
Steam-Assisted Ammonolysis of MoO2 as a Synthetic Pathway to Oxygenated δ-MoN

 , , , , and
, , , , and
Abstract
1. Introduction
2. Survey of Ammonolysis Outcomes
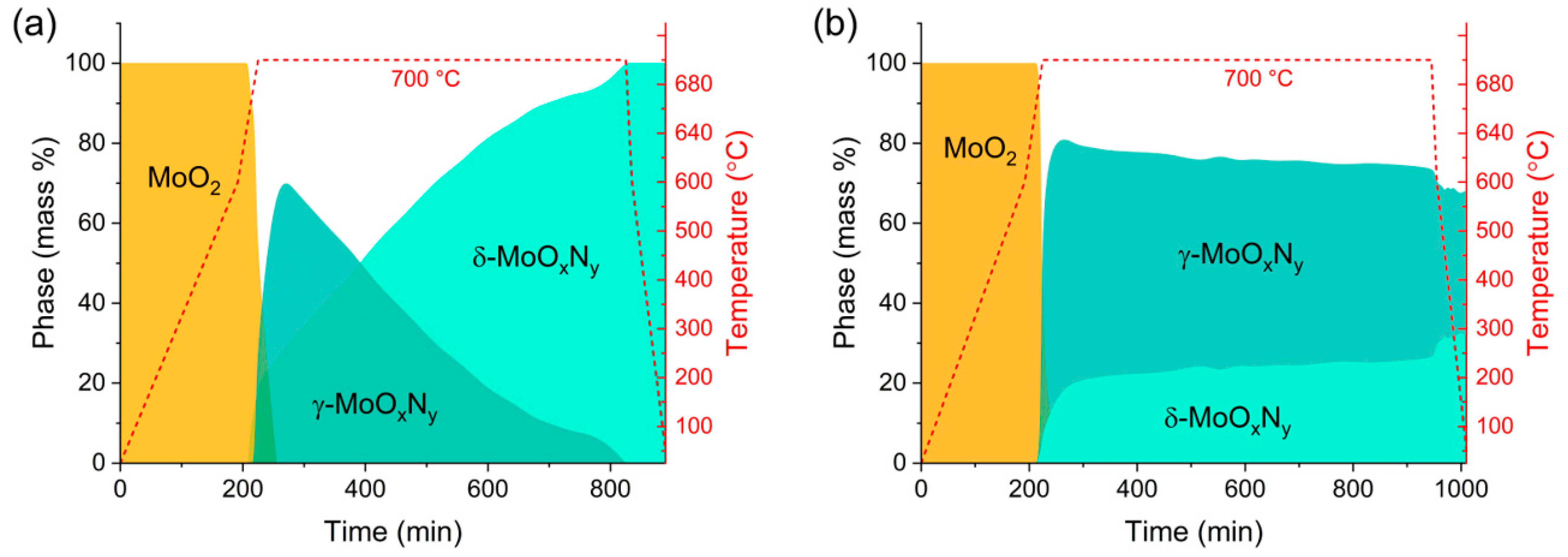
3. In Situ Transformation of MoO2 to Nitrogen-Bearing Phases
4. Physico-Chemical Characterization of δ-MoOxNy
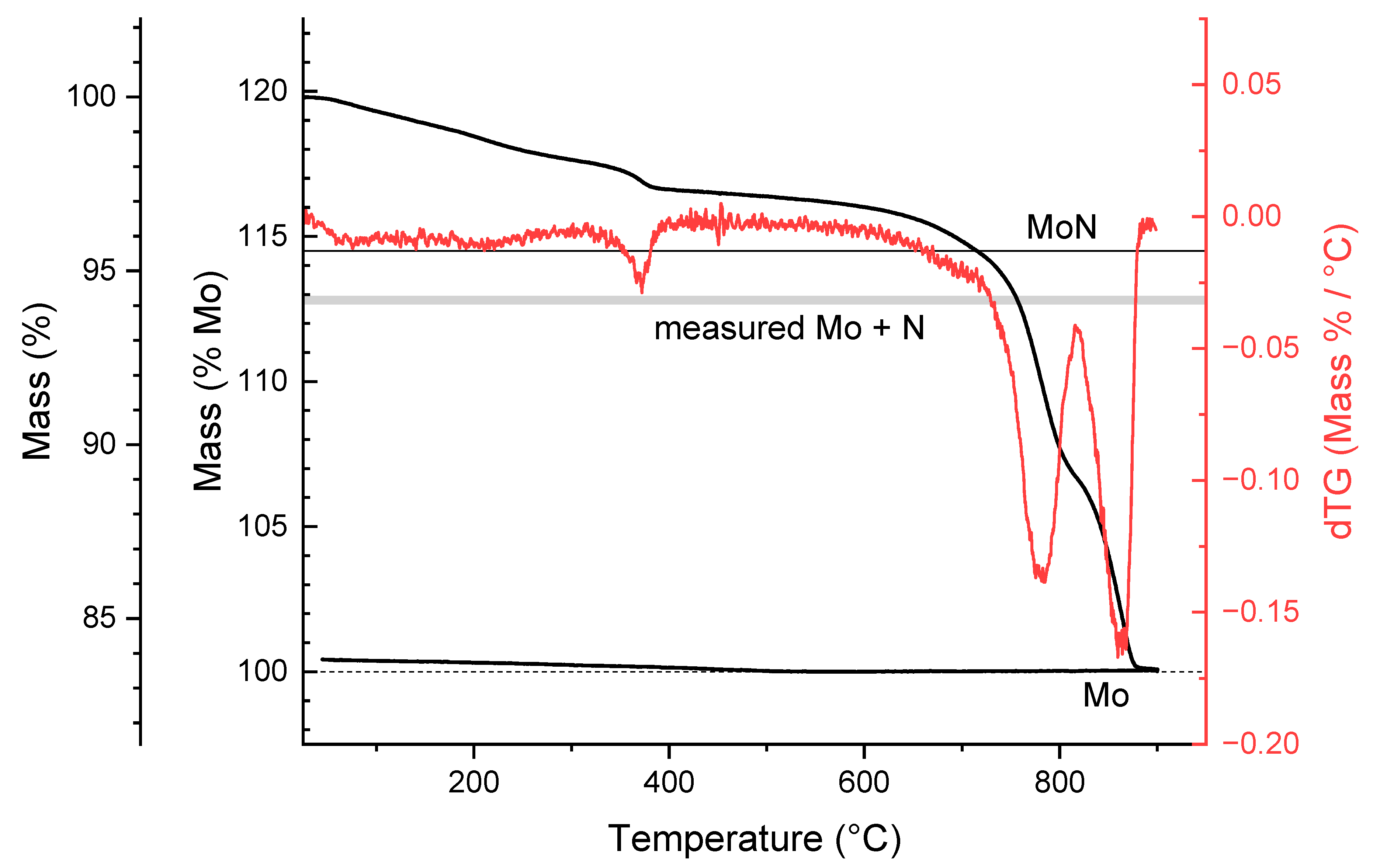
4.1. Chemical Analysis
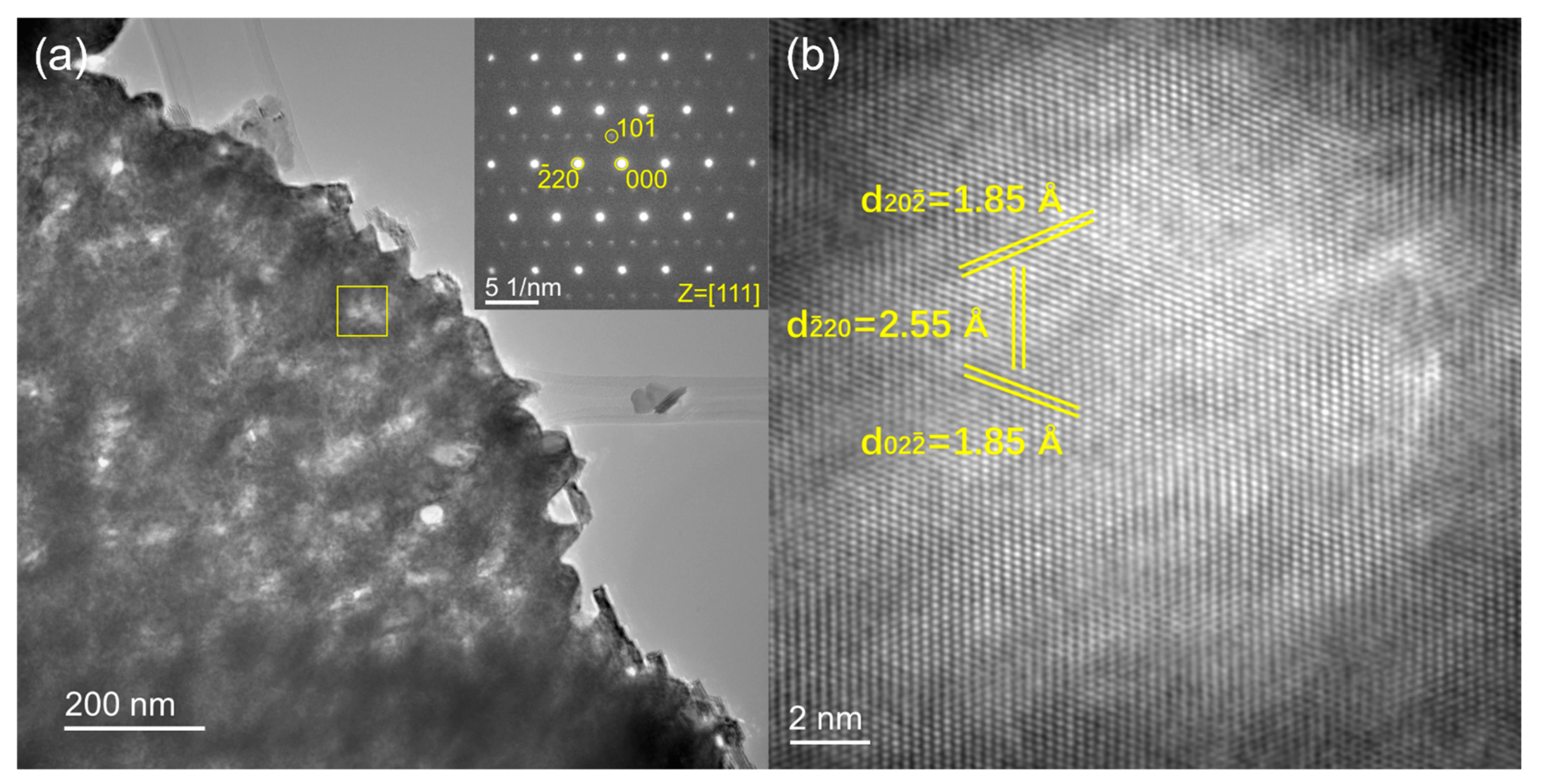
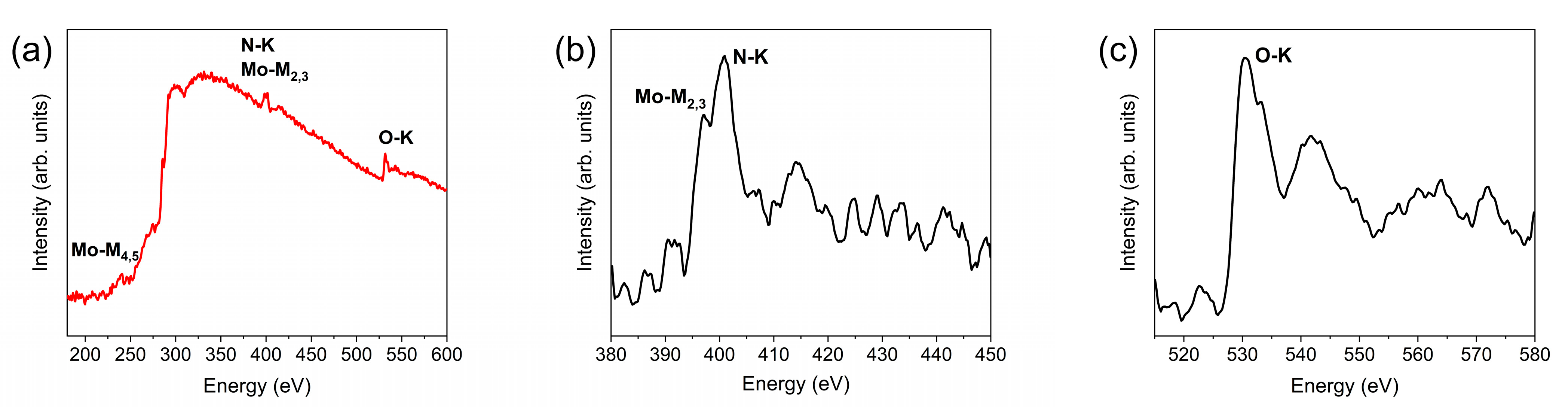
4.2. Electron Microscopy and Spectroscopy
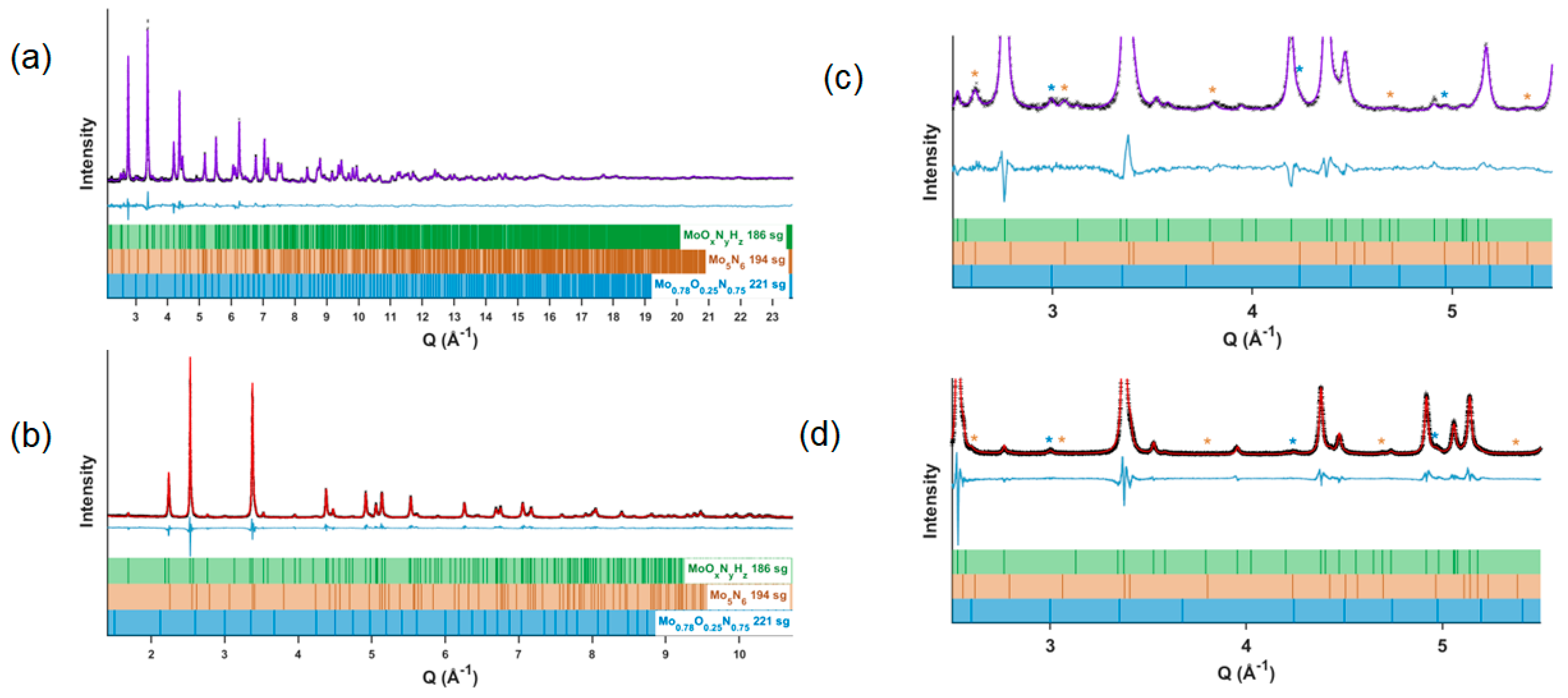
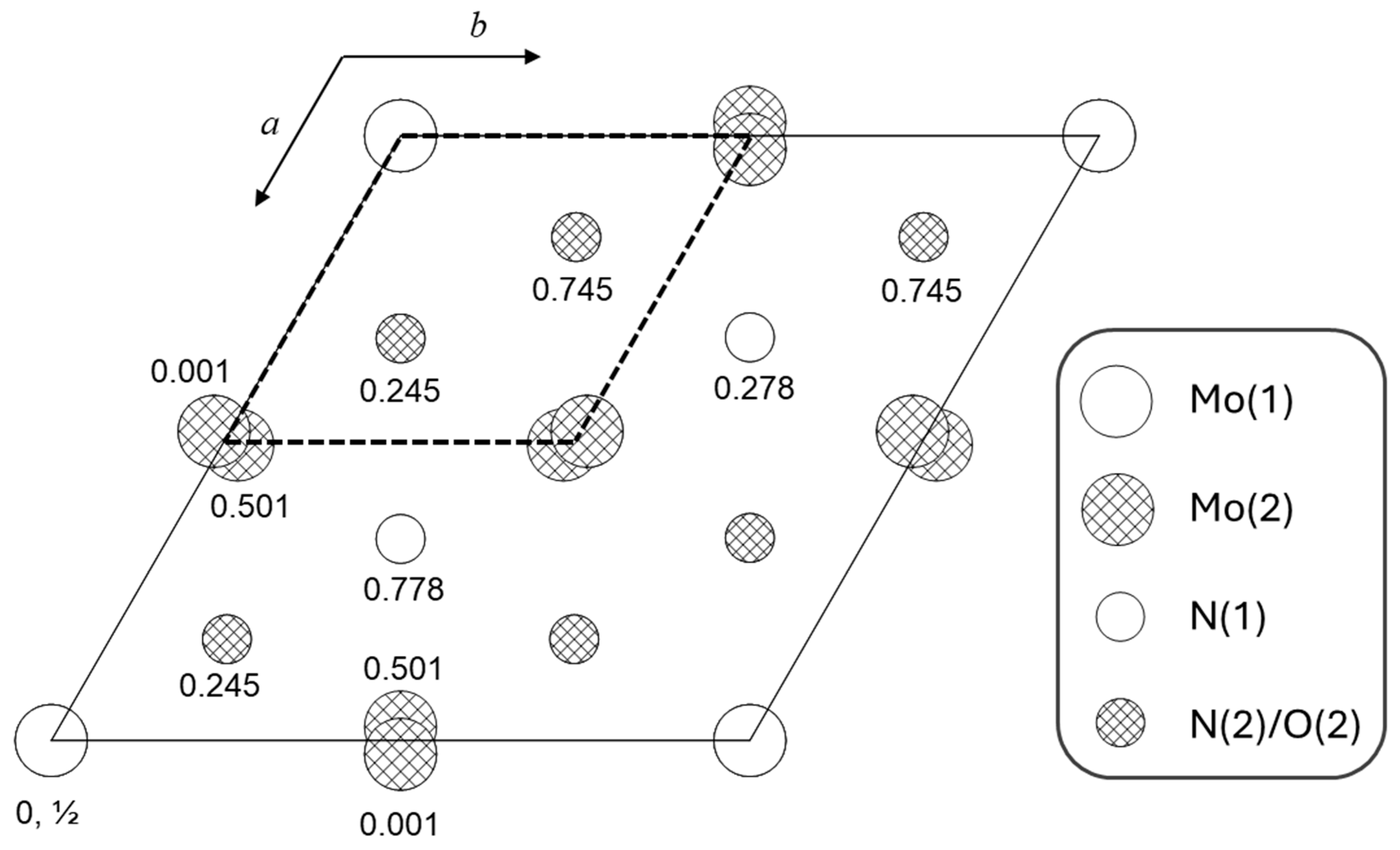
4.3. Neutron and X-Ray Diffraction Structure Refinement
5. Characterization Methods
6. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BET | Brunauer–Emmett–Teller |
| EELS | Electron Energy Loss Spectroscopy |
| ENLES | Electron-Energy-Loss Near Edge Structure |
| FWHM | Full Width at Half Maximum |
| GMS | Gatan Microscopy Suite |
| GSAS | General Structure Analysis System |
| NPD | Neutron Powder Diffraction |
| PGAA | Prompt Gamma Activation Analysis |
| SAED | Selected Area Electron Diffraction |
| STEM | Scanning Transmission Electron Microscopy |
| TEM | Transmission Electron Microscopy |
| TGA | Thermogravimetric Analysis |
| TOF | Time-of-Flight |
| XPS | X-Ray Photoemission Spectroscopy |
| XRD | X-Ray Diffraction |
| ZLP | Zero-Loss Peak |
References
- Fuertes, A. Synthetic approaches in oxynitride chemistry. Prog. Solid State Chem. 2018, 51, 63–70. [Google Scholar] [CrossRef]
- Choi, J.-G.; Curl, R.L.; Thompson, L.T. Molybdenum nitride catalysts: I. Influence of the synthesis factors on structural properties. J. Catal. 1994, 146, 218–227. [Google Scholar] [CrossRef]
- Bull, C.L.; McMillan, P.F.; Soignard, E.; Leinenweber, K. Determination of the crystal structure of δ-MoN by neutron diffraction. J. Solid State Chem. 2004, 177, 1488–1492. [Google Scholar] [CrossRef]
- Jaggers, C.H.; Michaels, J.N.; Stacy, A.M. Preparation of High-Surface-Area Transition-Metal Nitrides: Mo2N and MoN. Chem. Mater. 1990, 2, 150. [Google Scholar] [CrossRef]
- Volpe, L.; Boudart, M. Compounds of molybdenum and tungsten with high specific surface area: I. Nitrides. J. Solid State Chem. 1985, 59, 332–347. [Google Scholar] [CrossRef]
- Song, X.; Yi, W.; Li, J.; Kong, Q.; Bai, H.; Xi, G. Selective Preparation of Mo2N and MoN with High Surface Area for Flexible SERS Sensing. Nano Lett. 2021, 21, 4410–4414. [Google Scholar] [CrossRef]
- Pandey, S.A.; Zhang, C.; Ibrahim, D.H.; Goldfine, E.A.; Wenderott, J.K.; dos Reis, R.; Paul, R.L.; Spanopoulos, I.; Kanatzidis, M.; Bedzyk, M.J.; et al. Hidden Complexity in the Chemistry of Ammonolysis-Derived “γ-Mo2N”: An Overlooked Oxynitride Hydride. Chem. Mater. 2021, 33, 6671–6684. [Google Scholar] [CrossRef]
- Gouin, X.; Marchand, R.; L’Haridon, P.; Laurent, Y. Action de l’ammoniac sur l’oxyde de molybdene MoO3. Caractérisation physico-chimique de la phase oxynitrure de type Mo2N γ. J. Solid State Chem. 1994, 109, 175–180. [Google Scholar] [CrossRef]
- Lyutaya, M.D. Formation of nitrides of group 6 transition metals of the Periodic system. Poroshkovaya Metall. (Kiev) 1979, 3, 60–66. [Google Scholar]
- Miga, K.; Stanczyk, K.; Sayag, C.; Brodzki, D.; Djéga-Mariadassou, G. Bifunctional Behavior of Bulk MoOxNy and Nitrided Supported NiMo Catalyst in Hydrodenitrogenation of Indole. J. Catal. 1999, 183, 63–68. [Google Scholar] [CrossRef]
- Sayag, C.; Bugli, G.; Havil, P.; Djéga-Mariadassou, G. Surface Studies of Passivated Molybdenum Oxynitride. J. Catal. 1997, 167, 372–378. [Google Scholar] [CrossRef]
- Kreider, M.E.; Stevens, M.B.; Liu, Y.; Patel, A.M.; Statt, M.J.; Gibbons, B.M.; Gallo, A.; Ben-Naim, M.; Mehta, A.; Davis, R.C.; et al. Nitride or Oxynitride? Elucidating the Composition–Activity Relationships in Molybdenum Nitride Electrocatalysts for the Oxygen Reduction Reaction. Chem. Mater. 2020, 32, 2946–2960. [Google Scholar] [CrossRef]
- de Respinis, M.; Fravventura, M.; Abdi, F.F.; Schreuders, H.; Savenije, T.J.; Smith, W.A.; Dam, B.; van de Krol, R. Oxynitrogenography: Controlled Synthesis of Single-Phase Tantalum Oxynitride Photoabsorbers. Chem. Mater. 2015, 27, 7091–7099. [Google Scholar] [CrossRef]
- Orhan, E.; Tessier, F.; Marchand, R. Synthesis and energetics of yellow TaON. Solid State Sci. 2002, 4, 1071–1076. [Google Scholar] [CrossRef]
- Szymanski, N.J.; Walters, L.N.; Puggioni, D.; Rondinelli, J.M. Design of Heteroanionic MoON Exhibiting a Peierls Metal-Insulator Transition. Phys. Rev. Lett. 2019, 123, 6. [Google Scholar] [CrossRef]
- O’Hara, A.; Demkov, A.A. Nature of the metal-insulator transition in NbO2. Phys. Rev. B 2015, 91, 6. [Google Scholar] [CrossRef]
- Griesemer, S.D.; Baldassarri, B.; Zhu, R.; Shen, J.; Pal, K.; Park, C.W.; Wolverton, C. Wide-ranging predictions of new stable compounds powered by recommendation engines. Sci. Adv. 2025, 11, eadq1431. [Google Scholar] [CrossRef]
- Shen, J.H.; Hegde, V.I.; He, J.G.; Xia, Y.; Wolverton, C. High-Throughput Computational Discovery of Ternary Mixed-Anion Oxypnictides. Chem. Mater. 2021, 33, 9486–9500. [Google Scholar] [CrossRef]
- Zakutayev, A.; Bauers, S.R.; Lany, S. Experimental Synthesis of Theoretically Predicted Multivalent Ternary Nitride Materials. Chem. Mater. 2022, 34, 1418–1438. [Google Scholar] [CrossRef]
- Sharan, A.; Lany, S. Computational discovery of stable and metastable ternary oxynitrides. J. Chem. Phys. 2021, 154, 10. [Google Scholar] [CrossRef]
- Michie, C.W.; Claridge, J.B.; Clarke, S.J.; Rosseinsky, M.J. Zr4O5N2—Intergrowth of Fluorite and Bixbyite Anion Layers Formed by Coupled Site-Selective Anion and Vacancy Ordering. Chem. Mater. 2003, 15, 1547–1553. [Google Scholar] [CrossRef]
- Füglein, E.; Lerch, M.; Hock, R. Über Kristallstruktur und Hochtemperaturverhalten von Zr2ON2. Z. Für Anorg. Und Allg. Chem. 1997, 623, 304–308. [Google Scholar] [CrossRef]
- Clarke, S.J.; Michie, C.W.; Rosseinsky, M.J. Structure of Zr2ON2 by Neutron Powder Diffraction: The Absence of Nitride–Oxide Ordering. J. Solid State Chem. 1999, 146, 399–405. [Google Scholar] [CrossRef]
- Wang, C.-H.; Kennedy, B.J.; Menezes de Oliveira, A.L.; Polt, J.; Knight, K.S. The impact of anion ordering on octahedra distortion and phase transitions in SrTaO2N and BaTaO2N. Acta Crystallogr. Sect. B 2017, 73, 389–398. [Google Scholar] [CrossRef]
- Brauer, G.; Weidlein, J.R. Synthesis and Properties of Tantalum Oxide Nitride TaON. Angew. Chem.-Int. Edit. 1965, 4, 875. [Google Scholar] [CrossRef]
- Marchand, R.; Gouin, X.; Tessier, F.; Laurent, Y. New routes to molybdenum nitrides and oxynitrides: Preparation and characterization of new phases. In The Chemistry of Transition Metal Carbides and Nitrides; Oyama, S.T., Ed.; Blackie Academic and Professional: London, UK, 1996. [Google Scholar]
- Goldfine, E.A.; Wenderott, J.K.; Sweers, M.E.; Pandey, S.; Seitz, L.C.; Bedzyk, M.J.; Haile, S.M. Molybdenum Oxide Precursors that Promote the Low-Temperature Formation of High-Surface-Area Cubic Molybdenum (Oxy)nitride. Inorg. Chem. 2022, 61, 16760–16769. [Google Scholar] [CrossRef] [PubMed]
- Panda, R.N.; Kaskel, S. Synthesis and characterization of high surface area molybdenum nitride. J. Mater. Sci. 2006, 41, 2465–2470. [Google Scholar] [CrossRef]
- Zhang, C.; Goldfine, E.A.; He, K.; Wenderott, J.K.; Pandey, S.A.; dos Reis, R.; Shen, J.H.; Wolverton, C.; Bedzyk, M.J.; Poeppelmeier, K.R.; et al. Elucidating the Reaction Pathway in the Ammonolysis of MoO3 via In Situ Powder X-ray Diffraction and Transmission Electron Microscopy. Chem. Mater. 2024, 36, 1123–1135. [Google Scholar] [CrossRef]
- Ganin, A.Y.; Kienle, L.; Vajenine, G.V. Synthesis and characterisation of hexagonal molybdenum nitrides. J. Solid State Chem. 2006, 179, 2339–2348. [Google Scholar] [CrossRef]
- Powell, C.J. Practical guide for inelastic mean free paths, effective attenuation lengths, mean escape depths, and information depths in x-ray photoelectron spectroscopy. J. Vac. Sci. Technol. A 2020, 38, 14. [Google Scholar] [CrossRef]
- Choi, J.G.; Thompson, L.T. XPS study of as-prepared and reduced molybdenum oxides. Appl. Surf. Sci. 1996, 93, 143–149. [Google Scholar] [CrossRef]
- Bolzan, A.; Kennedy, B.; Howard, C. Neutron Powder Diffraction Study of Molybdenum and Tungsten Dioxides. Aust. J. Chem. 1995, 48, 1473–1477. [Google Scholar] [CrossRef]
- Leisegang, T.; Levin, A.A.; Walter, J.; Meyer, D.C. In situ X-ray analysis of MoO3 reduction. Cryst. Res. Technol. 2005, 40, 95–105. [Google Scholar] [CrossRef]
- Cao, B.; Neuefeind, J.C.; Adzic, R.R.; Khalifah, P.G. Molybdenum nitrides as oxygen reduction reaction catalysts: Structural and electrochemical studies. Inorg. Chem. 2015, 54, 2128–2136. [Google Scholar] [CrossRef]
- Toby, B.H.; Von Dreele, R.B. GSAS-II: The genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Egerton, R.F. Electron Energy-Loss Spectroscopy in the Electron Microscope, 3rd ed.; Springer: New York, NY, USA, 2011. [Google Scholar]
- Chuang, T.J.; Brundle, C.R.; Wandelt, K. X-ray Photoelectron-Spectroscopy Study of Chemical Changes in Oxide and Hydroxide Surfaces Induced by Ar+ Ion-Bombardment. Thin Solid Film. 1978, 53, 19–27. [Google Scholar] [CrossRef]
- Ruan, D.; Lin, R.; Jiang, K.; Yu, X.; Zhu, Y.; Fu, Y.; Wang, Z.; Yan, H.; Mai, W. High-Performance Porous Molybdenum Oxynitride Based Fiber Supercapacitors. ACS Appl. Mater Interfaces 2017, 9, 29699–29706. [Google Scholar] [CrossRef] [PubMed]
- Sing, M.; Neudert, R.; von Lips, H.; Golden, M.S.; Knupfer, M.; Fink, J.; Claessen, R.; Mücke, J.; Schmitt, H. The electronic structure of metallic K0.3MoO3 and insulating MoO3 from high-energy spectroscopy. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 60, 8559–8568. [Google Scholar] [CrossRef]
- Lajaunie, L.; Boucher, F.; Dessapt, R.; Moreau, P. Quantitative use of electron energy-loss spectroscopy Mo-M2,3 edges for the study of molybdenum oxides. Ultramicroscopy 2015, 149, 1–8. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Experiment Description | pNH3, atm | pAr, atm | pH2O, atm | Flow Rate, sccm | % δ-MoOxNy |
|---|---|---|---|---|---|---|
| 1 | MoO3—dry | 1.0 | 0 | 0 | 100 | 2 |
| 2 | MoO2—dry | 1.0 | 0min | 0 | 100 | 29 |
| 3 | H2MoO5—dry | 1.0 | 0 | 0 | 100 | 0 |
| 4 | MoO3—humid (1.5%) | 0.493 | 0.492 | 0.015 | 203 | 24 |
| 5 | MoO3—humid (2%) | 0.328 | 0.652 | 0.020 | 305 | 97 |
| 6 | MoO3—humid (2.5%) | 0.189 | 0.786 | 0.025 | 528 | 67 |
| 7 | MoO2—humid (2%) | 0.328 | 0.652 | 0.020 | 305 | 100 |
| 8 | H2MoO5—humid (2%) | 0.328 | 0.652 | 0.020 | 305 | 30 |
| 9 | MoO3—humid (2%)—slow ramp * | 0.328 | 0.652 | 0.020 | 305 | 36 |
| Thermogravimetric Analysis | |
|---|---|
| Mass loss under Ar at 400 °C (surface) | 2.00(1) wt.% |
| Mass loss under H2 at 400 °C (surface) | 2.66(1) wt.% |
| Mass loss under H2 at 700 °C (surface) | 4.15(3) wt.% |
| Mass loss under H2 at 900 °C | 16.53(6) wt.% |
| Mass loss under H2 from 700 to 900 °C (bulk) | 17.25(6) wt.% relative to mass at 700 °C |
| Combustion Analysis | |
| Absolute N content, as-synthesized | 11.7(3) mass % |
| Absolute N content, surface-desorbed | 11.0(3) mass % |
| Absolute H content, as-synthesized | 0.4(3) mass % |
| Absolute H content, surface-desorbed | 0.2(3) mass % |
| Prompt Gamma Activation Analysis | |
| Mo:N atomic ratio | 1.03(3) |
| Mo:H atomic ratio | 4.05(3) |
| BET Surface Area | 30.1(6) m2/g |
| Pycnometry (measured density) | 9.01(1) g/cm3 |
| X-ray Photoelectron Spectroscopy | |
| Mod+ binding energy | 228.7(1) eV |
| N:O | 1.34 |
| Electron Energy Loss Spectroscopy | |
| Mo edge peak | 396.7(6) eV |
| N edge peak | 400.9(6) eV |
| Space group | P63mc (186) | ||||
| Lattice parameters (Å) | a = b = 5.73835 (13) |c = 5.61840 (7) | ||||
| Composition | MoO0.108(8)N0.892(8)H0.012(5) | ||||
| Density (g/cm3) | 9.135 | ||||
| Site|Element | x | y | z | Occupancy | Uiso, × 102 Å2 |
| Site 2(a), 3m | Mo(1) | 0 | 0 | 0.0 (fixed) | 1 (fixed) | 0.14(2) |
| Site 6(c), m | Mo(2) | 0.48875(4) | 0.51125(4) | 0.0010(5) | 1 (fixed) | 0.038(6) |
| Site 2(b), 3m | N(1) | 1/3 | 2/3 | 0.2779(7) | 1 (fixed) | 0.259(4) |
| Site 6(c), m | N(2) + O(2) | 0.1676(2) | 0.8324(2) | 0.7453(7) | 0.856(10) N | 0.259(4) |
| (net occupancy fixed to 1) | 0.144(10) O | ||||
| Site 2(b), m | H(1) | 1/3 | 2/3 | 0.575(16) | 0.048(18) | 1.00 (fixed) |
| Secondary phase Mo5N6 P63/mmc (194) [35] | 9.71(8) wt.% | ||||
| Lattice parameters (Å) | a = b = 2.8394(3) | c = 11.1563(11) | ||||
| Density (g/cm3) | 8.148 | ||||
| Secondary phase Mo0.78O0.25N0.75 Pmm (221) [7] | 1.94(4) wt.% | ||||
| Lattice parameters (Å) | a = b = c = 4.1886(6) Å | ||||
| Density (g/cm3) | 8.031 | ||||
| Overall sample composition | MoO0.105(7)N0.905(7)H0.011(4) | ||||
| Residual wR (%) | |||||
| Neutron | 3.509%, 3019 observations | ||||
| X-ray | 15.009%, 40182 observations | ||||
| Combined | 7.830% | ||||
| Goodness of Fit | 1.62 | ||||
| Mo(1) | Distance, Å | Mo(2) | Distance, Å |
|---|---|---|---|
| N(2) × 3 | 2.197(3) | N(1) | 2.192(2) |
| N(2) × 3 | 2.162(3) | N(1) | 2.1676(17) |
| N(2) × 2 | 2.1933(16) | ||
| N(2) × 2 | 2.1491(17) | ||
| S | 0.047 | S | 0.052 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, S.; Goldfine, E.A.; Sinha, S.; Zhang, C.; Wenderott, J.K.; Kaczmarczyk, L.; Dabrowiecki, K.; Dravid, V.P.; González, G.B.; Haile, S.M. Steam-Assisted Ammonolysis of MoO2 as a Synthetic Pathway to Oxygenated δ-MoN. Materials 2025, 18, 2340. https://doi.org/10.3390/ma18102340
Pandey S, Goldfine EA, Sinha S, Zhang C, Wenderott JK, Kaczmarczyk L, Dabrowiecki K, Dravid VP, González GB, Haile SM. Steam-Assisted Ammonolysis of MoO2 as a Synthetic Pathway to Oxygenated δ-MoN. Materials. 2025; 18(10):2340. https://doi.org/10.3390/ma18102340
Chicago/Turabian StylePandey, Shobhit, Elise A. Goldfine, Shriya Sinha, Chi Zhang, Jill K. Wenderott, Lucien Kaczmarczyk, Ksawery Dabrowiecki, Vinayak P. Dravid, Gabriela B. González, and Sossina M. Haile. 2025. "Steam-Assisted Ammonolysis of MoO2 as a Synthetic Pathway to Oxygenated δ-MoN" Materials 18, no. 10: 2340. https://doi.org/10.3390/ma18102340
APA StylePandey, S., Goldfine, E. A., Sinha, S., Zhang, C., Wenderott, J. K., Kaczmarczyk, L., Dabrowiecki, K., Dravid, V. P., González, G. B., & Haile, S. M. (2025). Steam-Assisted Ammonolysis of MoO2 as a Synthetic Pathway to Oxygenated δ-MoN. Materials, 18(10), 2340. https://doi.org/10.3390/ma18102340