
The Modulation of the Pore Structure in Porous Carbon by Metal Salts and Its Application for Joining Silicon Carbide Ceramics


Abstract
1. Introduction
2. Experimental Procedure
2.1. Materials
2.2. Porous Carbon Material Synthesis
2.3. Experimental Methods for Joining
2.4. Characterization
3. Results and Discussion
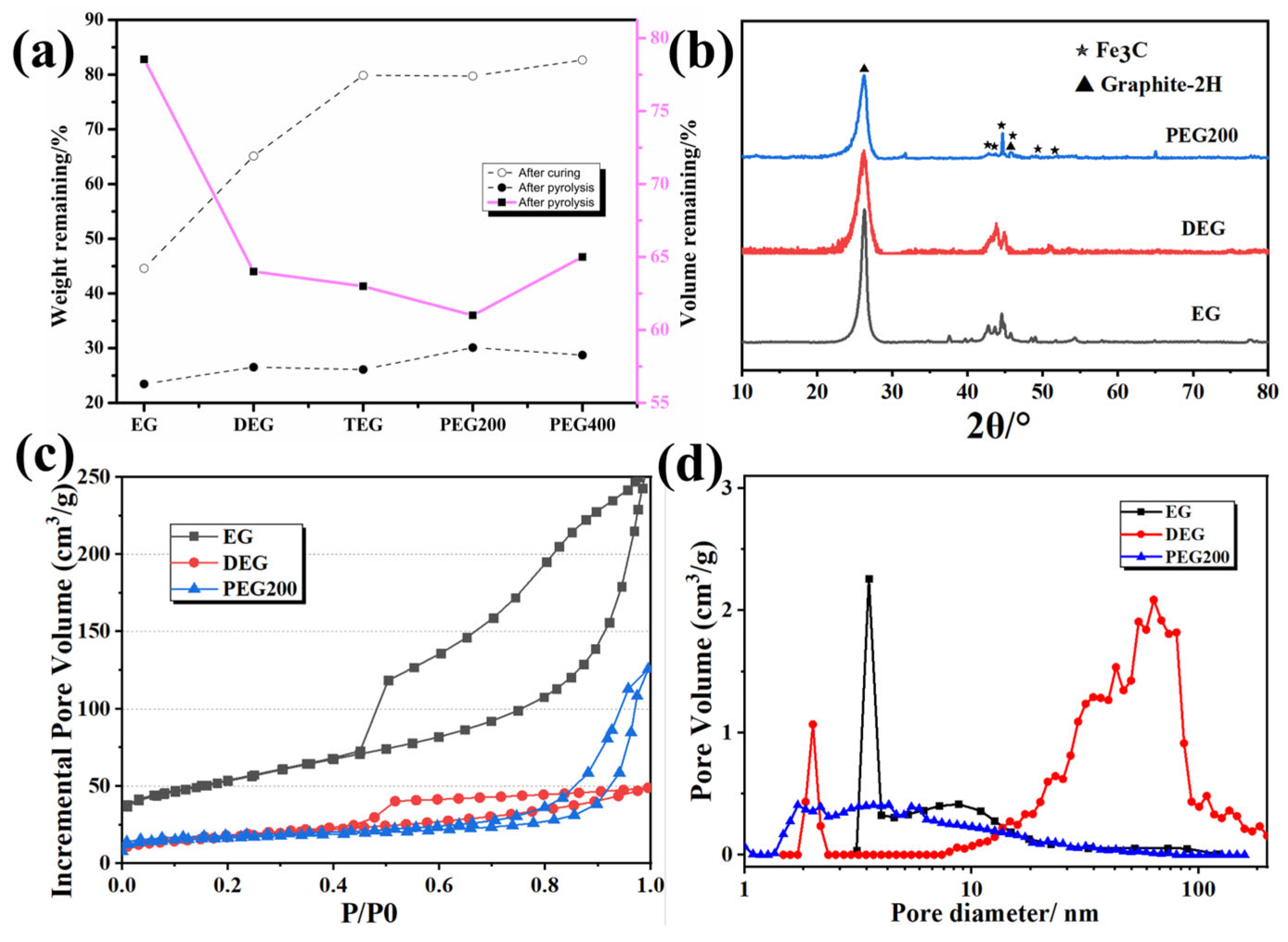
3.1. Influence of Solvent Type and Resin-Solvent Quality on Porous Carbon Pore Structure
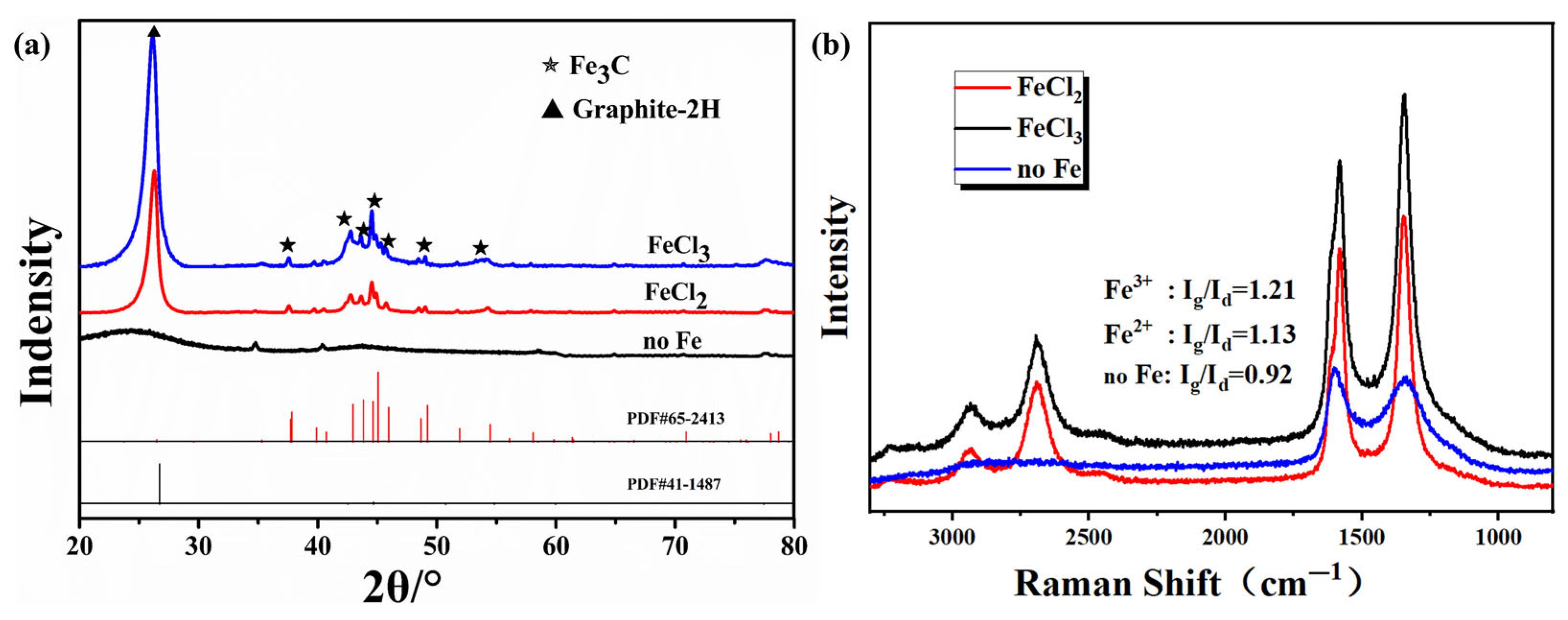
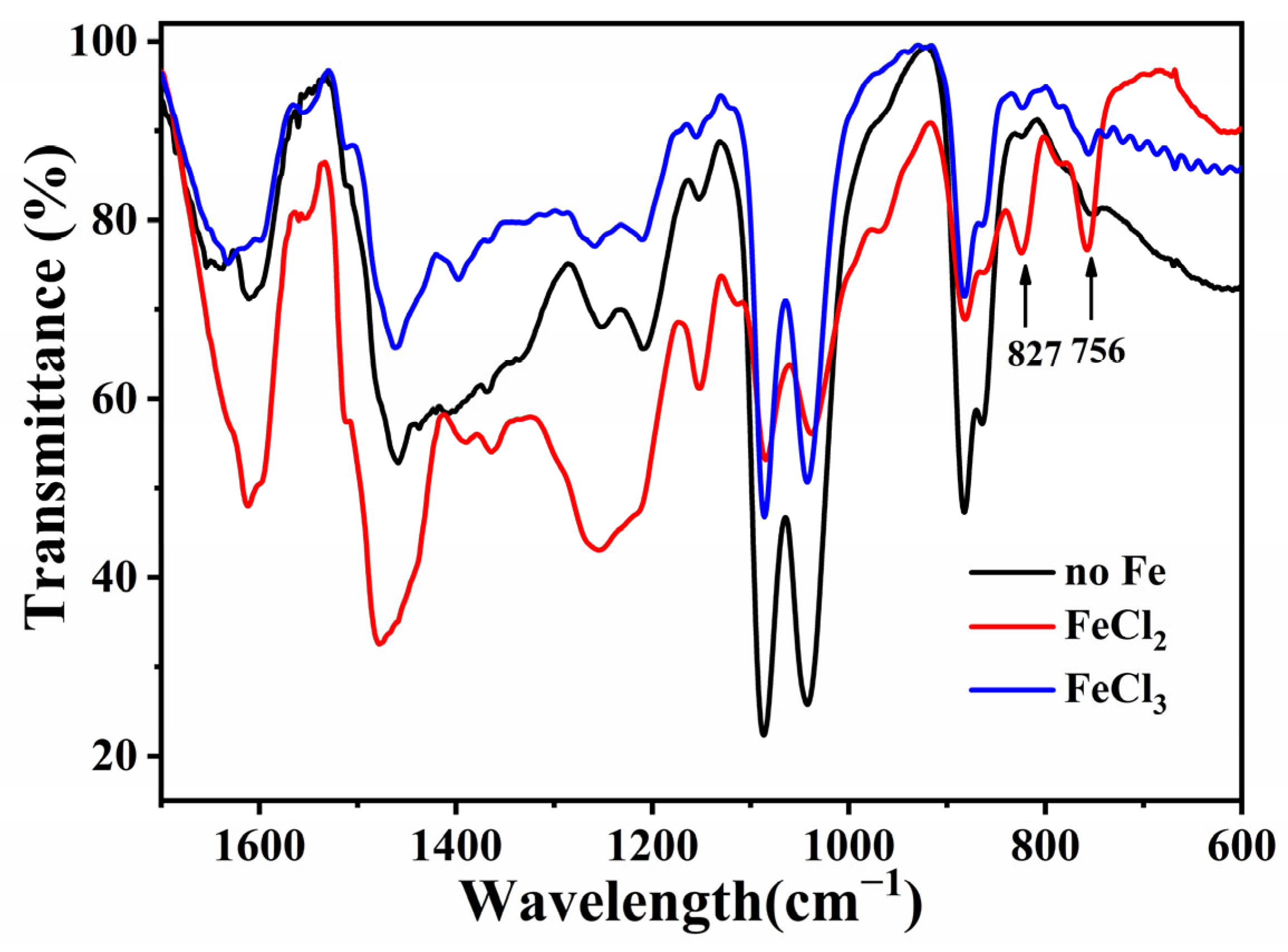
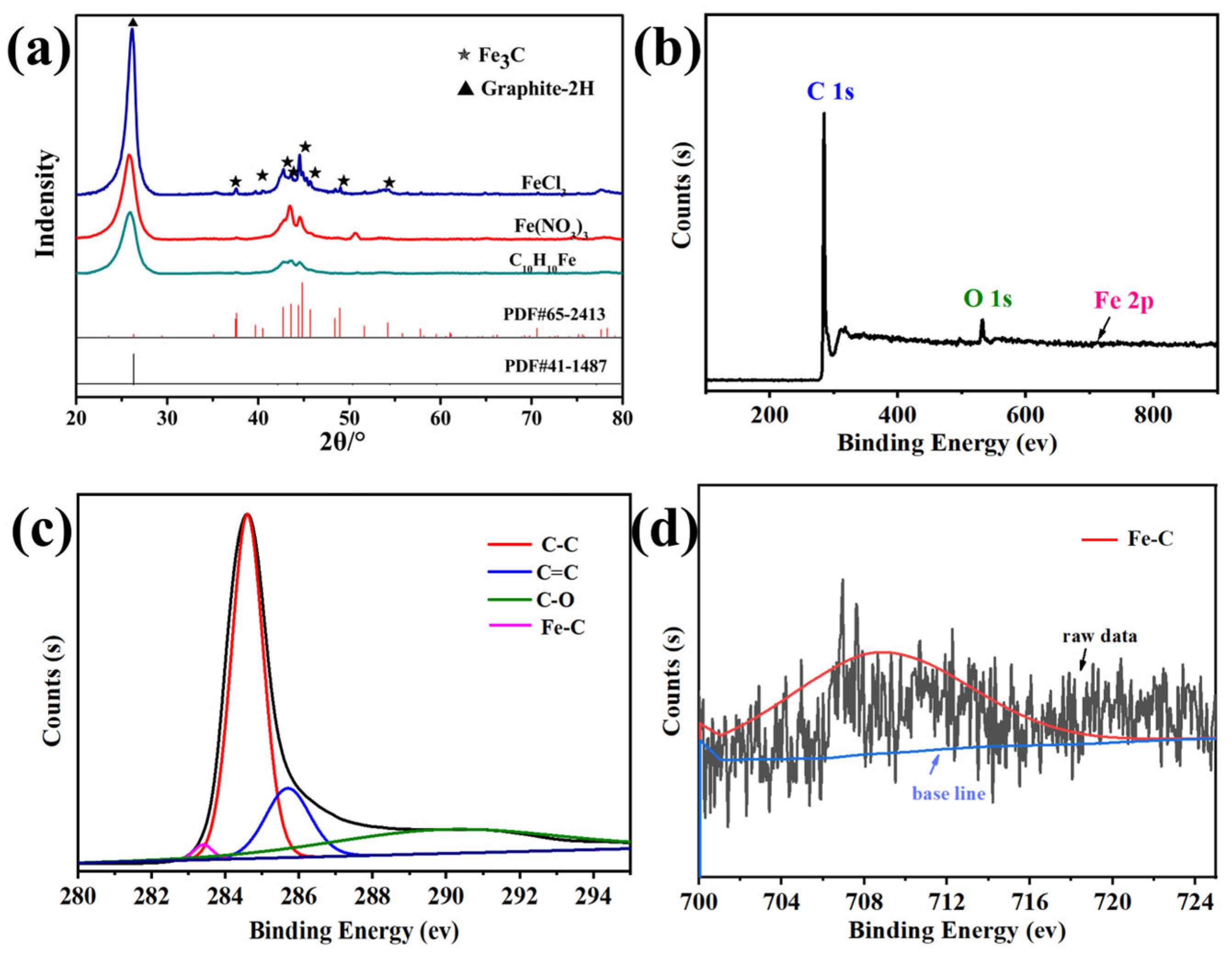
3.2. Influence of Fe Ion Valence State on Pore Structure
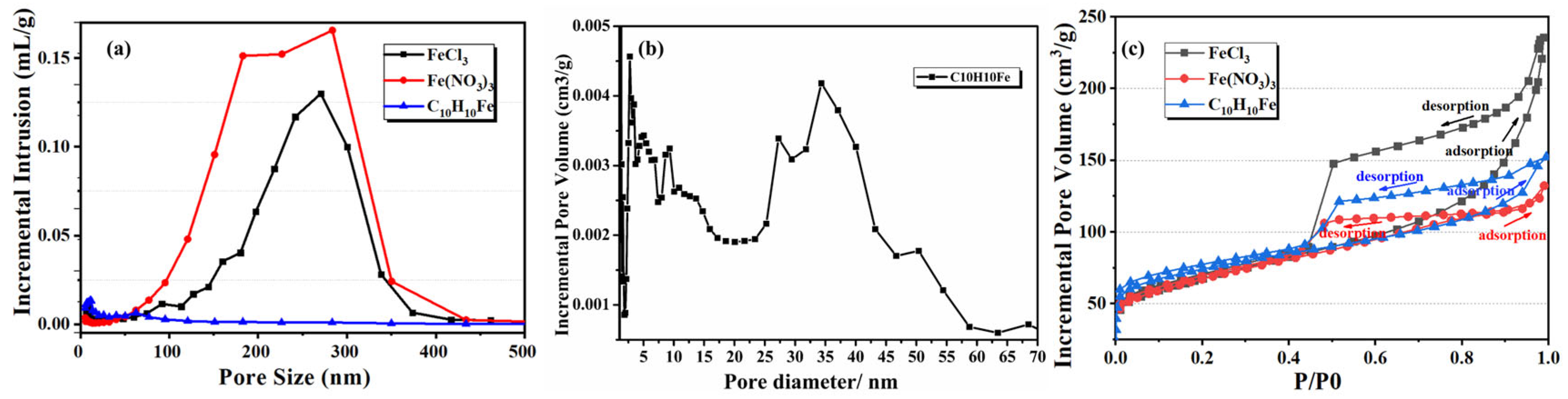
3.3. Influence of Coexisting Anion Type on Porous Carbon Pore Structure
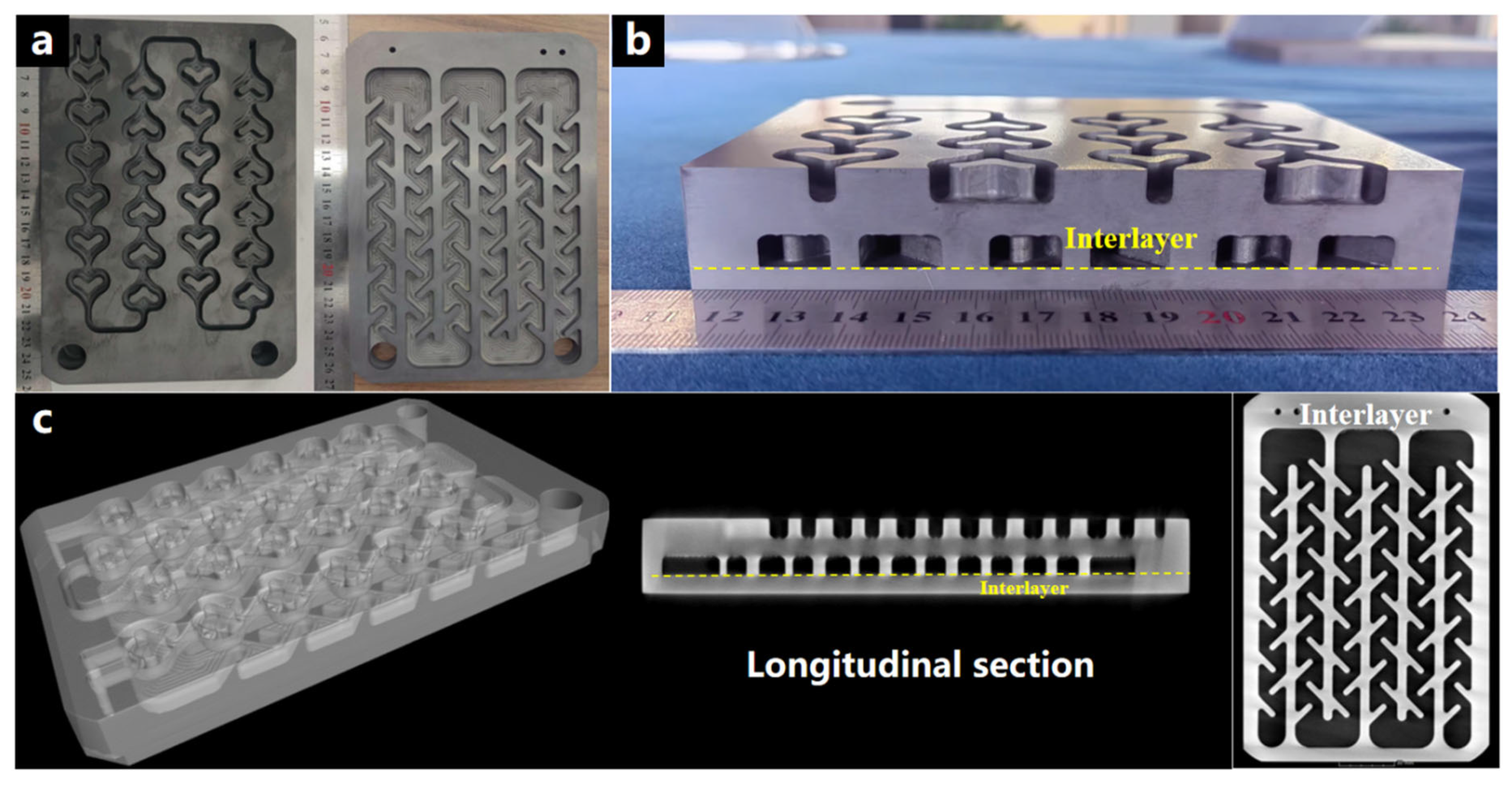
3.4. The Joining of Silicon Carbide Parts
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.; Cheng, C.; Thomas, A. Carbon-based microbial-fuel-cell electrodes: From conductive supports to active catalysts. Adv. Mater. 2017, 29, 1602547. [Google Scholar] [CrossRef] [PubMed]
- Eid, K.; Abdelhafiz, A.A.; Abdel-Azeim, S.; Varma, R.S.; Shibl, M.F. Scalable nitrogen-enriched porous sub-100 nm graphitic carbon nanocapsules for efficient oxygen reduction reaction in different media. Green Chem. 2023, 25, 6748–6758. [Google Scholar] [CrossRef]
- Lu, Q.; Eid, K.; Li, W. Heteroatom-Doped Porous Carbon-Based Nanostructures for Electrochemical CO2 Reduction. Nanomaterials 2022, 12, 2379. [Google Scholar] [CrossRef]
- Salah, B.; Abdelgawad, A.; El-Demellawi, J.K.; Lu, Q.; Xia, Z.; Abdullah, A.M.; Eid, K. Scalable one-pot fabrication of carbon-nanofiber-supported noble-metal-free nanocrystals for synergetic-dependent green hydrogen production: Unraveling electrolyte and support effects. ACS Appl. Mater. Interfaces 2024, 16, 18768–18781. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, Y.; Gu, W.; Zhou, M.; Tang, S.; Cao, J.; Zou, Z.; Ji, G. Research progress on nanostructure design and composition regulation of carbon spheres for the microwave absorption. Carbon 2022, 189, 617–633. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, M.; Xie, Z.; Hao, X.; Tang, S.; Wang, J.; Zou, Z.; Ji, G. Enhanced interfacial polarization of biomass-derived porous carbon with a low radar cross-section. J. Colloid Interface Sci. 2022, 612, 146–155. [Google Scholar] [CrossRef]
- Guan, X.; Yang, Z.; Zhu, Y.; Yang, L.; Zhou, M.; Wu, Y.; Yang, L.; Deng, T.; Ji, G. The controllable porous structure and s-doping of hollow carbon sphere synergistically act on the microwave attenuation. Carbon 2022, 188, 1–11. [Google Scholar] [CrossRef]
- Li, B.; Xiong, H.; Xiao, Y. Progress on synthesis and applications of porous carbon materials. Int. J. Electrochem. Sci. 2020, 15, 1363–1377. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Yang, Y.; Gao, S. A universal strategy for carbon–based ORR–active electrocatalyst: One porogen, two pore–creating mechanisms, three pore types. Nano Energy 2019, 62, 628–637. [Google Scholar] [CrossRef]
- Ding, J.P.; Zhang, H.P.; Li, X.D.; Yang, G.C. Adaptive local mean based empirical mode decomposition and its application in harmonic detections. Mater. Des. 2014, 141, 17–25. [Google Scholar] [CrossRef]
- Kapri, S.; Majee, R.; Bhattacharyya, S. Chemical Modifications of Porous Carbon Nanospheres Obtained from Ubiquitous Precursors for Targeted Drug Delivery and Live Cell Imaging. ACS Sustain. Chem. Eng. 2018, 6, 8503–8514. [Google Scholar] [CrossRef]
- Su, X.L.; Li, S.H.; Jiang, S.; Peng, Z.K.; Guan, X.X.; Zheng, X.C. Superior capacitive behavior of porous activated carbon tubes derived from biomass waste-cotonier strobili fibers. Adv. Powder Technol. 2018, 29, 2097–2107. [Google Scholar] [CrossRef]
- Wu, X.; Liu, J.; Wang, Y.; Zhao, Y.; Li, G.; Zhang, G. Fabrication of porous carbon nanosheets via Urea-Promoted activation of potassium citrate for enhanced supercapacitor performance. Chem. Eng. J. 2025, 512, 162487. [Google Scholar] [CrossRef]
- Li, F.; Zhu, M.; Chen, J.; Huang, C.; Zhu, Y.; Huang, Z. High-strength and low-silicon SiC ceramics prepared by extrusion molding 3D printing. J. Eur. Ceram. Soc. 2024, 44, 617–625. [Google Scholar] [CrossRef]
- Hucke, E.E. Methods of Producing Carbonaceous Bodies and the Products Thereof. U.S. Patent No. 3,859,421, 7 January 1975. [Google Scholar]
- Du, W.; Ma, B.; Thomas, J.; Singh, D. Concurrent reaction-bonded joining and densification of additively manufactured silicon carbide by liquid silicon infiltration. J. Eur. Ceram. Soc. 2023, 43, 2345–2353. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Q.; Zhu, Y.; Huang, Z. Joining of SiC Ceramic by Si–C Reaction Bonding Using Organic Resin as Carbon Precursor. Materials 2022, 15, 4242. [Google Scholar] [CrossRef]
- Seo, J.; Park, H.; Shin, K.; Baeck, S.H.; Rhym, Y.; Shim, S.E. Lignin-derived macroporous carbon foams prepared by using poly(methyl methacrylate) particles as the template. Carbon 2014, 76, 357–367. [Google Scholar] [CrossRef]
- Han, S.; Feng, Y.; Zhang, F.; Yang, C.; Yao, Z.; Zhao, W.; Qiu, F.; Yang, L.; Yao, Y.; Zhuang, X.; et al. Metal-Phosphide-Containing Porous Carbons Derived from an Ionic-Polymer Framework and Applied as Highly Efficient Electrochemical Catalysts for Water Splitting. Adv. Funct. Mater. 2015, 25, 3899–3906. [Google Scholar] [CrossRef]
- Xu, S.; Li, J.; Qiao, G.; Wang, H.; Lu, T. Pore structure control of mesoporous carbon monoliths derived from mixtures of phenolic resin and ethylene glycol. Carbon 2009, 47, 2103–2111. [Google Scholar] [CrossRef]
- Choi, S.H.; Kang, Y.C. Polystyrene-Templated Aerosol Synthesis of MoS2–Amorphous Carbon Composite with Open Macropores as Battery Electrode. ChemSusChem 2015, 8, 2260–2267. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, J.; Liu, Q.; Zhang, X. Macroporous interconnected hollow carbon nanofibers inspired by golden-toad eggs toward a binder-free, high-rate, and flexible electrode. Adv. Mater. 2016, 28, 7494–7500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tan, S.; Jiang, D.; Zhang, X. Preparation of porous carbon derived from mixtures of furfuryl resin and glycol with controlled pore size distribution. Carbon 2003, 41, 2065–2072. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, Z.; Zhou, Y. Effect of furfural alcohol/phenol-formaldehyde resin mass ratio on the properties of porous carbon. Mater. Lett. 2013, 109, 124–126. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhang, Y.; Zhou, Y.; Han, J. Preparation and characterization of porous carbons obtained from mixtures of furfuryl alcohol and phenol-formaldehyde resin. Mater. Chem. Phys. 2014, 143, 707–712. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhang, Y.; Zhou, Y. Effect of curing catalyst content on the pore structure of porous carbon obtained from phenolic resin and furfuryl alcohol. Mater. Lett. 2013, 110, 218–220. [Google Scholar] [CrossRef]
- Pizzi, A. Kinetics of the metal-catalyzed condensations of phenols and polyelavonoid tannins with formaldehyde. J. Polym. Sci. 1980, 18, 3447–3454. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, Y.; Pei, B.; Cai, P.; Huang, Z. Effect of FeCl2 on the pore structure of porous carbon obtained from phenol formaldehyde resin and ethylene glycol. Mater. Lett. 2018, 215, 50–52. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, Y.; Huang, Q.; Huang, Z. Facile synthesis of magnetic hierarchical porous carbon and the adsorption properties. Mater. Charact. 2022, 193, 112273. [Google Scholar] [CrossRef]
- Hashimoto, T.; Takenaka, M.; Jinnai, H. Morphology control of polymer blends by a chemical pinning of spinodal decomposition. Polymer 1989, 30, 177–179. [Google Scholar]
- Salmani, H.; Bilibin, A. Nanoprecipitation-miniemulsion polymerization combined method: A novel approach to synthesis drug loaded nanoparticles with tunable characteristics. Eur. Polym. J. 2016, 84, 631–641. [Google Scholar] [CrossRef]
- Chen, D.; Feng, C.; Han, Y.; Yu, B.; Chen, W.; Zhou, Z.; Chen, N.; Goodenough, J.B.; He, W. Origin of extra capacity in the solid electrolyte interphase near high-capacity iron carbide anodes for Li ion batteries. Energy Environ. Sci. 2020, 13, 2924–2937. [Google Scholar] [CrossRef]
- Urbonaite, S.; Hälldahl, L.; Svensson, G. Raman spectroscopy studies of carbide derived carbons. Carbon 2008, 46, 1942–1947. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M. Gas adsorption characterization of ordered organic− inorganic nanocomposite materials. Chem. Mater. 2001, 13, 3169–3183. [Google Scholar] [CrossRef]
- Ouchi, K. Infra-red study of structural changes during the pyrolysis of a phenol-formaldehyde resin. Carbon 1966, 4, 59–66. [Google Scholar] [CrossRef]
- Zhao, M.; Song, H.; Chen, X.; Lian, W. Large-scale synthesis of onion-like carbon nanoparticles by carbonization of phenolic resin. Acta Mater. 2007, 55, 6144–6150. [Google Scholar] [CrossRef]
- Behrendt, D.R.; Singh, M. Effect of carbon preform pore volume and infiltrants on the composition of reaction-formed silicon carbide materials. J. Mater. Synth. Process. 1994, 2, 117–123. [Google Scholar]
- Sung, H.-W.; Kim, Y.-H.; Kim, D.J. Joining of reaction bonded silicon carbide using self-infiltration of residual Si present in the RBSC. Ceram. Int. 2020, 46, 28800–28805. [Google Scholar] [CrossRef]
- Wu, X.; Pei, B.; Zhu, Y.; Huang, Z. Joining of the Cf/SiC composites by a one-step Si infiltration reaction bonding. Mater. Charact. 2019, 155, 109799. [Google Scholar] [CrossRef]
- Giuranno, D.; Sobczak, N.; Bruzda, G.; Nowak, R.; Polkowski, W.; Polkowska, A.; Kudyba, A.; Novakovic, R. Studies of the joining-relevant interfacial properties in the Si-Ti/C and Si-Ti/SiC systems. J. Mater. Eng. Perform. 2020, 29, 4864–4871. [Google Scholar] [CrossRef]
- Grasso, S.; Tatarko, P.; Rizzo, S.; Porwal, H.; Hu, C.; Katoh, Y.; Salvo, M.; Reece, M.J.; Ferraris, M. Joining of β-SiC by spark plasma sintering. J. Eur. Ceram. Soc. 2014, 34, 1681–1686. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent Type | Melting Point /°C | Viscosity /mPa·s | Average Molecular Weight | Bulk Density (g·cm−3) | Apparent Porosity (%) | Materials Compositions (wt%) |
|---|---|---|---|---|---|---|
| EG | 186.5 | 16 | 62.07 | 0.71 ± 0.03 | 52.9 ± 1.0 | 97.83 (C), 0.34 (Fe), 1.83 (O) |
| DEG | 245.5 | 28 | 106.12 | 0.98 ± 0.02 | 34.7 ± 0.9 | 97.24 (C), 0.38 (Fe), 1.83 (O) |
| TEG | 288.0 | 35 | 150.17 | 1.08 ± 0.04 | 28.1 ± 1.3 | 97.45 (C), 0.41 (Fe), 2.14 (O) |
| PEG200 | >250 | 31 | 200.00 | 1.19 ± 0.03 | 21.2 ± 1.1 | 98.23 (C), 0.40 (Fe), 1.37 (O) |
| PEG400 | >250 | 41 | 400.00 | 1.10 ± 0.02 | 16.5 ± 0.8 | 96.53 (C), 0.45 (Fe), 3.02 (O) |
| Resin/Solvent | Apparent Porosity (%) | Bulk Density (g·cm−3) | Carbon Residue Rate/% | Materials Compositions (wt%) |
|---|---|---|---|---|
| 7:3 | 34.2 ± 2.6 | 0.99 ± 0.02 | 34.83 ± 1.9 | 96.23 (C), 0.42 (Fe), 3.35 (O) |
| 5:5 | 52.9 ± 3.2 | 0.71 ± 0.04 | 25.53 ± 2.1 | 97.83 (C), 0.34 (Fe), 1.83 (O) |
| 1:2 | 66.5 ± 1.6 | 0.50 ± 0.03 | 19.48 ± 1.5 | 97.28 (C), 0.31 (Fe), 2.41 (O) |
| 2:8 | —— | —— | 5.61 ± 0.7 | 95.14 (C), 0.21 (Fe), 4.65 (O) |
| Sample | Apparent Porosity (%) | Average Aperture (nm) | Bulk Density (g·cm−3) | Vtotal (cm3·g−1) | Skeleton Density (g·cm−3) | Materials Compositions (wt%) |
|---|---|---|---|---|---|---|
| NoFe | 25.6 ± 1.1 | 14 ± 5 | 1.18 ± 0.08 | 0.21 ± 0.07 | 1.55 ± 0.04 | 98.11 (C), 1.89 (O) |
| FeCl2 | 63.3 ± 1.7 | 190 ± 15 | 0.73 ± 0.01 | 0.86 ± 0.02 | 1.91 ± 0.05 | 97.83 (C), 0.34 (Fe), 1.83 (O) |
| FeCl3 | 50.9 ± 1.3 | 248 ± 29 | 0.95 ± 0.02 | 0.75 ± 0.03 | 1.94 ± 0.03 | 97.51 (C), 0.45 (Fe), 2.04 (O) |
| Sample | Apparent Porosity (%) | Average Aperture (nm) | Bulk Density (g·cm−3) | Vtotal (cm3·g−1) | Skeleton Density (g·cm−3) | Materials Compositions (wt%) |
|---|---|---|---|---|---|---|
| FeCl3 | 50.9 ± 1.3 | 248 ± 29 | 0.95 ± 0.02 | 0.75 ± 0.03 | 1.94 ± 0.03 | 97.51 (C), 0.45 (Fe), 2.04 (O) |
| Fe(NO3)3 | 51.4 ± 1.9 | 228 ± 21 | 0.97 ± 0.01 | 0.67 ± 0.03 | 1.89 ± 0.05 | 97.42 (C), 0.46 (Fe), 2.12 (O) |
| C10H10Fe | 30.9 ± 1.9 | 8 ± 3 | 1.21 ± 0.02 | 0.24 ± 0.04 | 1.75 ± 0.02 | 97.51 (C), 0.45 (Fe), 2.04 (O) |
| no Fe | 25.6 ± 1.1 | 14 ± 5 | 1.18 ± 0.08 | 0.21 ± 0.07 | 1.55 ± 0.04 | 98.11 (C), 1.89 (O) |
| Joining Base Material | Interlayer Materials | Joining Condition | Joint Strength | Source |
|---|---|---|---|---|
| SiC-SiC | PF-FeCl3 | Silicon powder, 1600 °C, vac | Flexural strength 309 MPa; Weibull modulus 10.67 | This work |
| SiC-SiC | Parchment paper | 1450~1550 °C, vac | Flexural strength 243~246 MPa | [38] |
| Cf/SiC- Cf/SiC | PF | Silicon powder, 1600 °C, vac | Flexural strength 203 ± 24 MPa | [39] |
| SiCf/SiC- SiCf/SiC | Si-Ti/SiC | Si-Ti infiltration, 1350 °C 2 h | / | [40] |
| additive-manufactured SiC | graphite paper | Silicon powder, 1550 °C 2 h | / | [16] |
| SiC-SiC | / | Spark Plasma Sintering (SPS) 1900 °C, 5 min, 60 MPa, | Flexural strength 193 ± 21 MPa | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Liu, Z.; Pei, B.; Wu, H.; Huang, Z. The Modulation of the Pore Structure in Porous Carbon by Metal Salts and Its Application for Joining Silicon Carbide Ceramics. Materials 2025, 18, 2336. https://doi.org/10.3390/ma18102336
Wu X, Liu Z, Pei B, Wu H, Huang Z. The Modulation of the Pore Structure in Porous Carbon by Metal Salts and Its Application for Joining Silicon Carbide Ceramics. Materials. 2025; 18(10):2336. https://doi.org/10.3390/ma18102336
Chicago/Turabian StyleWu, Xishi, Zehua Liu, Bingbing Pei, Haibo Wu, and Zhengren Huang. 2025. "The Modulation of the Pore Structure in Porous Carbon by Metal Salts and Its Application for Joining Silicon Carbide Ceramics" Materials 18, no. 10: 2336. https://doi.org/10.3390/ma18102336
APA StyleWu, X., Liu, Z., Pei, B., Wu, H., & Huang, Z. (2025). The Modulation of the Pore Structure in Porous Carbon by Metal Salts and Its Application for Joining Silicon Carbide Ceramics. Materials, 18(10), 2336. https://doi.org/10.3390/ma18102336