

Development of a Polyethylene Glycol/Polymethyl Methacrylate-Based Binder System for a Borosilicate Glass Filler Suitable for Injection Molding
 ,
,
Abstract
:1. Introduction
- (a)
- feedstock preparation (compounding), including material selection;
- (b)
- comprehensive rheological characterization with respect to feedstock composition, shear rate, and temperature;
- (c)
- debinding, covering the steps of liquid pre-debinding and thermal debinding;
- (d)
- sintering, including characterization of the sintered part.
2. Materials and Methods
2.1. Material Selection
2.2. Compounding and Rheological Characterization
2.3. Glass Injection Molding
2.4. Debinding
2.5. Sintering and Further Densification by Hot Isostatic Pressing (HIP)
2.6. Sintered Glass Part Characterization
3. Results and Discussion
3.1. Feedstock Compounding and Melt Flow Behavior
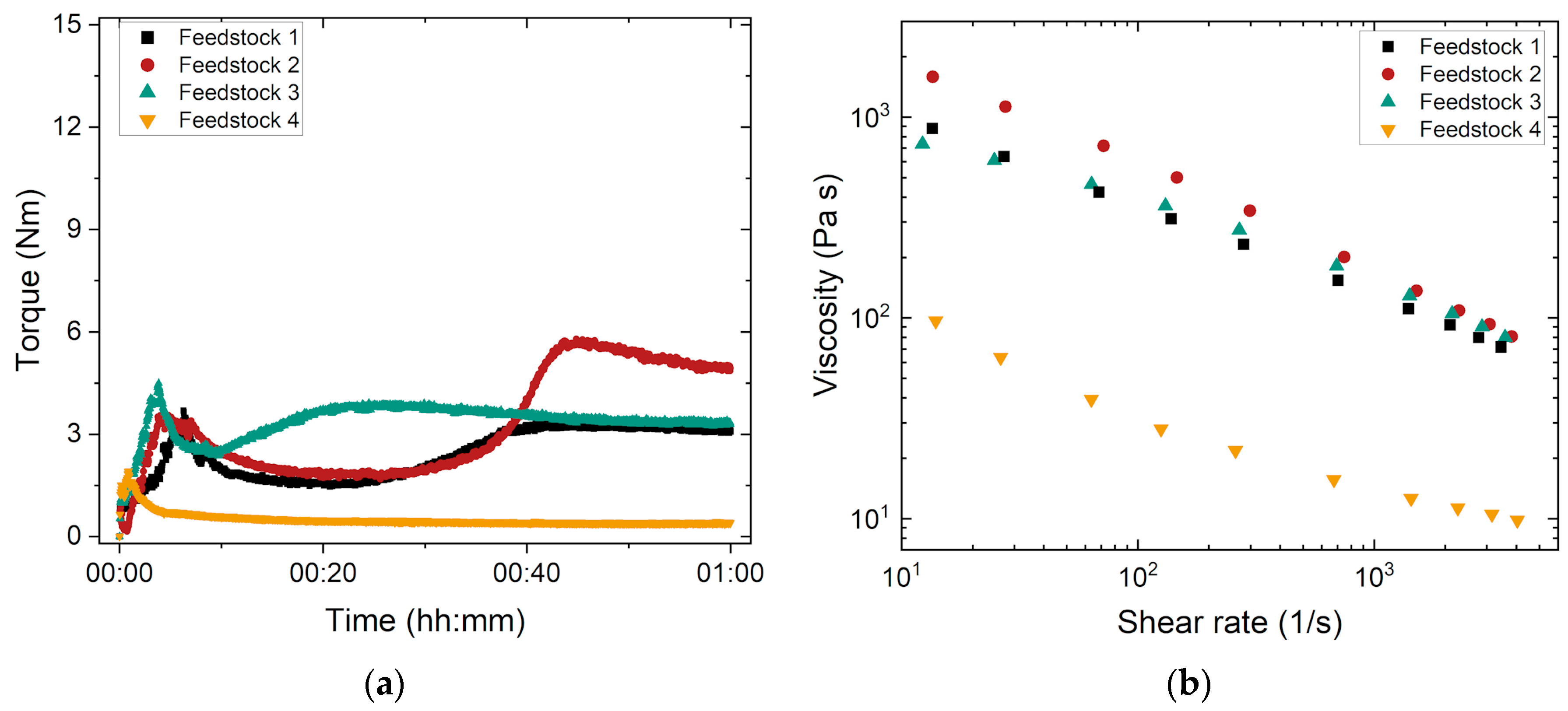
3.1.1. Influence of the PMMA’s Average Molecular Mass
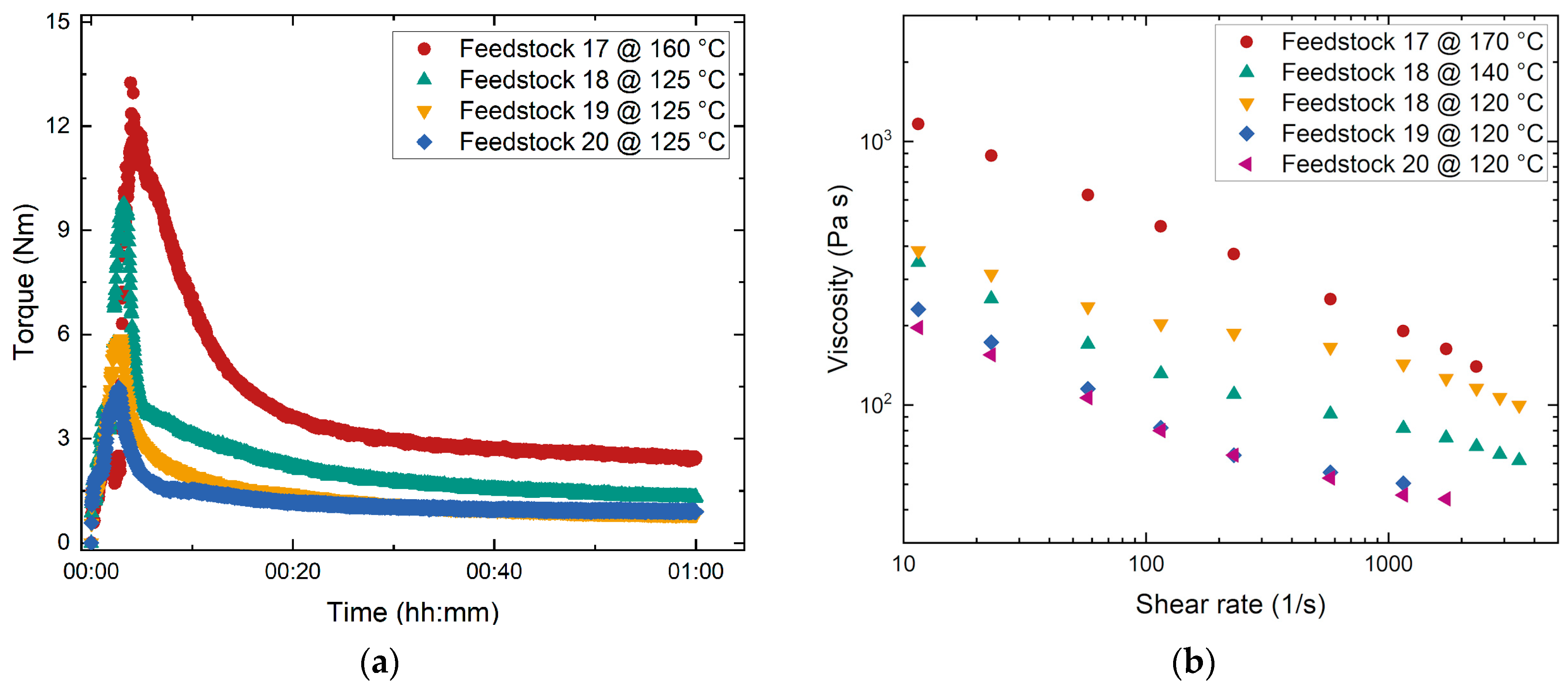
- Filling state: pronounced torque increase caused by filling all materials (PEG, PMMA, SA, and glass powder) into the kneader and pronounced friction between the solid glass particles prior to wetting.
- Mixing state: drop in the torque curve due to the disruption of agglomerates and particle wetting by the different binder components, especially the surfactant.
- Equilibrium (stationary) state with a stable final torque value reflecting a homogeneous feedstock and good moldability.
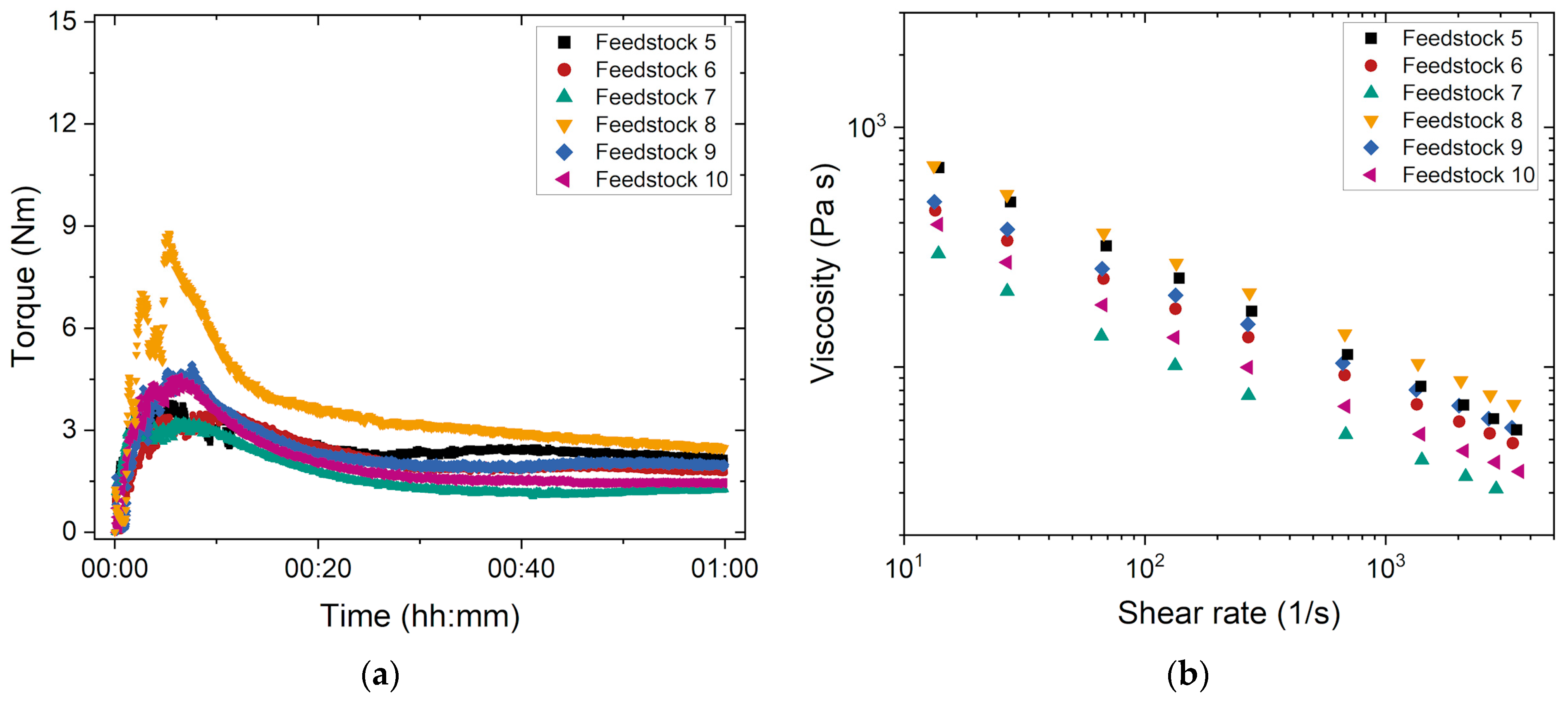
3.1.2. Influence of the PEG’s Average Molecular Mass and Stearic Acid Amount
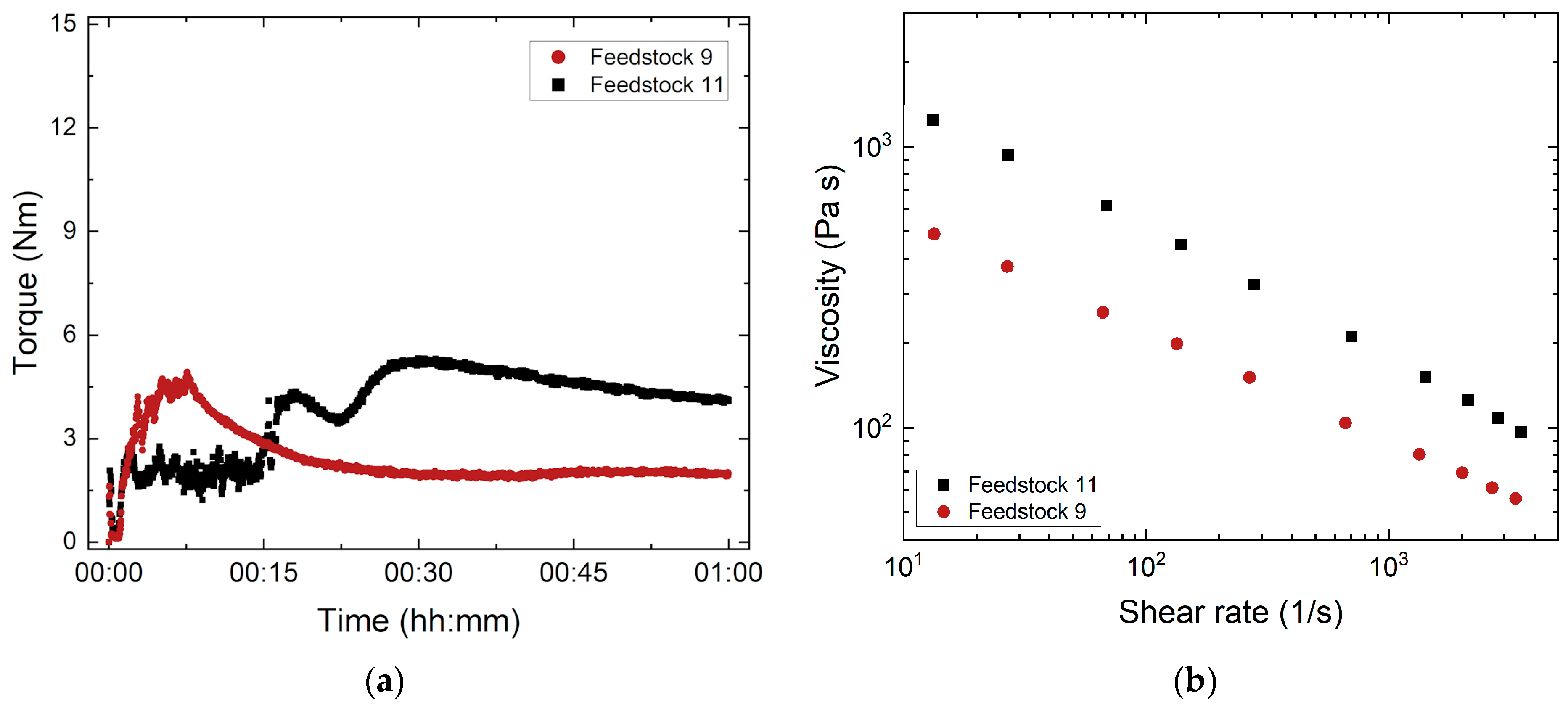
3.1.3. Influence of the PEG/PMMA Ratio
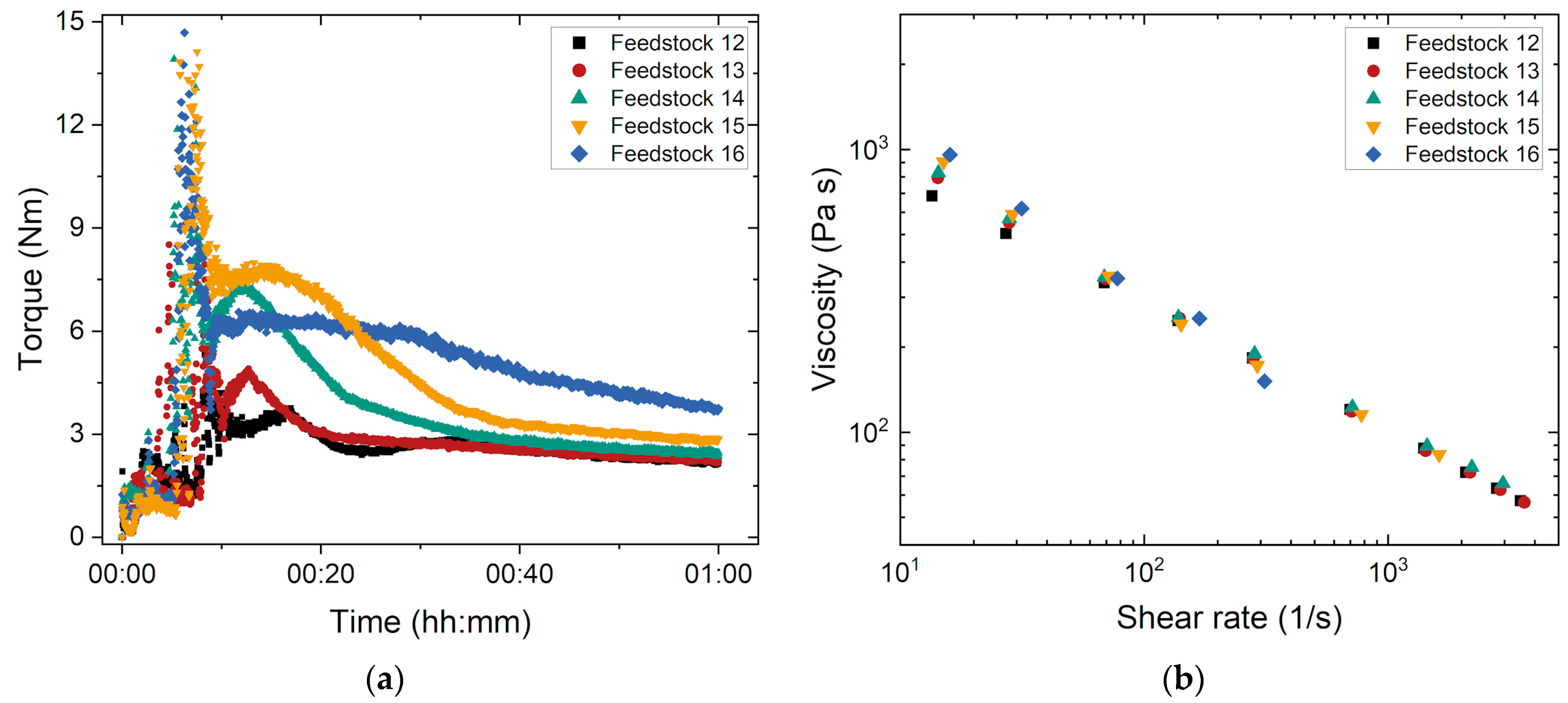
3.1.4. Influence of the Solid Load
3.1.5. Influence of the PMMA’s Average Molecular Mass–Revisited
3.2. Injection Molding
3.3. Debinding
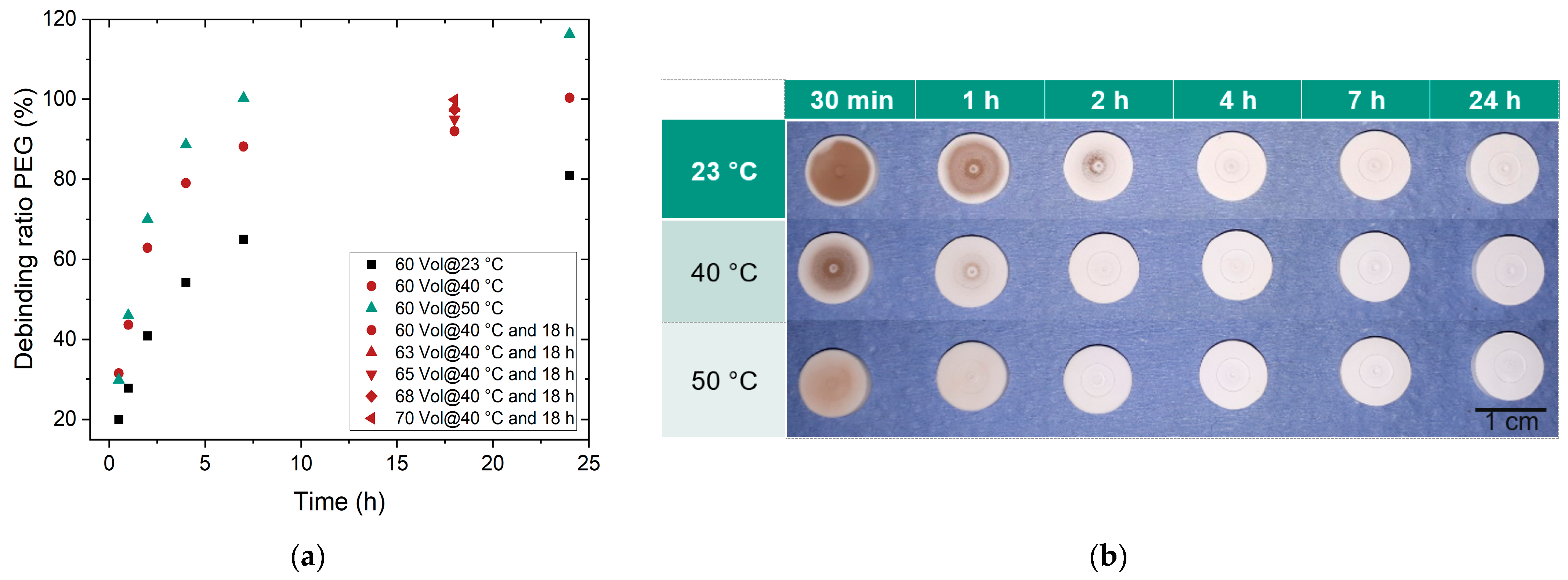
3.3.1. Liquid Pre-Debinding
3.3.2. Thermal Debinding
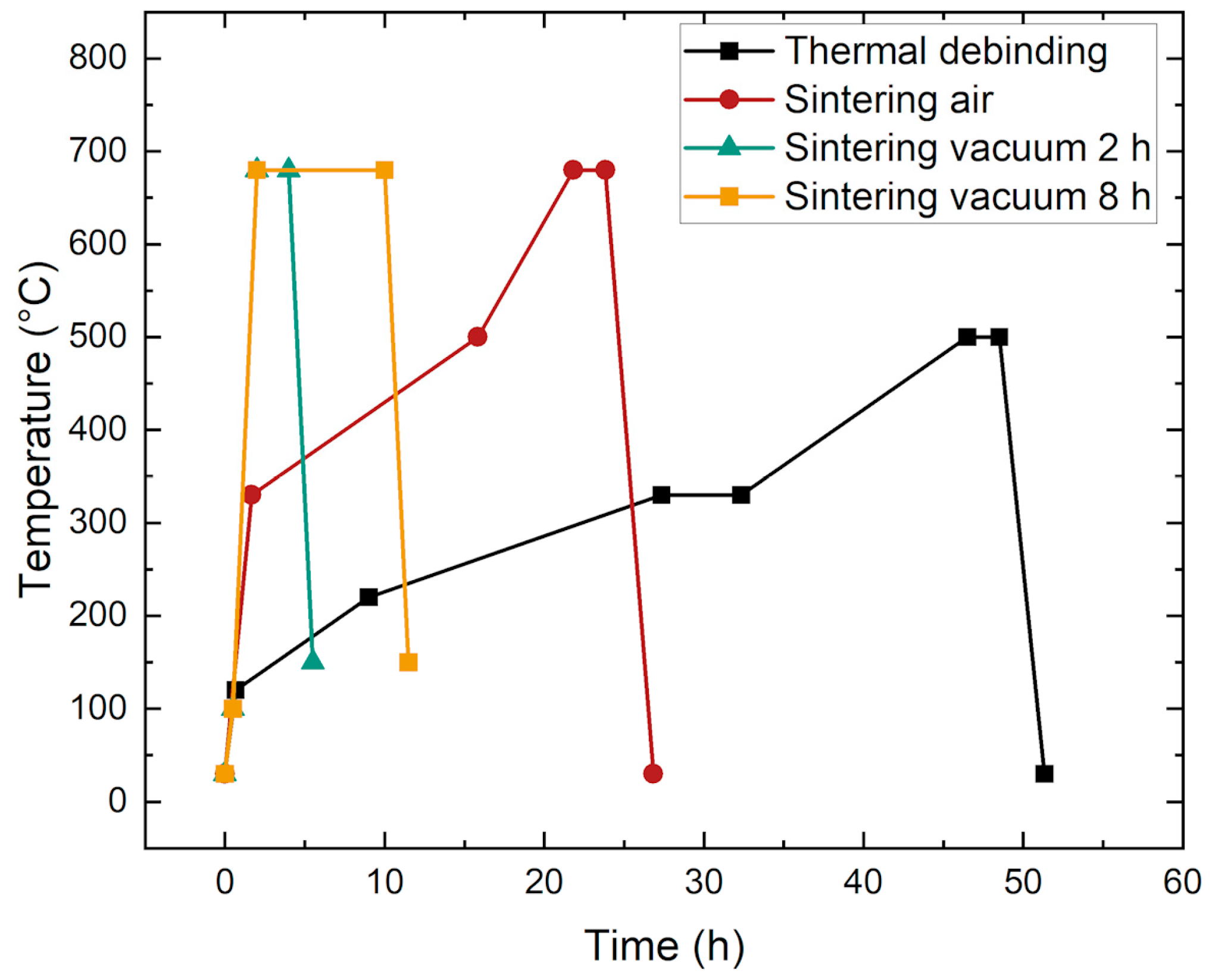
3.4. Sintering Process Development
3.5. Influence of Feedstock Composition, Debinding, and Sintering on Part Density
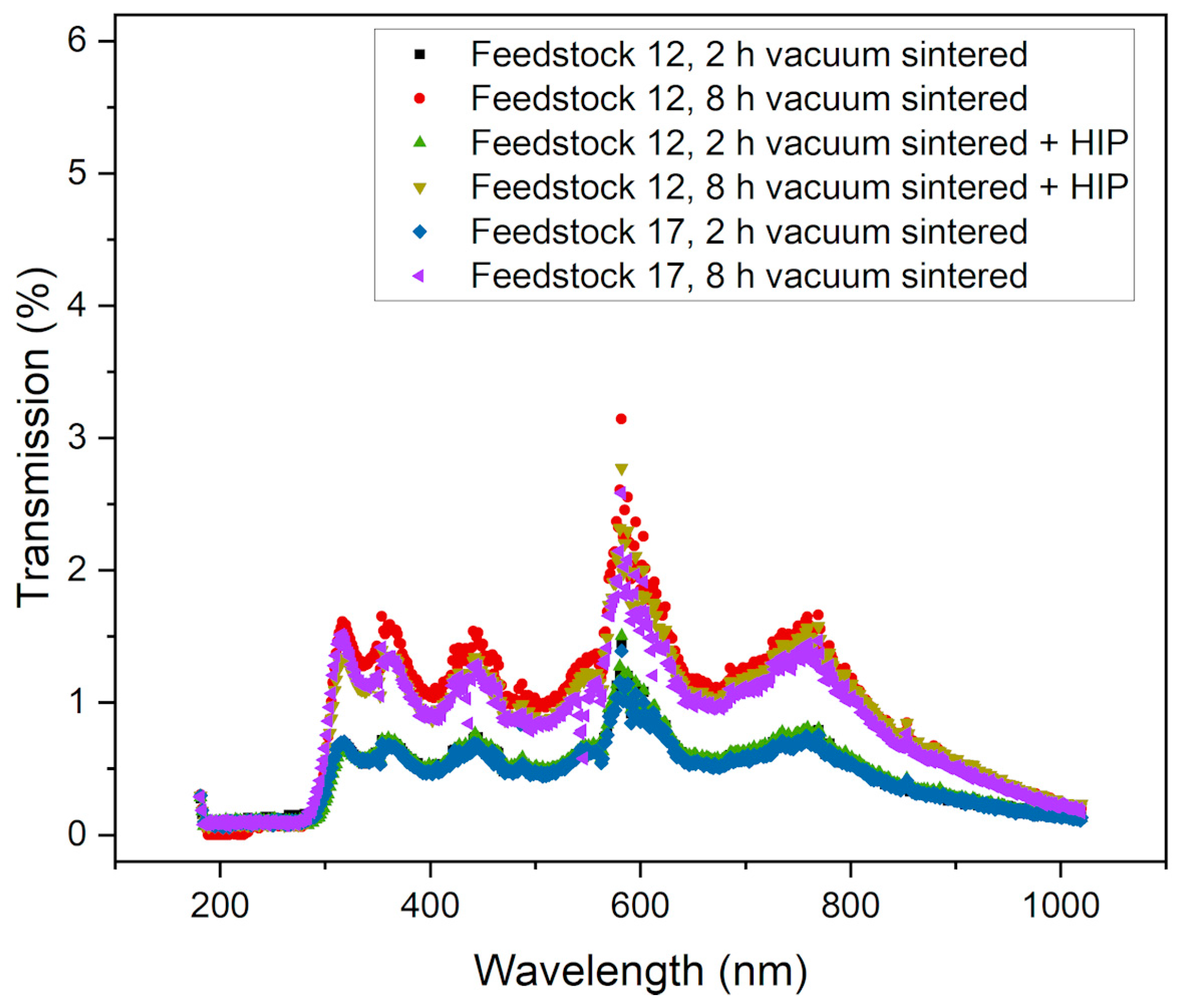
3.6. Optical Properties
4. Conclusions
- The glass injection molding process chain was evaluated systematically, from feedstock development to molding, to debinding, to sintering.
- In the first process step, feedstock development using the given binder components PEG, PMMA, and SA, the average molecular masses of PEG and PMMA, their ratios, and the SA content were varied to enable simple, fast, and reliable compounding as well as good molding. During replication, good mold filling as well as defect-free stable demolding were ensured by selecting suitable feedstock compositions. As regards the debinding procedure, it was found that a combination of liquid pre-debinding and thermal treatment was recommended. This could be verified after the final sinter process. In this step, vacuum sintering is also favorable to achieve the highest sinter densities. Sinter densities of around 99–100% of theoretical density could be achieved.
- An increase in the initial feedstock’s solid load does not result in any improvement in the final sinter densities and part appearance. This also holds when an additional thermal post-treatment by HIP takes place.
- Suitable feedstock systems with 60 Vol% glass filler, 25 mg/m2 SA, and PEG as well as PMMA having different average molecular weights in combination with two-step debinding and vacuum sintering can be recommended for further investigations.
- A certain translucency was measured, with optical transmission values reaching up to 3% in the visible range.
- The comprehensive investigations allow for a clear correlation between the feedstock composition and the influence of each individual binder component on compounding, molding, debinding, and sintering.
5. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mauro, J.C. Grand Challenges in Glass Science. Front. Mater. 2014, 1, 13. [Google Scholar] [CrossRef]
- Quintero, F.; Penide, J.; Riveiro, A.; Del Val, J.; Comesaña, R.; Lusquiños, F.; Pou, J. Continuous fiberizing by laser melting (Cofiblas): Production of highly flexible glass nanofibers with effectively unlimited length. Sci. Adv. 2020, 6, eaax7210. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.; Kamin’ski, J.; Maria Scalet, B.; Soria, A. Energy consumption and CO2 emissions of the European glass industry. Energy Policy 2011, 39, 142–155. [Google Scholar] [CrossRef]
- Butler, J.H.; Hooper, P.D. Chapter 15—Glass Waste. In Waste; Letcher, T.M., Vallero, D.A., Eds.; Academic Press: London, UK, 2019; pp. 307–322. [Google Scholar] [CrossRef]
- Fu, H.; Xu, H.; Liu, Y.; Yang, Z.; Kormakov, S.; Wu, D.; Sun, J. Overview of Injection Molding Technology for Processing Polymers and Their Composites. ES Mater. Manuf. 2020, 8, 3–23. [Google Scholar] [CrossRef]
- Czepiel, M.; Bankosz, M.; Sobczak-Kupiec, A. Advanced Injection Molding Methods: Review. Materials 2023, 16, 5802. [Google Scholar] [CrossRef] [PubMed]
- German, R.M. Powder Injection Molding; Metal Powder Industries Federation: Princeton, NJ, USA, 1990. [Google Scholar]
- German, R.M.; Bose, A. Injection Molding of Metals and Ceramics; Metal Powder Industries Federation: Princeton, NJ, USA, 1997. [Google Scholar]
- Yunn, H.S.; Muhamad, N.; Sulong, A.B. Micro Powder Injection Molding (µPIM): Review. Appl. Mech. Mater. 2011, 52–54, 91–96. [Google Scholar] [CrossRef]
- Piotter, V. A review of the current status of MicroMIM Part I. Powder Inject. Mould. Int. 2011, 5, 27–36. [Google Scholar]
- Piotter, V. A review of the current status of MicroMIM Part II. Powder Inject. Mould. Int. 2011, 5, 25–30. [Google Scholar]
- Moritz, T.; Lenk, R. Ceramic injection molding: A review of developments in production technology, materials and applications. Powder Inject. Mould. Int. 2009, 3, 23–34. [Google Scholar]
- Momeni, V.; Hufnagl, M.; Shahroodi, Z.; Gonzalez-Gutierrez, J.; Schuschnigg, S.; Kukla, C.; Holzer, C. Research Progress on Low-Pressure Powder Injection Molding. Materials 2022, 16, 379. [Google Scholar] [CrossRef]
- Mader, M.; Schlatter, O.; Heck, B.; Warmbold, A.; Dorn, A.; Zappe, H.; Risch, P.; Helmer, D.; Kotz, F.; Rapp, B.E. High-throughput injection molding of transparent fused silica glass. Science 2021, 372, 182–186. [Google Scholar] [CrossRef]
- Hidalgo, J.; Conteras, J.M.; Berzal, D.; Jiménez-Morales, A.; Torralba, J.M. Fabrication of Glass Components by Powder Injection Moulding (PIM) Recycling Glass Waster. In Proceedings of the PM 2010 World Congress, Florence, Italy, 10–14 October 2010. [Google Scholar]
- Giassi, L.; Hotza, D.; Alarcon, O.E.; Fredel, M.C.; Novaes de Oliveira, A.P. Sintering and Crystallization of LZSA Glass Powder Compacts Formed by Injection Molding. Am. Ceram. Soc. Bull. 2005, 84, 9301–9306. [Google Scholar]
- Enríquez, E.; Berges, C.; Fuertes, V.; Gallego, A.; Naranjo, J.A.; Herranz, G.; Fernández, J.F. Ceramic Injection Moulding of engineered glass-ceramics: Boosting the rare-earth free photoluminescence. Ceram. Int. 2020, 46, 9334–9341. [Google Scholar] [CrossRef]
- Hanemann, T.; Heldele, R. Fatty Acid Surfactant Structure-Feedstock Flow Properties: Correlation for High-Pressure Ceramic Injection Molding. Int. J. Appl. Ceram. Technol. 2011, 8, 1296–1304. [Google Scholar] [CrossRef]
- Thomas-Vielma, P.; Cervera, A.; Levenfeld, B.; Várez, A. Production of alumina parts by powder injection molding with a binder system based on high density polyethylene. J. Eur. Ceram. Soc. 2008, 28, 763–771. [Google Scholar] [CrossRef]
- Fayyaz, A.; Muhamad, N.; Sulong, A.B.; Rajabi, J.; Wong, Y.N. Fabrication of cemented tungsten carbide components by micro-powder injection moulding. J. Mater. Proc. Technol. 2014, 214, 1436–1444. [Google Scholar] [CrossRef]
- Xie, Z.P.; Wang, L.L.; Yang, X.F.; Zhang, Z.T. Water Debinding for Zirconia Powder Injection Molding. High-Perform. Ceram. V 2008, 368–372, 732–735. [Google Scholar] [CrossRef]
- Chakartnarodom, P.; Kongkajun, N.; Chuankrerkkul, N. Powder Injection Molding of Mullite: The Study of Binder Dissolution Behavior during Debinding Step Using Statistical Methods. Tradit. Adv. Ceram. II 2016, 690, 87–91. [Google Scholar] [CrossRef]
- Thavanayagam, G.; Pickering, K.L.; Swan, J.E.; Cao, P. Analysis of rheological behaviour of titanium feedstocks formulated with a water-soluble binder system for powder injection moulding. Powder Technol. 2015, 269, 227–232. [Google Scholar] [CrossRef]
- Hayat, M.D.; Goswami, A.; Matthews, S.; Li, T.; Yuan, X.; Cao, P. Modification of PEG/PMMA binder by PVP for titanium metal injection moulding. Powder Technol. 2017, 315, 243–249. [Google Scholar] [CrossRef]
- Hnatkova, E.; Hausnerova, B.; Hales, A.; Jiranek, L.; Derguti, F.; Todd, I. Processing of MIM feedstocks based on Inconel718 powder and partially water-soluble binder varying in PEG molecular weight. Powder Technol. 2017, 322, 439–446. [Google Scholar] [CrossRef]
- Basir, A.; Sulong, A.B.; Jamadon, N.H.; Muhamad, N.; Emeka, U.B. Process Parameters Used in Macro/Micro Powder Injection Molding: An Overview. Met. Mater. Int. 2021, 27, 2023–2045. [Google Scholar] [CrossRef]
- Hanemann, T.; Weber, O. Polymethylmethacrylate/polyethyleneglycol-based partially water soluble binder system for micro ceramic injection moulding. Microsyst. Technol. 2014, 30, 51–58. [Google Scholar] [CrossRef]
- Eickhoff, R.; Antusch, S.; Nötzel, D.; Hanemann, T. New Partially Water-Soluble Feedstocks for Additive Manufacturing of Ti6Al4V Parts by Material Extrusion. Materials 2023, 16, 3162. [Google Scholar] [CrossRef] [PubMed]
- Moritz, T.; Schilm, J.; Mannschatz, A.; Peschel, M. Glass Powder Injection Molding—Ceramic Processing Applied to Glass Components. Ceram. Appl. 2014, 2, 44–49. [Google Scholar]
- Schilm, J.; Moritz, T.; Wagner, D.; Schwarzer, E.; Weingarten, S.; Wätzig, K. Additive Manufacturing of Functionalized Glass Components. Ceram. Appl. 2019, 7, 28–34. [Google Scholar]
- Hanemann, T.; Heldele, R. Modern Alchemy: Ceramic Feedstock Optimization by Surfactant Screening. CFI Ceram. Forum Int. 2010, 87, E38–E40. [Google Scholar]
- Weber, O. Wasserlösliche Bindersysteme zum Minimieren von Pulver-Binder-Segregationseffekten im Mikropulverspritzguss. Ph.D. Dissertation, Albert-Ludwigs-Universität Freiburg im Breisgau, Freiburg im Breisgau, Germany, 2015. [Google Scholar] [CrossRef]
- Altstädt, V.; de Lucca Freitas, L.; Schubert, D.W. Rheological and mechanical properties of poly(α-methylstyrene-co-acrylonitrile)/poly(methylacrylate-co-methyl methacrylate) blends in miscible and phase-separated regimes of various morphologies part IV. influence of the morphology on the mechanical properties. Pure Appl. Chem. 2004, 76, 389–413. [Google Scholar]
- Zaky, M.T. Effect of solvent debinding variables on the shape maintenance of green molded bodies. J. Mater. Sci. 2004, 39, 3397–3402. [Google Scholar] [CrossRef]
- Thomas, Y.; Marple, B.R. Partially Water-Soluble Binder Formulation for Injection Molding Submicrometer Zirconia. Adv. Perform. Mater. 1998, 5, 25–41. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binder Component | Function | Supplier |
|---|---|---|
| PEG 4000 | Plasticizer | Carl Roth GmbH + Co., KG, Karlsruhe, Germany |
| PEG 8000 | Plasticizer | Carl Roth GmbH + Co., KG, Karlsruhe, Germany |
| PEG 20000 | Plasticizer | Carl Roth GmbH + Co., KG Karlsruhe, Germany |
| PMMA G77 | Backbone Polymer | Quinn Plastics GmbH, Mainz, Germany |
| PMMA G7E | Backbone Polymer | Röhm, Darmstadt, Germany |
| PMMA 120k | Backbone Polymer | Sigma-Aldrich, Taufkirchen, Germany |
| PMMA 15k | Backbone Polymer | Sigma-Aldrich, Taufkirchen, Germany |
| SA | Surfactant/Release agent | Carl Roth GmbH + Co., KG, Karlsruhe, Germany |
| Feedstock ID | PMMA Type | Average Molecular Mass MW (g/mol) |
|---|---|---|
| 1 | G77 | 88,000 [33] |
| 2 | G7E | 159,000 [28] |
| 3 | Sigma 120k | 120,000 1 |
| 4 | Sigma 15k | 15,000 1 |
| Feedstock ID | PEG Type | PEG/PMMA Ratio | SA Conc. (mg/m2) |
|---|---|---|---|
| 5 | 8000 1 | 65:35 | 4.4 |
| 6 | 8000 1 | 65:35 | 10 |
| 7 | 8000 1 | 65:35 | 20 |
| 8 | 20,000 1 | 65:35 | 4.4 |
| 9 | 20,000 1 | 65:35 | 10 |
| 10 | 20,000 1 | 65:35 | 20 |
| Feedstock ID | PEG Type | PMMA Type | PEG/PMMA Ratio | SA Conc. (mg/m2) |
|---|---|---|---|---|
| 9 | 20,000 | G7E | 65:35 | 10 |
| 11 | 20,000 | G7E | 50:50 | 10 |
| Feedstock ID | Solid Load (Vol%) | PEG Type | PMMA Type | PEG/PMMA Ratio | SA Conc. (mg/m2) |
|---|---|---|---|---|---|
| 12 | 60 | 20,000 | G7E | 50:50 | 25 |
| 13 | 63 | 20,000 | G7E | 50:50 | 25 |
| 14 | 65 | 20,000 | G7E | 50:50 | 25 |
| 15 | 68 | 20,000 | G7E | 50:50 | 25 |
| 16 | 70 | 20,000 | G7E | 50:50 | 25 |
| Feedstock ID | Solid Load (Vol%) | PEG Type | PMMA Type | PEG/PMMA Ratio | SA Conc. (mg/m2) |
|---|---|---|---|---|---|
| 17 | 60 | 20,000 | Sigma 120k | 50:50 | 25 |
| 18 | 60 | 20,000 | Sigma 15k | 50:50 | 25 |
| 19 | 60 | 8000 | Sigma 15k | 50:50 | 25 |
| 20 | 60 | 4000 | Sigma 15k | 50:50 | 25 |
| Feedstock System | Applied Feedstocks | Melt Injection Temperature (°C) | Injection Speed (mm/s) | Cooling Time (s) |
|---|---|---|---|---|
| PMMA G77 based | 1 | 170 | 50 | 30 |
| PMMA G7E-based | 2, 11–16 | 170 | 50 | 30 |
| PMMA G7E-based | 5–10 | 170 | 50 | 120–180 |
| PMMA 120k-based | 3, 17 | 160–170 | 50–65 | 20–30 |
| PMMA 15k-based | 18–20 | 105–120 | 50–65 | 20–30 |
| Feedstock ID | Debinding Method | Sinter Density (% th.) |
|---|---|---|
| 2 | thermal | 95.7 |
| 2 | liquid, thermal | 98.8 |
| 3 | thermal | 92.3 |
| 3 | liquid, thermal | 98.1 |
| 5 | thermal | 96.9 |
| 5 | liquid, thermal | 98.7 |
| 6 | thermal | 97.2 |
| 6 | liquid, thermal | 98.9 |
| 7 | thermal | 97.3 |
| 7 | liquid, thermal | 98.8 |
| 9 | thermal | 97.3 |
| 9 | liquid, thermal | 98.9 |
| 10 | thermal | 97.2 |
| 10 | liquid, thermal | 99.0 |
| 11 | thermal | 96.9 |
| 11 | liquid, thermal | 98.9 |
| 12 | thermal | 96.8 |
| 12 | liquid, thermal | 99.1 |
| 13 | liquid, thermal | 99.0 |
| 14 | liquid, thermal | 99.1 |
| 15 | liquid, thermal | 98.7 |
| 16 | liquid, thermal | 98.7 |
| 17 | liquid, thermal | n.a., see Table 9 |
| 18 | thermal | 98.4 |
| 20 | liquid, thermal | n.a., see Table 9 |
| Feedstock ID | Sintering Method | Sinter Density (% th.) |
|---|---|---|
| 12 | 2 h, 680 °C, vacuum | 99.8 ± 0.1 |
| 12 | 2 h, 680 °C, vacuum, 1 h, 550 °C, HIP | 100.4 ± 0.03 |
| 12 | 8 h, 680 °C, vacuum | 99.6 ± 0.04 |
| 12 | 8 h, 680 °C, vacuum, 1 h, 550 °C, HIP | 100.3 ± 0.06 |
| 17 | 2 h, 680 °C, vacuum | 99.8 ± 0.05 |
| 17 | 2 h, 680 °C, vacuum, 1 h, 550 °C, HIP | 100.4 ± 0.04 |
| 17 | 8 h, 680 °C, vacuum | 99.5 ± 0.09 |
| 17 | 8 h, 680 °C, vacuum, 1 h, 550 °C, HIP | 100.3 ± 0.06 |
| 20 | 2 h, 680 °C, vacuum | 99.4 ± 0.3 |
| 20 | 2 h, 680 °C, vacuum, 1 h, 550 °C, HIP | 100.1 ± 0.06 |
| 20 | 8 h, 680 °C, vacuum | 99.5 ± 0.07 |
| 20 | 8 h, 680 °C, vacuum, 1 h, 550 °C, HIP | 100.3 ± 0.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zürn, M.; Schrage, A.; Antusch, S.; Bohn, N.; Holzer, P.; Hanemann, T. Development of a Polyethylene Glycol/Polymethyl Methacrylate-Based Binder System for a Borosilicate Glass Filler Suitable for Injection Molding. Materials 2024, 17, 1396. https://doi.org/10.3390/ma17061396
Zürn M, Schrage A, Antusch S, Bohn N, Holzer P, Hanemann T. Development of a Polyethylene Glycol/Polymethyl Methacrylate-Based Binder System for a Borosilicate Glass Filler Suitable for Injection Molding. Materials. 2024; 17(6):1396. https://doi.org/10.3390/ma17061396
Chicago/Turabian StyleZürn, Martin, Annika Schrage, Steffen Antusch, Nicole Bohn, Peter Holzer, and Thomas Hanemann. 2024. "Development of a Polyethylene Glycol/Polymethyl Methacrylate-Based Binder System for a Borosilicate Glass Filler Suitable for Injection Molding" Materials 17, no. 6: 1396. https://doi.org/10.3390/ma17061396