
Derivatives of Phenyl Pyrimidine and of the Different Donor Moieties as Emitters for OLEDs
 , and
, and
Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials and Methods
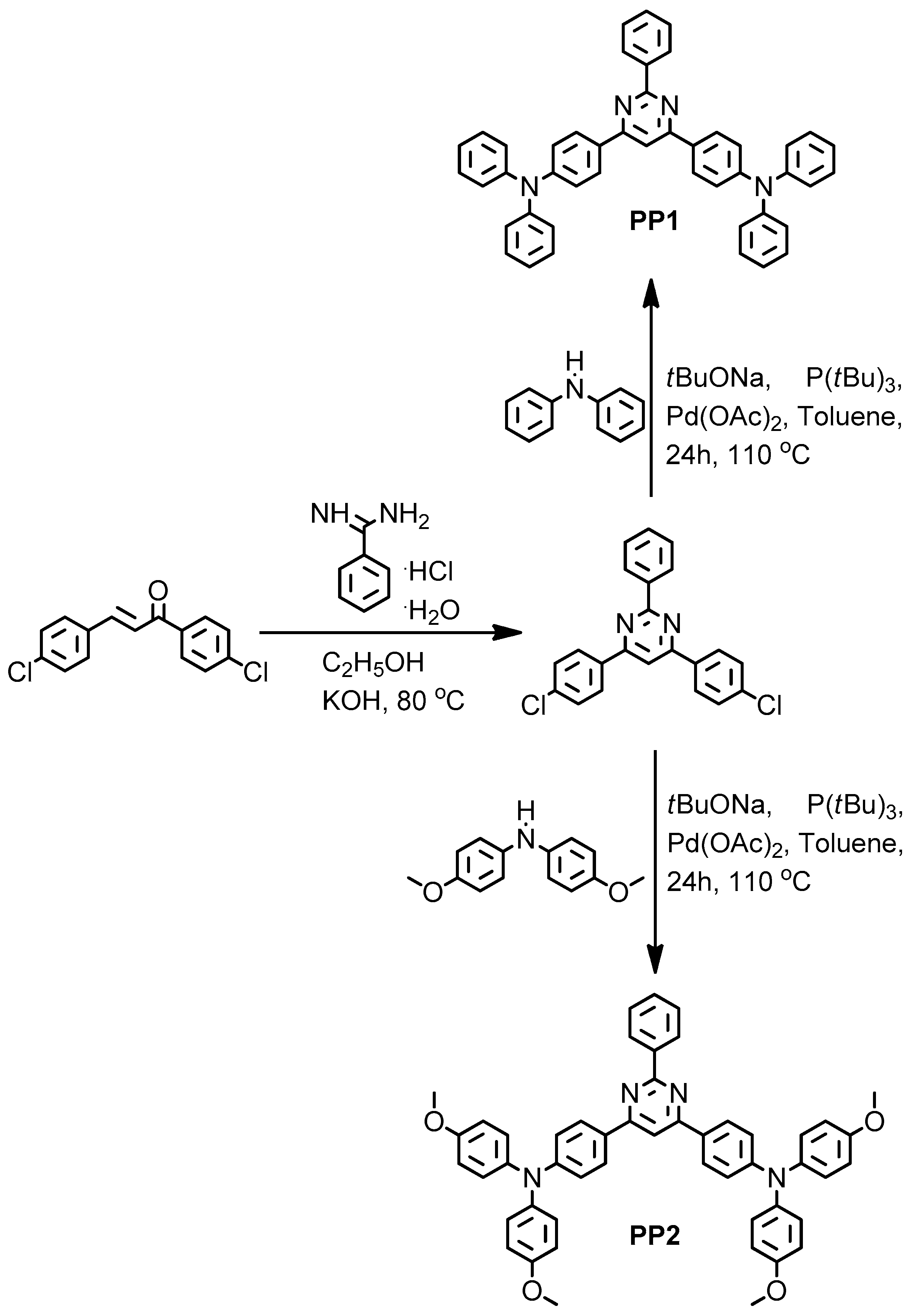
2.1.1. 4,6-Bis(4-Chlorophenyl)-2-phenylpyrimidine
2.1.2. 4,6-Bis(4-Diphenylamino-phenyl)-2-phenylpyrimidine (PP1)
2.1.3. 4,6-Bis((4-Di(4-metoxyphenil)amino)-phenil)-2-phenilpyrimidine (PP2)
3. Results and Discussion
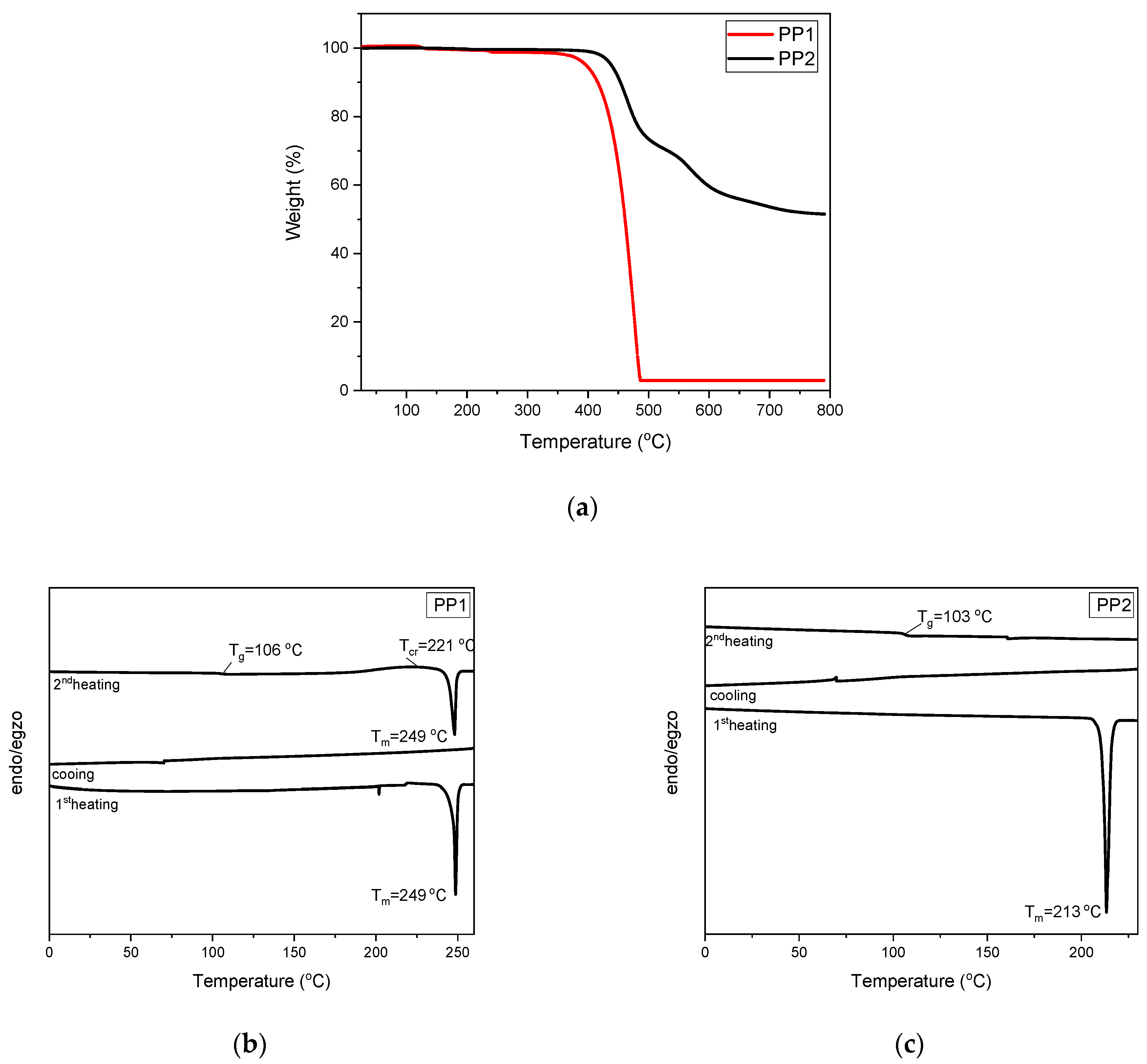
3.1. Synthesis and Thermal Properties
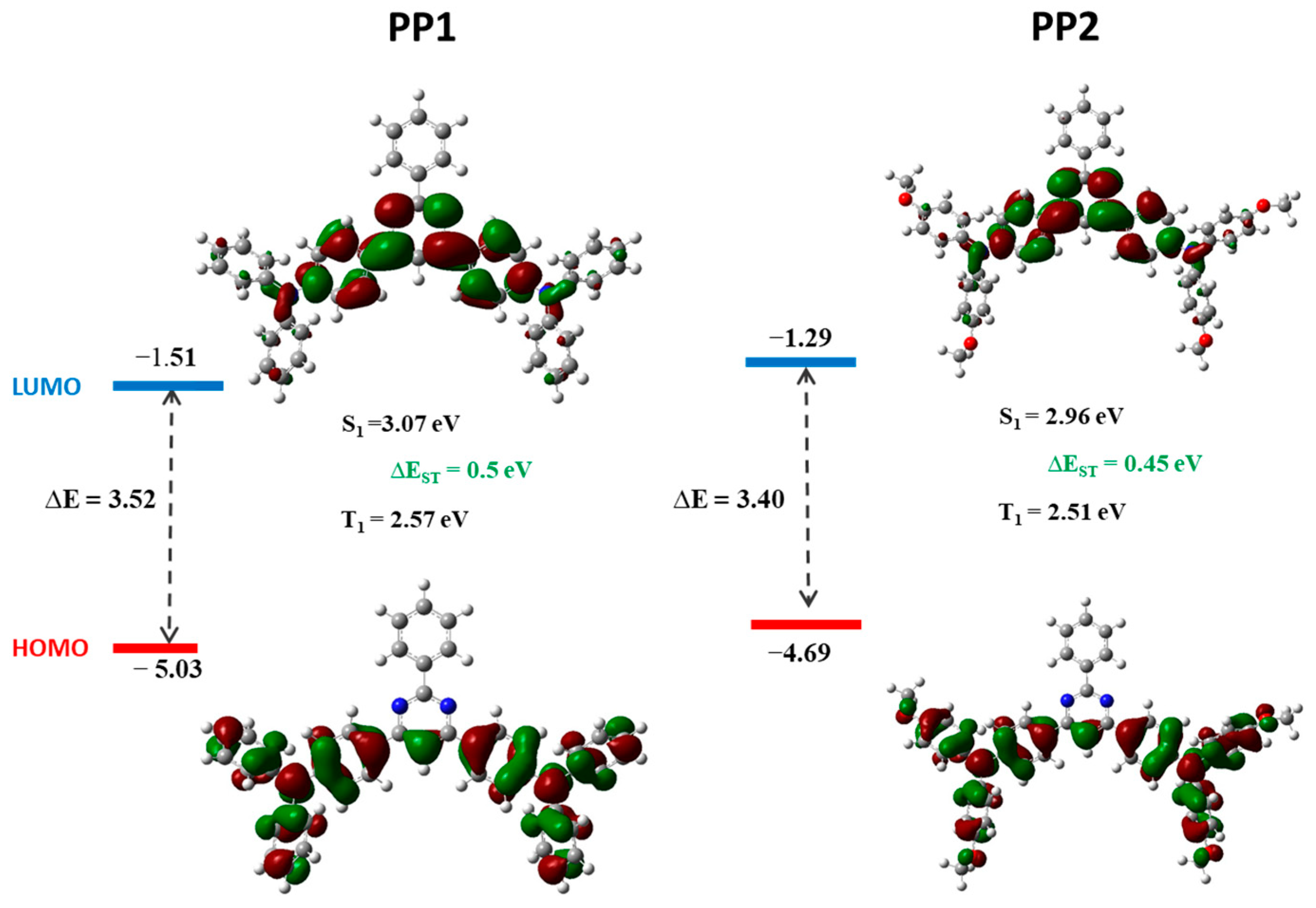
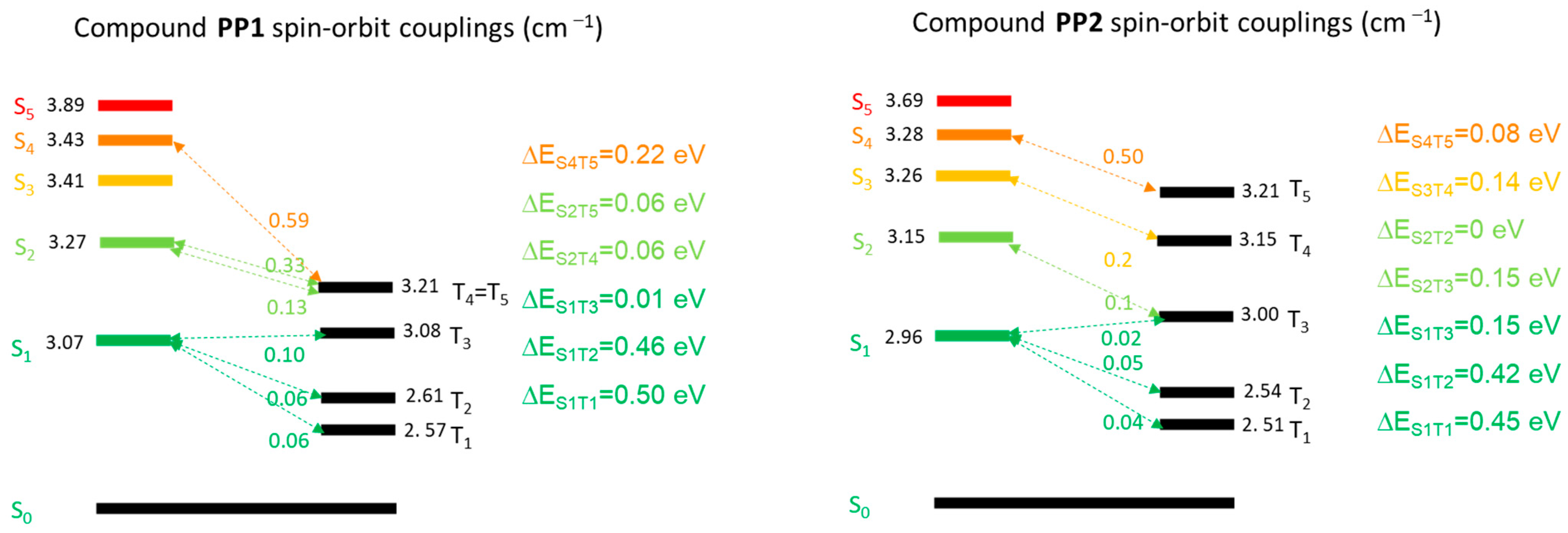
3.2. Theoretical Calculations, Electrochemical, and Photoelectric Properties
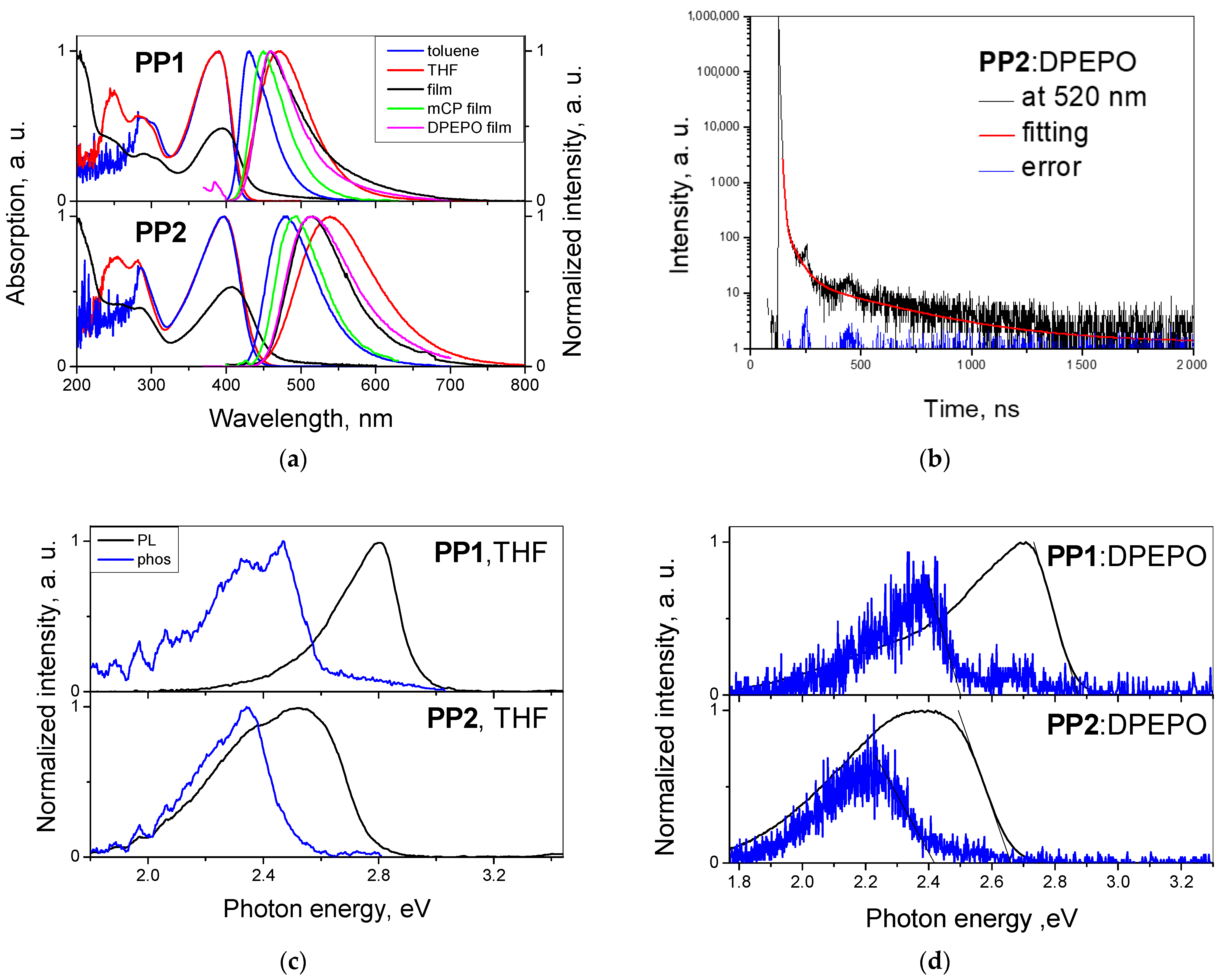
3.3. Photophysical Properties
3.4. Charge-Transporting Properties
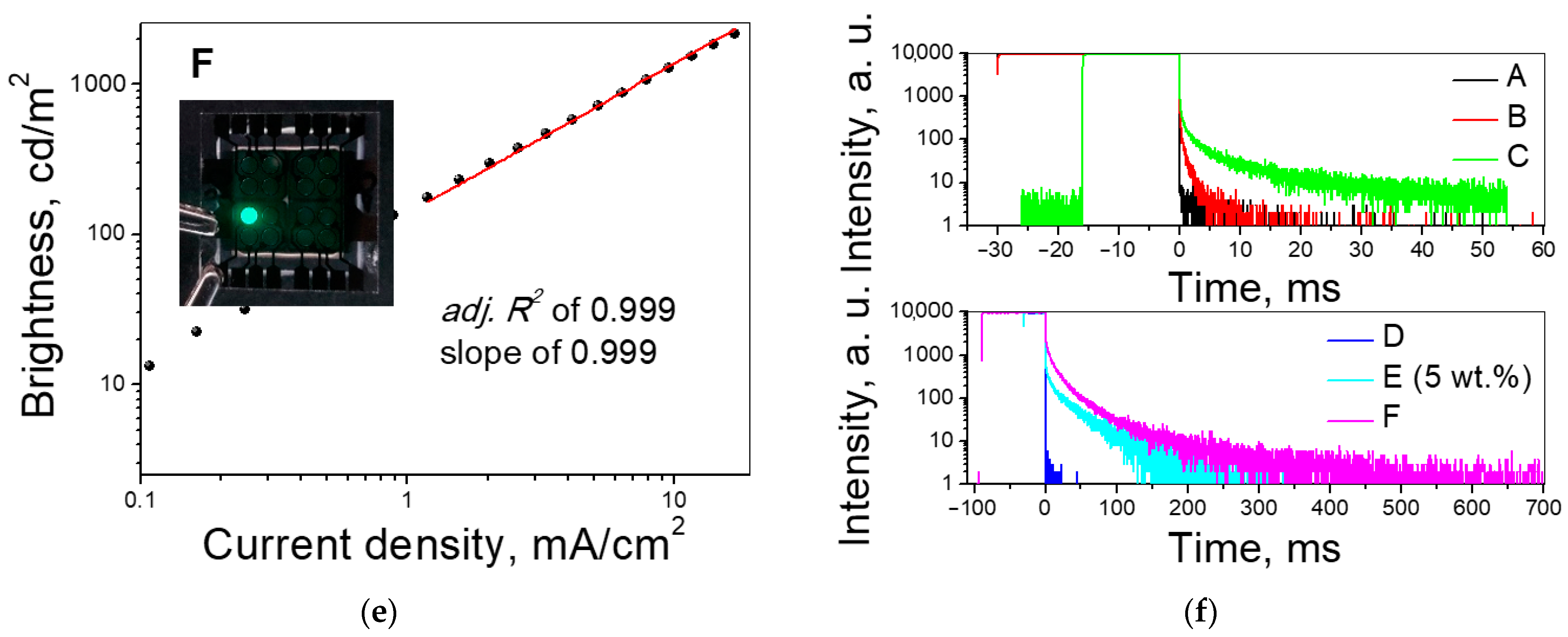
3.5. Electroluminescent Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nayak, D.; Choudhary, R.B. A Survey of the Structure, Fabrication, and Characterization of Advanced Organic Light Emitting Diodes. Microelectron. Reliab. 2023, 144, 114959. [Google Scholar]
- Pathak, S.K.; Yang, C. Chapter 10—Aggregation-Induced Emission Luminogens for Organic Light-Emitting Diodes. Mater. Today 2022, 2022, 315–372. [Google Scholar] [CrossRef]
- Méhes, G.; Nomura, H.; Zhang, Q.; Nakagawa, T.; Adachi, C. Enhanced Electroluminescence Efficiency in a Spiro-Acridine Derivative through Thermally Activated Delayed Fluorescence. Angew. Chem. Int. Ed. 2012, 51, 11311–11315. [Google Scholar] [CrossRef] [PubMed]
- Kastrati, A.; Oswald, F.; Scalabre, A.; Fromm, K.M. Photophysical Properties of Anthracene Derivatives. Photochem 2023, 3, 227–273. [Google Scholar] [CrossRef]
- Kim, K.H.; Ahn, E.S.; Huh, J.S.; Kim, Y.H.; Kim, J.J. Design of Heteroleptic Ir Complexes with Horizontal Emitting Dipoles for Highly Efficient Organic Light-Emitting Diodes with an External Quantum Efficiency of 38%. Chem. Mater. 2016, 28, 7505–7510. [Google Scholar] [CrossRef]
- Lee, J.; Chen, H.F.; Batagoda, T.; Coburn, C.; Djurovich, P.I.; Thompson, M.E.; Forrest, S.R. Deep Blue Phosphorescent Organic Light-Emitting Diodes with Very High Brightness and Efficiency. Nat. Mater. 2016, 15, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Yersin, H.; Finkenzeller, W.J. Triplet Emitters for Organic Light-Emitting Diodes: Basic Properties. In Highly Efficient OLEDs with Phosphorescent Materials; Wiley, John Wiley & Sons GmbH: Hoboken, NJ, USA, 2008. [Google Scholar]
- Hayduk, M.; Riebe, S.; Voskuhl, J. Frontispiece: Phosphorescence through Hindered Motion of Pure Organic Emitters. Chem. A Eur. J. 2018, 24, 12221–12230. [Google Scholar] [CrossRef]
- CP, K.P.; Naveen, K.P.; Hur, J. Acceptor–donor–acceptor based thermally activated delayed fluorescent materials: Structure–property insights and electroluminescence performances. Mater. Chem. Front. 2024, 8, 769–784. [Google Scholar]
- Endo, A.; Sato, K.; Yoshimura, K.; Kai, T.; Kawada, A.; Miyazaki, H.; Adachi, C. Efficient Up-Conversion of Triplet Excitons into a Singlet State and Its Application for Organic Light Emitting Diodes. Appl. Phys. Lett. 2011, 98, 083302. [Google Scholar] [CrossRef]
- Chapran, M.; Pander, P.; Vasylieva, M.; Wiosna-Salyga, G.; Ulanski, J.; Dias, F.B.; Data, P. Realizing 20% External Quantum Efficiency in Electroluminescence with Efficient Thermally Activated Delayed Fluorescence from an Exciplex. ACS Appl. Mater. Interfaces 2019, 11, 13460–13471. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, T.; Wakikawa, Y.; Ikoma, T. Mechanism of Intersystem Crossing of Thermally Activated Delayed Fluorescence Molecules. J. Phys. Chem. A 2015, 119, 3415–3418. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Yao, L.; Yang, B.; Ma, Y. Reverse Intersystem Crossing from Upper Triplet Levels to Excited Singlet: A “hot Excition” Path for Organic Light-Emitting Diodes. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2015, 373, 20140318. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, S.; Li, H.; Chen, R.; Jin, L.; Yuan, K.; Li, H.; Lu, P.; Yang, B.; Huang, W. Breaking the Efficiency Limit of Fluorescent OLEDs by Hybridized Local and Charge-Transfer Host Materials. J. Phys. Chem. Lett. 2018, 9, 5240–5245. [Google Scholar] [CrossRef]
- Yanai, N.; Kozue, M.; Amemori, S.; Kabe, R.; Adachi, C.; Kimizuka, N. Increased Vis-to-UV Upconversion Performance by Energy Level Matching between a TADF Donor and High Triplet Energy Acceptors. J. Mater. Chem. C 2016, 4, 6447–6451. [Google Scholar] [CrossRef]
- Gudem, M.; Kowalewski, M. Triplet-Triplet Annihilation Dynamics of Naphthalene. Chem. A Eur. J. 2022, 28, e202200781. [Google Scholar] [CrossRef] [PubMed]
- Zhan, G.; Liu, Z.; Bian, Z.; Huang, C. Recent Advances in Organic Light-Emitting Diodes Based on Pure Organic Room Temperature Phosphorescence Materials. Front. Chem. 2019, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H.; Suzuki, H.; Fukushima, T.; Shizu, K.; Suzuki, K.; Kubo, S.; Komino, T.; Oiwa, H.; Suzuki, F.; Wakamiya, A.; et al. Purely Organic Electroluminescent Material Realizing 100% Conversion from Electricity to Light. Nat. Commun. 2015, 6, 8476. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, H.; Kido, J. Multifunctional Materials in High-Performance OLEDs: Challenges for Solid-State Lighting. Chem. Mater. 2011, 23, 621–630. [Google Scholar] [CrossRef]
- Komatsu, R.; Sasabe, H.; Kido, J. Recent Progress of Pyrimidine Derivatives for High-Performance Organic Light-Emitting Devices. J. Photonics Energy 2018, 8, 032108. [Google Scholar] [CrossRef]
- Bouihi, F.; Schmaltz, B.; Mathevet, F.; Kreher, D.; Faure-Vincent, J.; Yildirim, C.; Elhakmaoui, A.; Bouclé, J.; Akssira, M.; Tran-Van, F.; et al. D-π-A-Type Pyrazolo [1,5-a]Pyrimidine-Based Hole-Transporting Materials for Perovskite Solar Cells: Effect of the Functionalization Position. Materials 2022, 15, 7992. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Davidova, E.; Boukalova, S.; Kovarova, J.; Bajzikova, M.; Coelho, A.; Terp, M.G.; Ditzel, H.J.; Rohlena, J.; Neuzil, J. Replication and Ribosomal Stress Induced by Targeting Pyrimidine Synthesis and Cellular Checkpoints Suppress P53-Deficient Tumors. Cell Death Dis. 2020, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Garavaglia, M.; Rossi, E.; Landini, P. The Pyrimidine Nucleotide Biosynthetic Pathway Modulates Production of Biofilm Determinants in Escherichia Coli. PLoS ONE 2012, 7, e31252. [Google Scholar] [CrossRef]
- Rodella, F.; Saxena, R.; Bagnich, S.; Banevičius, D.; Kreiza, G.; Athanasopoulos, S.; Juršėnas, S.; Kazlauskas, K.; Köhler, A.; Strohriegl, P. Low Efficiency Roll-off Blue TADF OLEDs Employing a Novel Acridine–Pyrimidine Based High Triplet Energy Host. J. Mater. Chem. C 2021, 9, 17471–17482. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, T.; Zhan, L.; Zhong, C.; Gong, S.; Jiang, N.; Lu, Z.H.; Yang, C. Optimizing Optoelectronic Properties of Pyrimidine-Based TADF Emitters by Changing the Substituent for Organic Light-Emitting Diodes with External Quantum Efficiency Close to 25 % and Slow Efficiency Roll-Off. Chem. A Eur. J. 2016, 22, 10860–10866. [Google Scholar] [CrossRef]
- Jang, J.S.; Lee, H.L.; Lee, K.H.; Lee, J.Y.; Hong, W.P. A Pyrimidine-5-Carbonitrile Acceptor Combined with Anortho-Linked Donor for Long Lifetime through Facilitated Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C 2021, 9, 2408–2415. [Google Scholar] [CrossRef]
- Li, B.; Li, Z.; Hu, T.; Zhang, Y.; Wang, Y.; Yi, Y.; Guo, F.; Zhao, L. Highly Efficient Blue Organic Light-Emitting Diodes from Pyrimidine-Based Thermally Activated Delayed Fluorescence Emitters. J. Mater. Chem. C 2018, 6, 2351–2359. [Google Scholar] [CrossRef]
- Park, I.S.; Komiyama, H.; Yasuda, T. Pyrimidine-Based Twisted Donor-Acceptor Delayed Fluorescence Molecules: A New Universal Platform for Highly Efficient Blue Electroluminescence. Chem. Sci. 2017, 8, 953–960. [Google Scholar] [CrossRef]
- Sarma, M.; Tsai, W.L.; Lee, W.K.; Chi, Y.; Wu, C.C.; Liu, S.H.; Chou, P.T.; Wong, K.T. Anomalously Long-Lasting Blue PhOLED Featuring Phenyl-Pyrimidine Cyclometalated Iridium Emitter. Chem 2017, 3, 461–476. [Google Scholar] [CrossRef]
- Ma, H.; Liu, D.; Li, J.; Mei, Y.; Li, D.; Ding, Y.; Wei, W. Sky-Blue Iridium Complexes with Pyrimidine Ligands for Highly Efficient Phosphorescent Organic Light-Emitting Diodes. New J. Chem. 2020, 44, 8743–8750. [Google Scholar] [CrossRef]
- Jiang, B.; Zhao, C.; Ning, X.; Zhong, C.; Ma, D.; Yang, C. Using Simple Fused-Ring Thieno[2,3-d]pyrimidine to Construct Orange/Red Ir(III) Complexes: High-Performance Red Organic Light-Emitting Diodes with EQEs up to Nearly 28%. Adv. Opt. Mater. 2018, 6, 1800108. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, F.; Liu, H.; Yan, Y.; Li, X.; Wang, S. Impact of Peripheral Groups on Pyrimidine Acceptor-Based HLCT Materials for Efficient Deep Blue OLED Devices. J. Mater. Chem. C 2022, 10, 9953–9960. [Google Scholar] [CrossRef]
- Tsiko, U.; Volyniuk, D.; Andruleviciene, V.; Leitonas, K.; Sych, G.; Bezvikonnyi, O.; Jasinskas, V.; Gulbinas, V.; Stakhira, P.; Grazulevicius, J.V. Triphenylamino or 9-Phenyl Carbazolyl-Substituted Pyrimidine-5-Carbonitriles as Bipolar Emitters and Hosts with Triplet Harvesting Abilities. Mater. Today Chem. 2022, 25, 100955. [Google Scholar] [CrossRef]
- Pérez-Caaveiro, C.; Moreno Oliva, M.; López Navarrete, J.T.; Sestelo, J.P.; Martínez, M.; Sarandeses, L.A. Synthesis of D−A−A and D−A−D Pyrimidineπ-Systems Using Triorganoindium Reagents: Optical, Vibrational, and Electrochemical Studies. J. Org. Chem. 2019, 84, 8870–8885. [Google Scholar] [CrossRef]
- Parra, R.D.; Zeng, X.C. Hydrogen Bonding and Cooperative Effects in Mixed Dimers and Trimers of Methanol and Trifluoromethanol: An Ab Initio Study. J. Chem. Phys. 1999, 110, 6329–6338. [Google Scholar] [CrossRef]
- Muzomwe, M.; Maes, G.; Kasende, O.E. Theoretical DFT(B3LYP)/6-31+G(d) Study on the Prediction of the Preferred Interaction Site of 3-methyl-4-pyrimidone with Different Proton Donors. Nat. Sci. 2012, 4, 286–297. [Google Scholar] [CrossRef]
- Samir, B.; Kalalian, C.; Roth, E.; Salghi, R.; Chakir, A. Gas-Phase UV Absorption Spectra of Pyrazine, Pyrimidine and Pyridazine. Chem. Phys. Lett. 2020, 751, 137469. [Google Scholar] [CrossRef]
- Stener, M.; Decleva, P.; Holland, D.M.P.; Shaw, D.A. A Study of the Valence Shell Electronic States of Pyrimidine and Pyrazine by Photoabsorption Spectroscopy and Time-Dependent Density Functional Theory Calculations. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 075203. [Google Scholar] [CrossRef]
- Bolovinos, A.; Tsekeris, P.; Philis, J.; Pantos, E.; Andritsopoulos, G. Absolute Vacuum Ultraviolet Absorption Spectra of Some Gaseous Azabenzenes. J. Mol. Spectrosc. 1984, 103, 240–256. [Google Scholar] [CrossRef]
- da Silva, F.F.; Almeida, D.; Martins, G.; Milosavljević, A.R.; Marinković, B.P.; Hoffmann, S.V.; Mason, N.J.; Nunes, Y.; Garcia, G.; Limão-Vieira, P. The Electronic States of Pyrimidine Studied by VUV Photoabsorption and Electron Energy-Loss Spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 6717–6731. [Google Scholar] [CrossRef] [PubMed]
- Vigante, B.; Leitonas, K.; Volyniuk, D.; Andruleviciene, V.; Simokaitiene, J.; Ivanova, A.; Bucinskas, A.; Grazulevicius, J.V.; Arsenyan, P. Synthesis of Linear and V-Shaped Carbazolyl-Substituted Pyridine-3,5-Dicarbonitriles Exhibiting Efficient Bipolar Charge Transport and E-Type Fluorescence. Chem. A Eur. J. 2019, 25, 3325–3336. [Google Scholar] [CrossRef]
- Dias, F.B.; Santos, J.; Graves, D.R.; Data, P.; Nobuyasu, R.S.; Fox, M.A.; Batsanov, A.S.; Palmeira, T.; Berberan-Santos, M.N.; Bryce, M.R.; et al. The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Adv. Sci. 2016, 3, 1600080. [Google Scholar] [CrossRef]
- Pivrikas, A.; Sariciftci, N.S.; Juška, G.; Österbacka, R. A Review of Charge Transport and Recombination in Polymer/Fullerene Organic Solar Cells. Prog. Photovolt. Res. Appl. 2007, 15, 677–696. [Google Scholar] [CrossRef]
- Fishchuk, I.I.; Kadashchuk, A.K.; Vakhnin, A.; Korosko, Y.; Bässler, H.; Souharce, B.; Scherf, U. Transition from Trap-Controlled to Trap-to-Trap Hopping Transport in Disordered Organic Semiconductors. Phys. Rev. B 2006, 73, 115210. [Google Scholar] [CrossRef]
- Qiao, X.; Yuan, P.; Ma, D.; Ahamad, T.; Alshehri, S.M. Electrical Pumped Energy Up-Conversion: A Non-Linear Electroluminescence Process Mediated by Triplet-Triplet Annihilation. Org. Electron. 2017, 46, 1–6. [Google Scholar] [CrossRef]
- Shukla, A.; Hasan, M.; Banappanavar, G.; Ahmad, V.; Sobus, J.; Moore, E.G.; Kabra, D.; Lo, S.C.; Namdas, E.B. Controlling Triplet–Triplet Upconversion and Singlet-Triplet Annihilation in Organic Light-Emitting Diodes for Injection Lasing. Commun. Mater. 2022, 3, 27. [Google Scholar] [CrossRef]
- Klimash, A.; Prlj, A.; Yufit, D.S.; Mallick, A.; Curchod, B.F.E.; McGonigal, P.R.; Skabara, P.J.; Etherington, M.K. From Phosphorescence to Delayed Fluorescence in One Step: Tuning Photophysical Properties by Quaternisation of an Sp2-Hybridised Nitrogen Atom. J. Mater. Chem. C 2022, 10, 9484. [Google Scholar] [CrossRef]
- Kang, J.; Son, J.B.; Kim, G.W.; Bae, S.; Min, K.S.; Sul, S.; Jeon, W.S.; Jang, J.; Park, G.S.; Shin, J.K.; et al. Time-Resolved Electroluminescence Study for the Effect of Charge Traps on the Luminescence Properties of Organic Light-Emitting Diodes. Phys. Status Solidi Appl. Mater. Sci. 2020, 217, 2000081. [Google Scholar] [CrossRef]
- Barth, S.; Müller, P.; Riel, H.; Seidler, P.F.; Rieß, W.; Vestweber, H.; Bässler, H. Electron Mobility in Tris(8-Hydroxy-Quinoline)Aluminum Thin Films Determined via Transient Electroluminescence from Single- and Multilayer Organic Light-Emitting Diodes. J. Appl. Phys. 2001, 89, 3711–3719. [Google Scholar] [CrossRef]
- Liu, D.; He, Y.; Qiu, W.; Peng, X.; Li, M.; Li, D.; Pu, J.; Yang, J.; Gan, Y.; Yang, G.; et al. Management of Host–Guest Triplet Exciton Distribution for Stable, High-Efficiency, Low Roll-Off Solution-Processed Blue Organic Light-Emitting Diodes by Employing Triplet-Energy-Mediated Hosts. Adv. Funct. Mater. 2023, 33, 2301327. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hočevar, T.; Demčar, J. Computation of Graphlet Orbits for Nodes and Edges in Sparse Graphs. J. Stat. Softw. 2016, 71, 1–24. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Td, °C | Tm, °C | Tg, °C | Tcr, °C |
|---|---|---|---|---|
| PP1 | 397 | 249 | 106 | 221 |
| PP2 | 438 | 213 | 103 | - |
| Eox ½ vs. FC, V | IPCV, eV | IPEP, eV | EACV, eV | Egtheo, eV | |
|---|---|---|---|---|---|
| PP1 | 0.48 | 5.28 | 5.82 | 2.33 | 3.52 |
| PP2 | 0.28 | 5.08 | 5.63 | 2.26 | 3.40 |
| Compound | Sample | λUVmax, nm | Egopt, eV | λFLmax, nm | Stokes Shift, nm | Lifetime, ns | PLQY, % | ES1, eV | ET1, eV | ΔEst, eV |
|---|---|---|---|---|---|---|---|---|---|---|
| PP1 | Toluene | -/390 | 2.95 | 431 | 41 | - | 80 | - | - | - |
| THF | 284/390 | 2.91 | 470 | 80 | - | - | 2.93 | 2.61 | 0.32 | |
| Film | 396 | 3.13 | 460 | 64 | 1.46/8.20 | 32 | - | - | - | |
| 20 wt.% mCP film | - | - | 450 | 2.94/9.28 | 62 | - | - | - | ||
| 20 wt.% DPEPO film | - | - | 466 | 2.56/6.86 DF: 218.86 (at 460 nm), 381.59 (at 530 nm) | 41 | 2.87 | 2.5 | 0.37 | ||
| PP2 | Toluene | -/397 | 2.82 | 479 | 82 | - | 63 | - | - | - |
| THF | 281/397 | 2.79 | 539 | 142 | - | - | 2.77 | 2.49 | 0.28 | |
| Film | 407 | 2.51 | 513 | 106 | 2.08/6.16 | 23 | - | - | - | |
| 20 wt.% mCP film | - | - | 494 | 3.41/7.14 | 34 | - | - | - | ||
| 20 wt.% DPEPO film | - | - | 509 | 3.65/7.89 DF: 372.21 (at 521 nm) | 26 | 2.66 | 2.42 | 0.24 |
| OLED | EML | CEmax,cd/A | PEmax, lm/W | EQEmax, % | 1931 CIEx,y |
|---|---|---|---|---|---|
| A | PP1 | 3.5 | 3.0 | 2.3 | (0.21, 0.27) |
| B | PP1:mCBP | 5.2 | 3.5 | 4.4 | (0.15, 0.15) |
| C | PP1:DPEPO | 11.4 | 7.3 | 9.0 | (0.15, 0.23) |
| D | PP2 | 8.2 | 5.7 | 2.8 | (0.28, 0.57) |
| E | PP2:mCBP | 13.1 | 7.9 | 6.7 | (0.20, 0.46) |
| F | PP2:DPEPO | 27.6 | 11.4 | 10.6 | (0.28, 0.56) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starykov, H.; Bezvikonnyi, O.; Leitonas, K.; Simokaitiene, J.; Volyniuk, D.; Skuodis, E.; Keruckiene, R.; Grazulevicius, J.V. Derivatives of Phenyl Pyrimidine and of the Different Donor Moieties as Emitters for OLEDs. Materials 2024, 17, 1357. https://doi.org/10.3390/ma17061357
Starykov H, Bezvikonnyi O, Leitonas K, Simokaitiene J, Volyniuk D, Skuodis E, Keruckiene R, Grazulevicius JV. Derivatives of Phenyl Pyrimidine and of the Different Donor Moieties as Emitters for OLEDs. Materials. 2024; 17(6):1357. https://doi.org/10.3390/ma17061357
Chicago/Turabian StyleStarykov, Hryhorii, Oleksandr Bezvikonnyi, Karolis Leitonas, Jurate Simokaitiene, Dmytro Volyniuk, Eigirdas Skuodis, Rasa Keruckiene, and Juozas Vidas Grazulevicius. 2024. "Derivatives of Phenyl Pyrimidine and of the Different Donor Moieties as Emitters for OLEDs" Materials 17, no. 6: 1357. https://doi.org/10.3390/ma17061357