

Graphene Oxide-Anchored Cu–Co Catalysts for Efficient Electrochemical Nitrate Reduction

 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Catalyst Preparation
2.1. Chemical Reagent
2.2. Preparation of Functional RGO Precursor
2.3. Preparation of CuxCox/RGO
3. Experimental Section
3.1. Material Characterization
3.2. In Situ FTIR Test
3.3. Electrochemical Test
3.4. Method for Detection of Substance Concentration
3.5. Relevant Formulas for Calculation
4. Results and Discussion
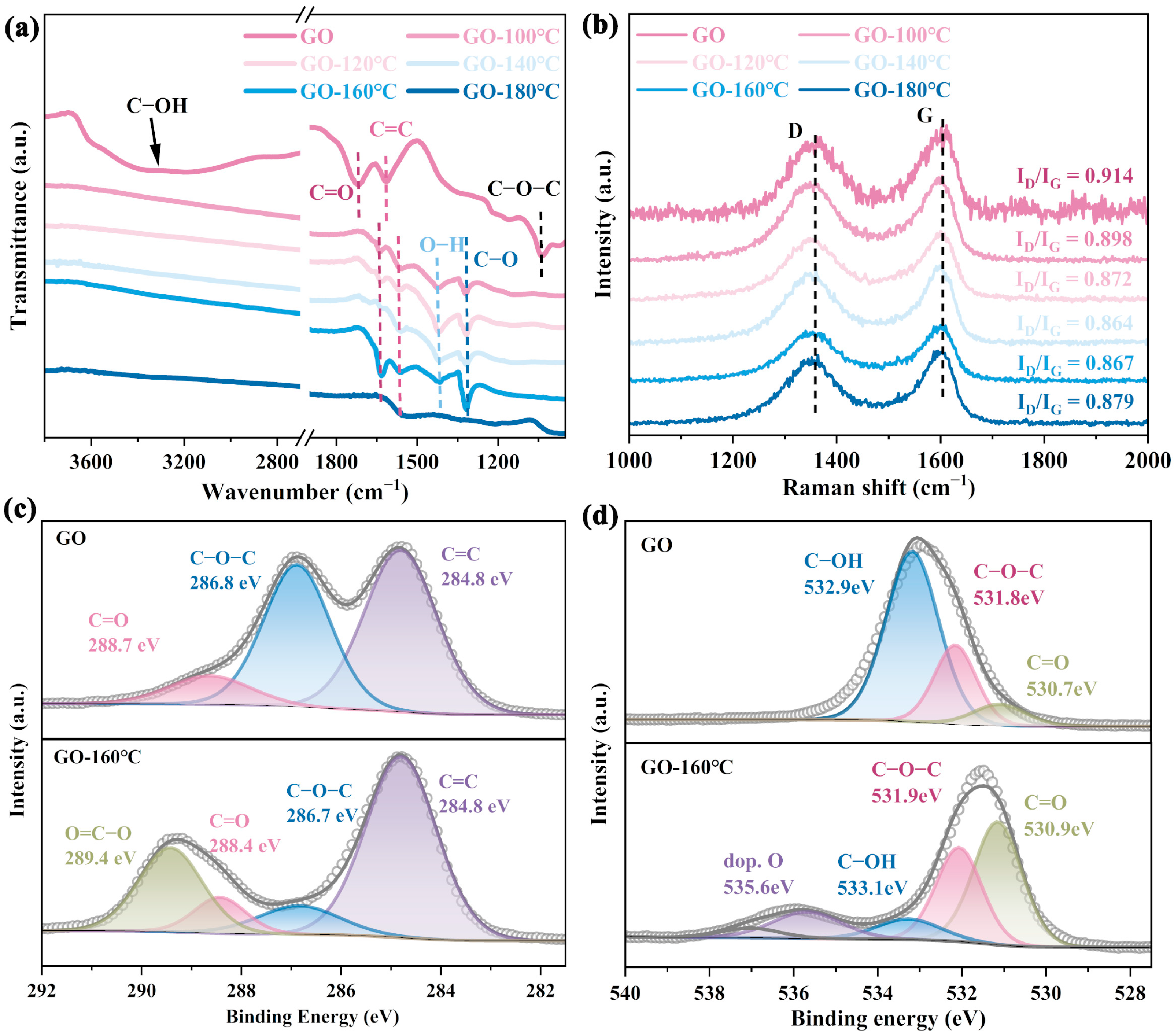
4.1. Regulation of Surface Oxygen-Containing Functional Groups on GO
4.2. Preparation and Characterization of CuxCox/RGO
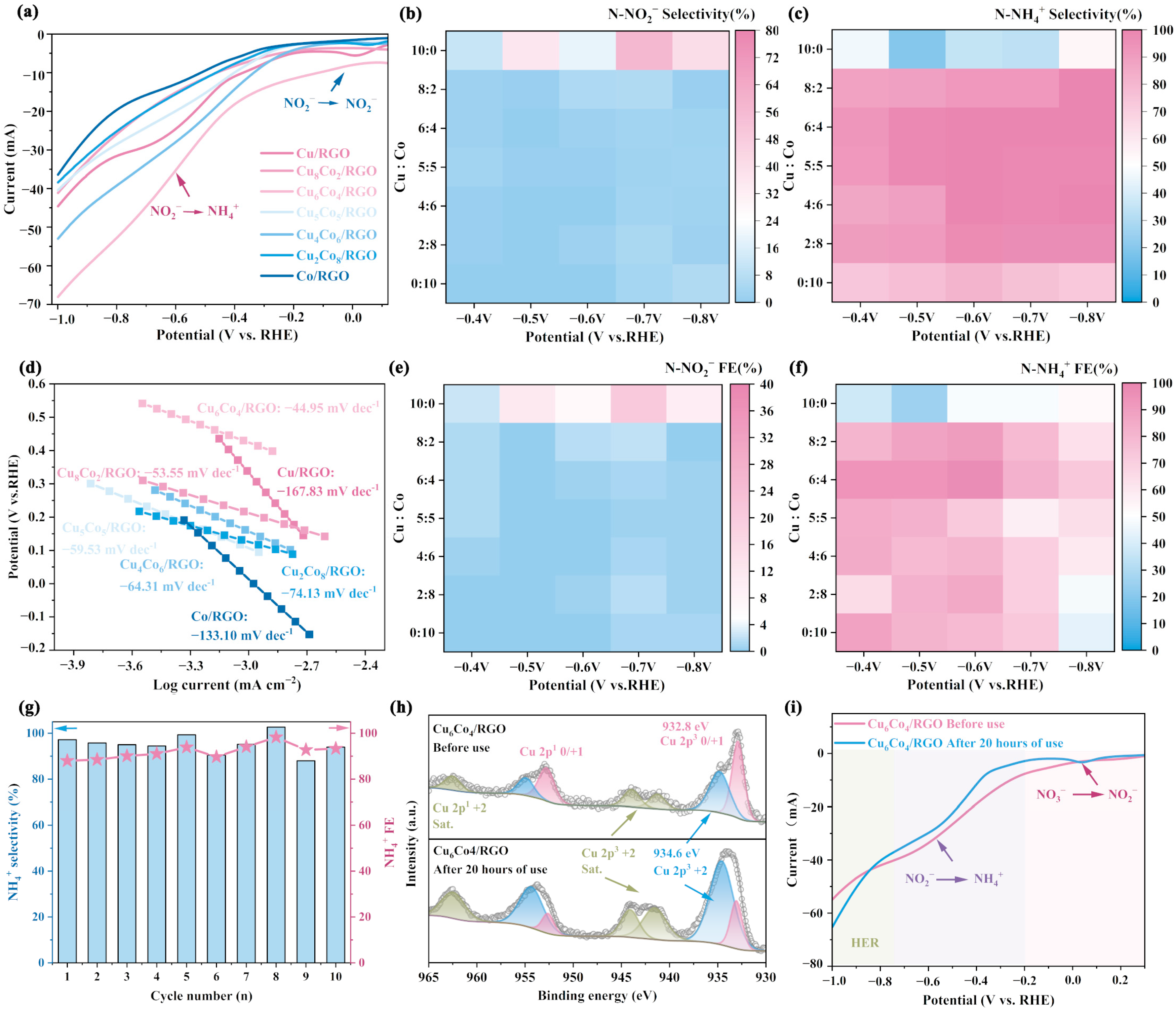
4.3. Performance Study of Catalysts for Nitrate Reduction to Ammonia
4.4. Mechanism of Electro Denitrification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jia, S.; Wu, L.; Liu, H.; Wang, R.; Sun, X.; Han, B. Nitrogenous Intermediates in NOx-involved Electrocatalytic Reactions. Angew. Chem. 2024, 136, e202400033. [Google Scholar] [CrossRef]
- Shao, J.; Jing, H.; Wei, P.; Fu, X.; Pang, L.; Song, Y.; Ye, K.; Li, M.; Jiang, L.; Ma, J.; et al. Electrochemical Synthesis of Ammonia from Nitric Oxide Using a Copper–Tin Alloy Catalyst. Nat. Energy 2023, 8, 1273–1283. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, K.; Wang, H.; Bian, Z. Advances in Electrocatalytic Nitrate Reduction to Ammonia over Cu-Based Catalysts. Environ. Res. 2025, 274, 121123. [Google Scholar] [CrossRef]
- Sekhar, S.J.; Al-Shahri, A.S.A.; Glivin, G.; Le, T.; Mathimani, T. A Critical Review of the State-of-the-Art Green Ammonia Production Technologies-Mechanism, Advancement, Challenges, and Future Potential. Fuel 2024, 358. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.S.S.; Venkateshalu, S.; Lee, J.; Lee, K.; Jin, K. Electrochemical Nitrogen Fixation for Green Ammonia: Recent Progress and Challenges. Adv. Sci. 2023, 10, 2300951. [Google Scholar] [CrossRef]
- Ma, N.; Zhao, W.; Wang, W.; Li, X.; Zhou, H. Large Scale of Green Hydrogen Storage: Opportunities and Challenges. Int. J. Hydrogen Energy 2024, 50, 379–396. [Google Scholar] [CrossRef]
- Yang, X.; Yu, H.; Zhang, L.; Liu, X.; Ding, X. Electrocatalytic Nitrogen Oxidation Reaction: Potential Cornerstones for New Green Nitrogen Economy. Sci. China-Mater. 2025, 68, 744–754. [Google Scholar] [CrossRef]
- Tang, X.; Liu, W.; Lei, C.; Ling, Y.; Kang, S. Mechanism of Key Intermediates Regulation in Electrocatalytic Nitrate-to-Ammonia Conversion Driven by Polarized Electric Field. Nano Energy 2025, 135, 110708. [Google Scholar] [CrossRef]
- Cao, Y.; Yuan, S.; Meng, L.; Wang, Y.; Hai, Y.; Su, S.; Ding, W.; Liu, Z.; Li, X.; Luo, M. Recent Advances in Electrocatalytic Nitrate Reduction to Ammonia: Mechanism Insight and Catalyst Design. ACS Sustain. Chem. Eng. 2023, 11, 7965–7985. [Google Scholar] [CrossRef]
- Zhou, Y.; Duan, R.; Li, H.; Zhao, M.; Ding, C.; Li, C. Boosting Electrocatalytic Nitrate Reduction to Ammonia via Promoting Water Dissociation. ACS Catal. 2023, 13, 10846–10854. [Google Scholar] [CrossRef]
- He, W.; Zhang, J.; Dieckhöfer, S.; Varhade, S.; Brix, A.C.; Lielpetere, A.; Seisel, S.; Junqueira, J.R.C.; Schuhmann, W. Splicing the Active Phases of Copper/Cobalt-Based Catalysts Achieves High-Rate Tandem Electroreduction of Nitrate to Ammonia. Nat. Commun. 2022, 13, 1129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, A.; Wang, Z.; Huang, L.; Li, J.; Li, F.; Wicks, J.; Luo, M.; Nam, D.-H.; Tan, C.-S.; et al. Enhanced Nitrate-to-Ammonia Activity on Copper–Nickel Alloys via Tuning of Intermediate Adsorption. J. Am. Chem. Soc. 2020, 142, 5702–5708. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Meng, Z.; Zhu, Y.; Sun, X.; Deng, X.; Shi, M.; Hao, Q.; Kang, X.; Dai, T.; Zhong, H.; et al. Gram-level NH3 Electrosynthesis via NOx Reduction on a Cu Activated Co Electrode. Angew. Chem. Int. Ed. 2024, 63, e202315238. [Google Scholar] [CrossRef]
- Abdel-Rahman, L.H.; Mohammed, K.M.H.; Ahmed, E.A.-H.; Abdelghani, A.A.; Nafady, A. Anchoring of Cu(II) Schiff Base Complex to the Surface of Graphene Oxide: Access to an Efficient Catalyst for Application in Water Remediation. J. Inorg. Organomet. Polym. 2025, 35, 72–86. [Google Scholar] [CrossRef]
- Pishgahinejad, S.; Aghamohammadi, H.; Hassanzadeh, N. Copper-Coated Graphite Electrodes for the Facile Preparation of Copper Oxide Particles Anchored on Functionalized Holey Graphene Sheets. J. Electroanal. Chem. 2023, 931, 117204. [Google Scholar] [CrossRef]
- Yousaf, S.; Zulfiqar, S.; Shahid, M.; Jamil, A.; Shakir, I.; Agboola, P.O.; Warsi, M.F. Electrochemical Energy Storage Properties Studies of Cu0.2Ni0.8O-Reduced Graphene Oxide Nano-Hybrids. Ceram. Int. 2020, 46, 14304–14310. [Google Scholar] [CrossRef]
- Nandihalli, N. Microwave-Driven Synthesis and Modification of Nanocarbons and Hybrids in Liquid and Solid Phases. J. Energy Storage 2025, 111, 115315. [Google Scholar] [CrossRef]
- Palma, V.; Barba, D.; Cortese, M.; Martino, M.; Renda, S.; Meloni, E. Microwaves and Heterogeneous Catalysis: A Review on Selected Catalytic Processes. Catalysts 2020, 10, 246. [Google Scholar] [CrossRef]
- Guo, T. Application of Graphene Technology in the Removal of Heavy Metal Ions in Wastewater. Highlights Sci. Eng. Technol. 2023, 73, 464–470. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.; Tian, L.; Tang, Y.; Shi, Z.; Xiao, Y.; Wu, Z.; Zhu, Y.; Guo, Q.; Peng, Z. Preparation of Functionalized Graphene Oxide Composite Spheres and Removal of Cu2+ and Pb2+ from Wastewater. Water Air Soil Pollut. 2022, 233, 512. [Google Scholar] [CrossRef]
- Kotsyubynsky, V.; Khimyak, Y.Z.; Zapukhlyak, R.; Boychuk, V.; Turovska, L.; Hoi, V. NaOH-Assisted Hydrothermal Reduction of Graphene Oxide. J. Phys. Condens. Matter 2024, 36, 495701. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cao, M. Study on the Difference in Adsorption Performance of Graphene Oxide and Carboxylated Graphene Oxide for Cu(II), Pb(II) Respectively and Mechanism Analysis. Diam. Relat. Mater. 2022, 129, 109332. [Google Scholar] [CrossRef]
- Askari, M.J.; Kallick, J.D.; McCrory, C.C.L. Selective Reduction of Aqueous Nitrate to Ammonium with an Electropolymerized Chromium Molecular Catalyst. J. Am. Chem. Soc. 2024, 146, 7439–7455. [Google Scholar] [CrossRef] [PubMed]
- Fan, K.; Xie, W.; Li, J.; Sun, Y.; Xu, P.; Tang, Y.; Li, Z.; Shao, M. Active Hydrogen Boosts Electrochemical Nitrate Reduction to Ammonia. Nat. Commun. 2022, 13, 7958. [Google Scholar] [CrossRef]
- Lin, P.; Chen, R.; Xu, S.; Xia, X.; Zhao, F.; Ren, X.; Lu, Y.; Gao, L.; Bao, J.; Liu, A. Efficient Co and GO Co-Doped TiO2 Catalysts for the Electrochemical Reduction of Nitrate to Ammonia. Catal. Sci. Technol. 2025, 15, 1445–1455. [Google Scholar] [CrossRef]
- You, Y.; Wei, M.; Yang, L.; Wang, J.; Zhang, Y.; Xu, J. Multifunctional MoSe2@rGO Coating on the Cathode versus the Separator as an Efficient Polysulfide Barrier for High-Performance Lithium-Sulfur Battery. Appl. Surf. Sci. 2020, 527, 146785. [Google Scholar] [CrossRef]
- Gautam, M.; Kanade, S.; Kale, B.B. Electrochemical Energy Storage and Conversion Applications of Graphene Oxide: A Review. Energy Fuels 2023, 37, 17134–17160. [Google Scholar] [CrossRef]
- Hassanpour, A.; Gauthier, M.A.; Sun, S. Ion-Retention Properties of Graphene Oxide/Zinc Oxide Nanocomposite Membranes at Various pH and Temperature Conditions. Sci. Rep. 2024, 14, 1443. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, Y.; Wang, J.; Peng, T.; Xu, J.; Yang, B.; Tang, K. Reduced Graphene Oxide/Carbon Nanotube Composites as Electrochemical Energy Storage Electrode Applications. Nanoscale Res. Lett. 2018, 13, 181. [Google Scholar] [CrossRef]
- Aukštakojytė, R.; Niaura, G.; Bukauskas, V.; Barkauskas, J.; Pauliukaitė, R.; Gaidukevič, J. Bismarck Brown-Assisted Hydrothermal Synthesis of Nitrogen-Modified Reduced Graphene Oxide for Selective Electrochemical Detection of Dopamine. Surf. Interfaces 2024, 46, 104041. [Google Scholar] [CrossRef]
- Wang, J.; Ren, P.; Ren, F.; Zhu, G.; Sun, A.; You, C. Preparation of Highly Thermally Conductive Epoxy Composites via Constructing a Vertically Aligned Foam of Cetyltrimethylammonium Bromide–Graphene@polydopamine–Multi-Walled Carbon Nanotubes. J. Mater. Sci. 2021, 56, 7951–7965. [Google Scholar] [CrossRef]
- Van Tuan, P.; Thi Ha, T.; Hung, N.D.; Tan, V.T.; Thi Quynh Hoa, T.; Thi Ha, D.; Ha, L.T.; Khiem, T.N. Reduction of Graphene Oxide (GO) to Reduced Graphene Oxide (rGO) at Different Hydrothermal Temperatures and Enhanced Photodegradation of Zinc Oxide/rGO Composites. Phys. Scr. 2024, 99, 015912. [Google Scholar] [CrossRef]
- Kumuda, S.; Gandhi, U.; Mangalanathan, U.; Rajanna, K. Synthesis and Characterization of Graphene Oxide and Reduced Graphene Oxide Chemically Reduced at Different Time Duration. J. Mater. Sci. Mater. Electron. 2024, 35, 637. [Google Scholar] [CrossRef]
- Zheng, Y.; Ren, Y.; Yang, J.; Huang, Y.; Liu, Y. Rational Design of Electrocatalytic System to Selective Transform Nitrate to Nitrogen. Chemosphere 2023, 341, 140016. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Zhang, Z.; Mo, Z.; Wang, C.; Gao, S. Flower-like Open-Structured Polycrystalline Copper with Synergistic Multi-Crystal Plane for Efficient Electrocatalytic Reduction of Nitrate to Ammonia. Nano Energy 2022, 97, 107124. [Google Scholar] [CrossRef]
- Wang, D.; Fan, G.; Luan, D.; Guo, Y.; Gu, X.; Lou, X.W. Ru-Incorporation-Induced Phase Transition in Co Nanoparticles for Low-Concentration Nitric Oxide Electroreduction to Ammonia at Low Potential. Adv. Mater. 2024, 36, 2408580. [Google Scholar] [CrossRef]
- Yang, K.; Han, S.-H.; Cheng, C.; Guo, C.; Li, T.; Yu, Y. Unveiling the Reaction Mechanism of Nitrate Reduction to Ammonia Over Cobalt-Based Electrocatalysts. J. Am. Chem. Soc. 2024, 146, 12976–12983. [Google Scholar] [CrossRef]
- Niu, Z.; Fan, S.; Li, X.; Chen, G. Electrocatalytic Co-Upcycling of Nitrite and Ethylene Glycol over Cobalt–Copper Oxides. Adv. Energy Mater. 2024, 14, 2303515. [Google Scholar] [CrossRef]
- Bai, L.; Franco, F.; Timoshenko, J.; Rettenmaier, C.; Scholten, F.; Jeon, H.S.; Yoon, A.; Rüscher, M.; Herzog, A.; Haase, F.T.; et al. Electrocatalytic Nitrate and Nitrite Reduction toward Ammonia Using Cu2O Nanocubes: Active Species and Reaction Mechanisms. J. Am. Chem. Soc. 2024, 146, 9665–9678. [Google Scholar] [CrossRef]
- Qin, D.; Song, S.; Liu, Y.; Wang, K.; Yang, B.; Zhang, S. Enhanced Electrochemical Nitrate-to-Ammonia Performance of Cobalt Oxide by Protic Ionic Liquid Modification. Angew. Chem. Int. Ed. 2023, 62, e202304935. [Google Scholar] [CrossRef]
- Yu, J.; Qin, Y.; Wang, X.; Zheng, H.; Gao, K.; Yang, H.; Xie, L.; Hu, Q.; He, C. Boosting Electrochemical Nitrate-Ammonia Conversion via Organic Ligands-Tuned Proton Transfer. Nano Energy 2022, 103, 107705. [Google Scholar] [CrossRef]
- Zhang, J.; He, W.; Quast, T.; Junqueira, J.R.C.; Saddeler, S.; Schulz, S.; Schuhmann, W. Single-entity Electrochemistry Unveils Dynamic Transformation during Tandem Catalysis of Cu2O and Co3O4 for Converting NO3− to NH3. Angew. Chem. Int. Ed. 2023, 62, e202214830. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, S.; Wu, Z.; Jiang, M.; Kuang, M.; Liu, Y.; Luo, W.; Zhang, D.; Yang, J. Relay Catalysis of Fe and Co with Multi-Active Sites for Specialized Division of Labor in Electrocatalytic Nitrate Reduction Reaction. Adv. Funct. Mater. 2024, 34, 2403838. [Google Scholar] [CrossRef]
- Hu, Q.; Qi, S.; Huo, Q.; Zhao, Y.; Sun, J.; Chen, X.; Lv, M.; Zhou, W.; Feng, C.; Chai, X.; et al. Designing Efficient Nitrate Reduction Electrocatalysts by Identifying and Optimizing Active Sites of Co-Based Spinels. J. Am. Chem. Soc. 2024, 146, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xie, L.; Liang, J.; Ren, Y.; Wang, Y.; Zhang, L.; Yue, L.; Li, T.; Luo, Y.; Li, N.; et al. Ambient Ammonia Synthesis via Electrochemical Reduction of Nitrate Enabled by NiCo2O4 Nanowire Array. Small 2022, 18, 2106961. [Google Scholar] [CrossRef]
- Yang, Q.; Bu, Y.; Pu, S.; Chu, L.; Huang, W.; Zhu, X.; Liu, C.; Fang, G.; Cui, P.; Zhou, D.; et al. Matched Kinetics Process Over Fe2O3-Co/NiO Heterostructure Enables Highly Efficient Nitrate Electroreduction to Ammonia. Angew. Chem. Int. Ed. 2024, 63, e202400428. [Google Scholar] [CrossRef]
- Fang, L.; Wang, S.; Lu, S.; Yin, F.; Dai, Y.; Chang, L.; Liu, H. Efficient Electroreduction of Nitrate via Enriched Active Phases on Copper-Cobalt Oxides. Chin. Chem. Lett. 2024, 35, 108864. [Google Scholar] [CrossRef]
- Xue, Y.; Yu, Q.; Ma, Q.; Chen, Y.; Zhang, C.; Teng, W.; Fan, J.; Zhang, W. Electrocatalytic Hydrogenation Boosts Reduction of Nitrate to Ammonia over Single-Atom Cu with Cu(I)-N3C1 Sites. Environ. Sci. Technol. 2022, 56, 14797–14807. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Lee, C.; Luo, W.; Dong, J.; Liang, Z.; Chen, M.; Hu, E.; Zhang, M.; Debbie Soo, X.Y.; et al. Balanced NOx− and Proton Adsorption for Efficient Electrocatalytic NOx− to NH3 Conversion. ACS Nano 2023, 17, 23637–23648. [Google Scholar] [CrossRef]
- Rao, T.; Zhan, J.; Du, Y.; Zhang, L.; Yu, F. Tuning Nitrogen Configurations in Nitrogen-Doped Graphene Encapsulating Fe3C Nanoparticles for Enhanced Nitrate Electroreduction. ChemSusChem 2025, 18, e202402460. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, X.; Liu, X.; Liu, J.; Wang, M.; Ding, T.; Yan, L.; Zhang, Z.; Shi, G. Electrochemical Synthesis of Metasequoia-Like Reduced Graphene Oxide Coated Cobalt-Silver Catalyst for Stable and Efficient Electrocatalytic Nitrate Reduction to Ammonia. Small 2025, 21, 2408566. [Google Scholar] [CrossRef]
- Li, Y.; Ji, S.; Fan, Y.; Duan, T.; Zhang, Y. The Charge Redistribution Induced by Cu-Co Bimetallic Synergies Efficiently Promotes Electrochemical Reduction to Ammonia. Appl. Surf. Sci. 2025, 696, 162950. [Google Scholar] [CrossRef]
- Li, S.; Yan, J.; Liu, M.; Su, H. Localized Enriching Nitrate/Proton on Reconstituted Fe Nanoparticles Boosting Electrocatalytic Nitrate Reduction to Ammonia. J. Energy Chem. 2025, 103, 682–691. [Google Scholar] [CrossRef]
- He, L.; Yao, F.; Zhong, Y.; Tan, C.; Hou, K.; Pi, Z.; Chen, S.; Li, X.; Yang, Q. Achieving High-Performance Electrocatalytic Reduction of Nitrate by N-Rich Carbon-Encapsulated Ni-Cu Bimetallic Nanoparticles Supported Nickel Foam Electrode. J. Hazard. Mater. 2022, 436, 129253. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.E.; Kim, J.Y.; Youn, D.H. Enhanced Electrochemical Nitrate Reduction to Ammonia with Nanostructured Mo2C on Carbon Nanotube-Reduced Graphene Oxide Hybrid Support. Dalton Trans. 2024, 53, 18141–18147. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liang, Y.; Bu, M.; Dai, Z.; Shi, J. V2O4 Nanowires/rGO Hybrid for Electrocatalytic Nitrogen Fixation. Catal. Lett. 2024, 154, 1795–1805. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, J.; Zheng, M.; Jin, X.; Shen, Z.; Li, Z.; Wang, Y.; Wang, Q.; Wang, X.; Wei, H.; et al. Fe/Cu Diatomic Catalysts for Electrochemical Nitrate Reduction to Ammonia. Nat. Commun. 2023, 14, 3634. [Google Scholar] [CrossRef]
- Sun, W.; Sahin, N.E.; Sun, D.; Wu, X.; Munoz, C.; Thakare, J.; Aulich, T.; Zhang, J.; Hou, X.; Oncel, N.; et al. One-Pot Synthesis of Ruthenium-Based Nanocatalyst Using Reduced Graphene Oxide as Matrix for Electrochemical Synthesis of Ammonia. ACS Appl. Mater. Interfaces 2023, 15, 1115–1128. [Google Scholar] [CrossRef]
- Bai, Z.; Li, X.; Ding, L.; Qu, Y.; Chang, X. Artificial Cu-Ni Catalyst towards Highly Efficient Nitrate-to-Ammonia Conversion. Sci. China Mater. 2023, 66, 2329–2338. [Google Scholar] [CrossRef]
- Li, T.; Tang, C.; Guo, H.; Wu, H.; Duan, C.; Wang, H.; Zhang, F.; Cao, Y.; Yang, G.; Zhou, Y. In Situ Growth of Fe2O3 Nanorod Arrays on Carbon Cloth with Rapid Charge Transfer for Efficient Nitrate Electroreduction to Ammonia. ACS Appl. Mater. Interfaces 2022, 14, 49765–49773. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, Z.; Jabeen, S.; Liu, N.; Liu, Y.; Cheng, Y.; Li, Y.; Yu, J.; Wu, X.; Yan, N.; et al. Mechanochemical Route to Fabricate an Efficient Nitrate Reduction Electrocatalyst. Nano Res. 2024, 17, 4889–4897. [Google Scholar] [CrossRef]
- Hu, Q.; Yang, K.; Peng, O.; Li, M.; Ma, L.; Huang, S.; Du, Y.; Xu, Z.-X.; Wang, Q.; Chen, Z.; et al. Ammonia Electrosynthesis from Nitrate Using a Ruthenium–Copper Cocatalyst System: A Full Concentration Range Study. J. Am. Chem. Soc. 2024, 146, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Han, S.; Yang, R.; Li, T.; Li, W.; Wang, Y.; Yu, Y.; Zhang, B. Linear Adsorption Enables NO Selective Electroreduction to Hydroxylamine on Single Co Sites. Angew. Chem. Int. Ed. 2023, 62, e202305184. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, H.; Zhang, Y.; Ding, L.; Li, X.; Zhao, Z.; Qu, Y.; Xia, Y.; Chang, X. Graphene Oxide-Anchored Cu–Co Catalysts for Efficient Electrochemical Nitrate Reduction. Materials 2025, 18, 2495. https://doi.org/10.3390/ma18112495
Lan H, Zhang Y, Ding L, Li X, Zhao Z, Qu Y, Xia Y, Chang X. Graphene Oxide-Anchored Cu–Co Catalysts for Efficient Electrochemical Nitrate Reduction. Materials. 2025; 18(11):2495. https://doi.org/10.3390/ma18112495
Chicago/Turabian StyleLan, Haosheng, Yi Zhang, Le Ding, Xin Li, Zhanhong Zhao, Yansen Qu, Yingjie Xia, and Xinghua Chang. 2025. "Graphene Oxide-Anchored Cu–Co Catalysts for Efficient Electrochemical Nitrate Reduction" Materials 18, no. 11: 2495. https://doi.org/10.3390/ma18112495
APA StyleLan, H., Zhang, Y., Ding, L., Li, X., Zhao, Z., Qu, Y., Xia, Y., & Chang, X. (2025). Graphene Oxide-Anchored Cu–Co Catalysts for Efficient Electrochemical Nitrate Reduction. Materials, 18(11), 2495. https://doi.org/10.3390/ma18112495