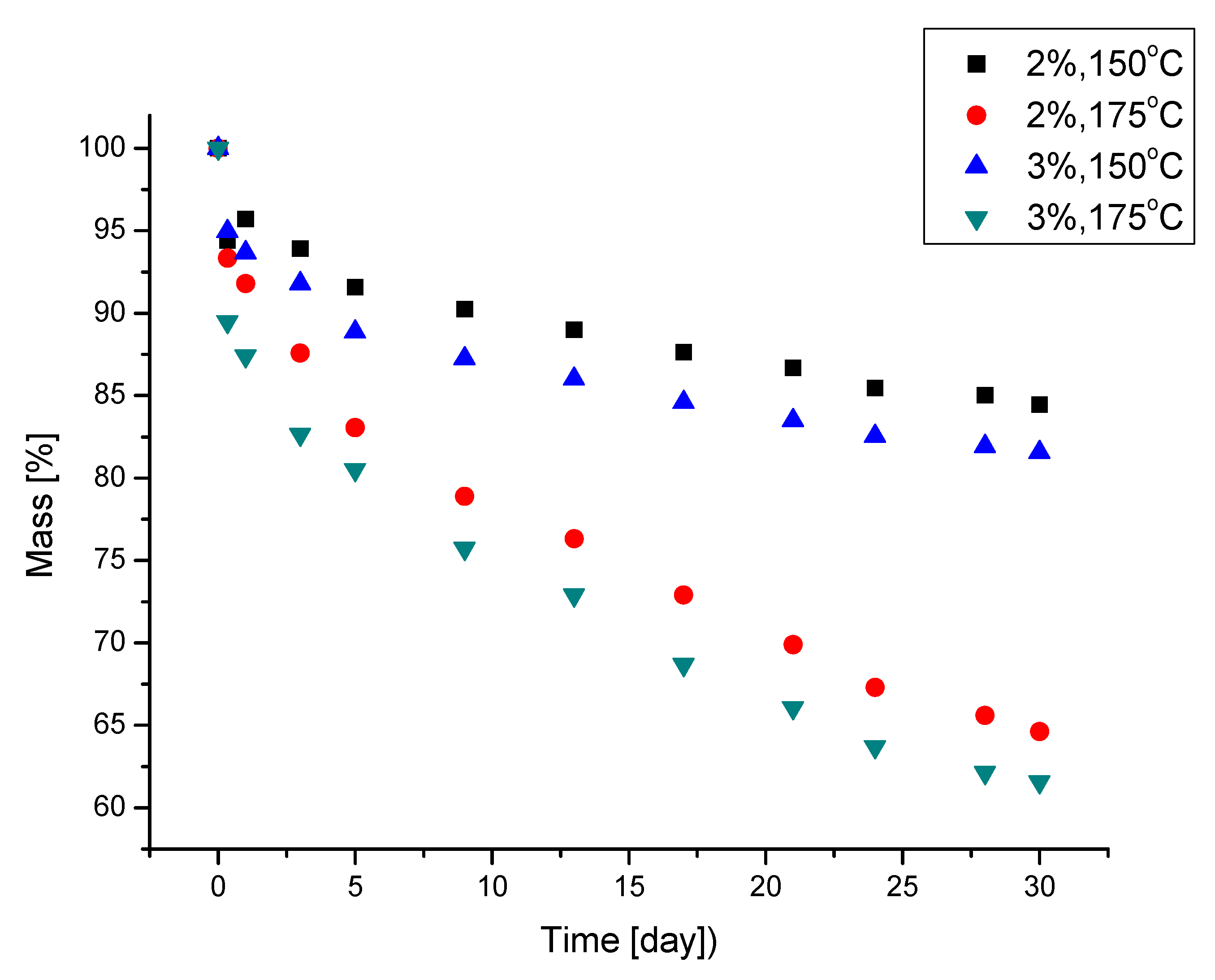
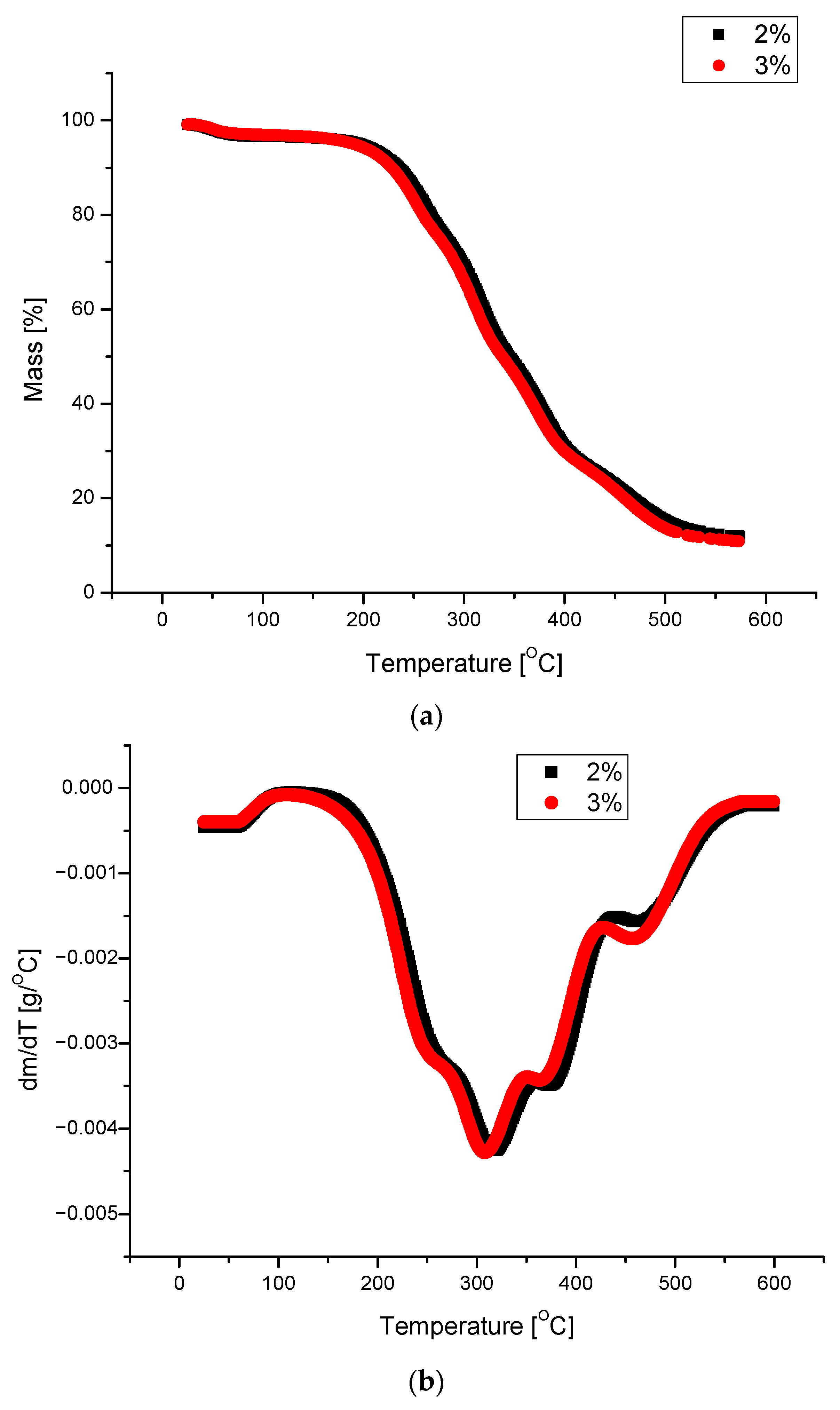
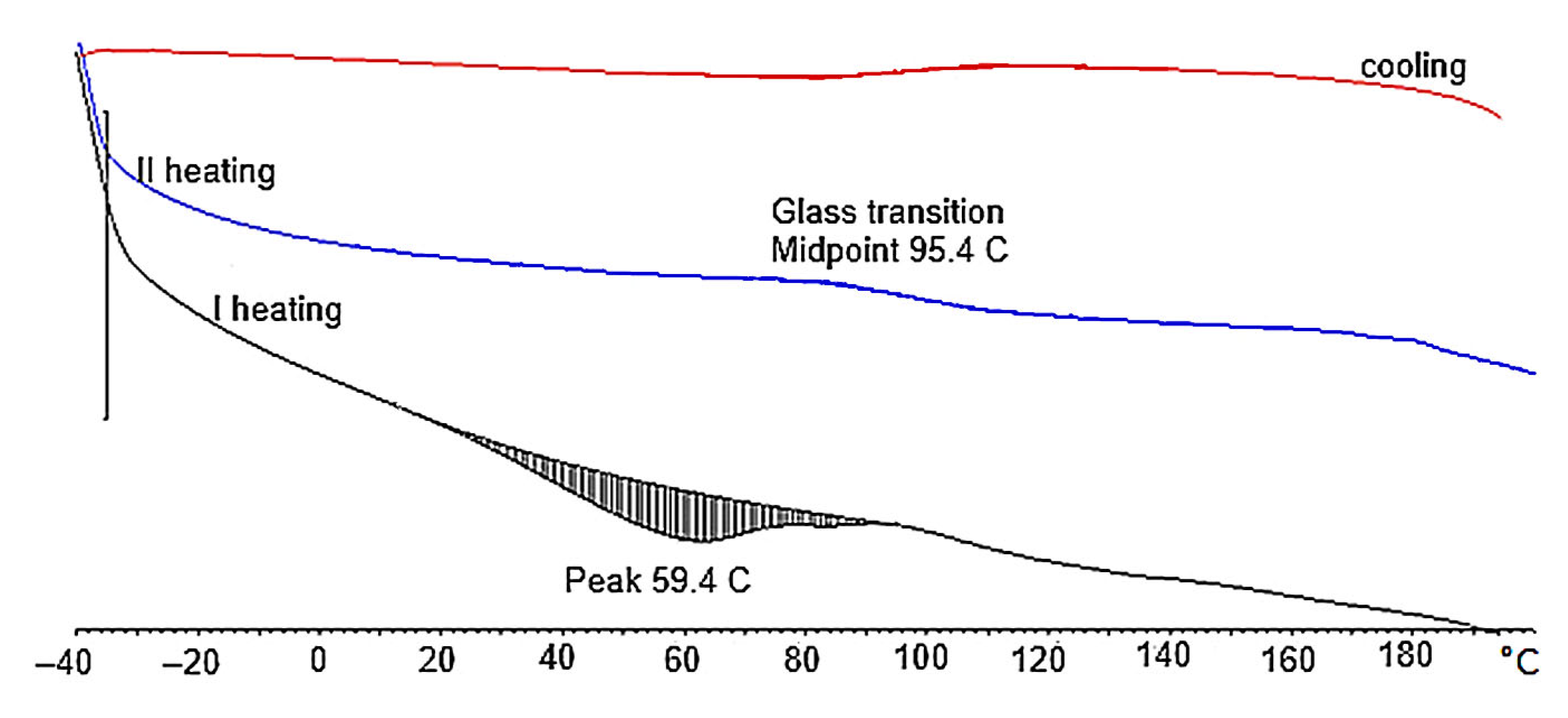
| Amount of Water/100 g of Polyol | T5% [°C] | T10% [°C] | T25% [°C] | T50% [°C] | Tmax of Decomposition [°C] | Tg |
|---|---|---|---|---|---|---|
| 2 | 197 | 235 | 280 | 346 | 320 | 95.5 |
| 3 | 190 | 228 | 277 | 338 | 310 | 92.5 |
| Element | |||
|---|---|---|---|
| C | H | O | N |
| 0.6236 | 0.0628 | 0.2484 | 0.2484 |
| Sample | BODX | BOD28 | Sample Mass [g] | TOD Counts | TOD [mg/dm3] | Dt [%] |
|---|---|---|---|---|---|---|
| Foam—cube | 87.4 | 28.7 | 0.20 | 10.5216 | 52.61 | 54.6 |
| Foam—powder | 1590 | 100.3 | 0.20 | 10.5216 | 52.61 | 100.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lubczak, J. Derivatization of PVA into Polyols Suitable for Fabrication of Rigid Polyurethane Foams—Preliminary Studies and Perspectives. Materials 2025, 18, 2780. https://doi.org/10.3390/ma18122780
Lubczak J. Derivatization of PVA into Polyols Suitable for Fabrication of Rigid Polyurethane Foams—Preliminary Studies and Perspectives. Materials. 2025; 18(12):2780. https://doi.org/10.3390/ma18122780
Chicago/Turabian StyleLubczak, Jacek. 2025. "Derivatization of PVA into Polyols Suitable for Fabrication of Rigid Polyurethane Foams—Preliminary Studies and Perspectives" Materials 18, no. 12: 2780. https://doi.org/10.3390/ma18122780
APA StyleLubczak, J. (2025). Derivatization of PVA into Polyols Suitable for Fabrication of Rigid Polyurethane Foams—Preliminary Studies and Perspectives. Materials, 18(12), 2780. https://doi.org/10.3390/ma18122780