

Investigating the Influence of Impurity Defects on the Adsorption Behavior of Hydrated Sc3+ on the Kaolinite (001) Surface Using Density Functional Theory

Abstract
1. Introduction
2. Theoretical Methods and Models
2.1. Calculation Methods and Parameters
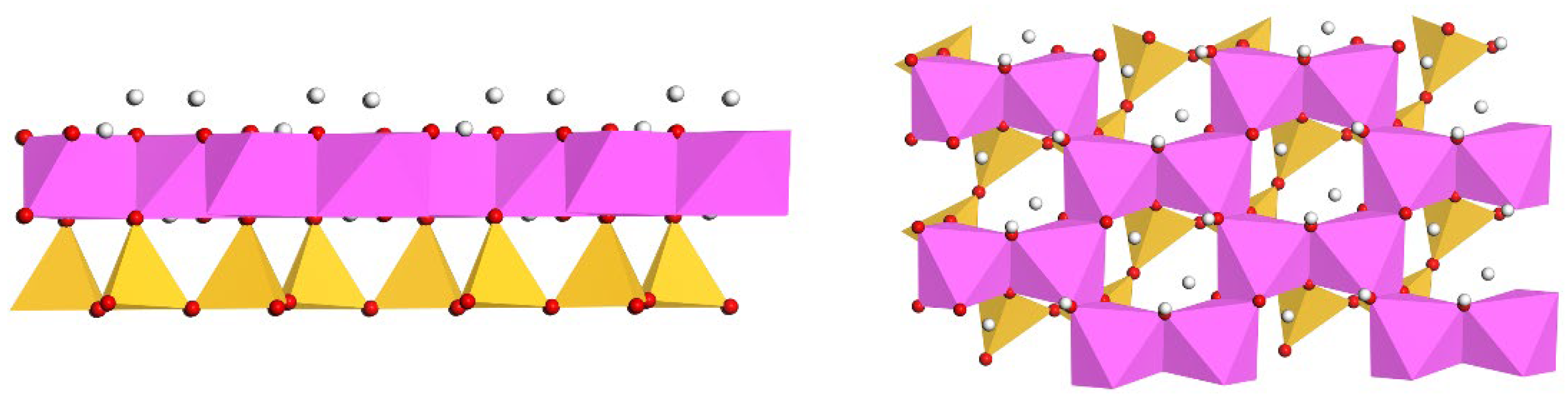
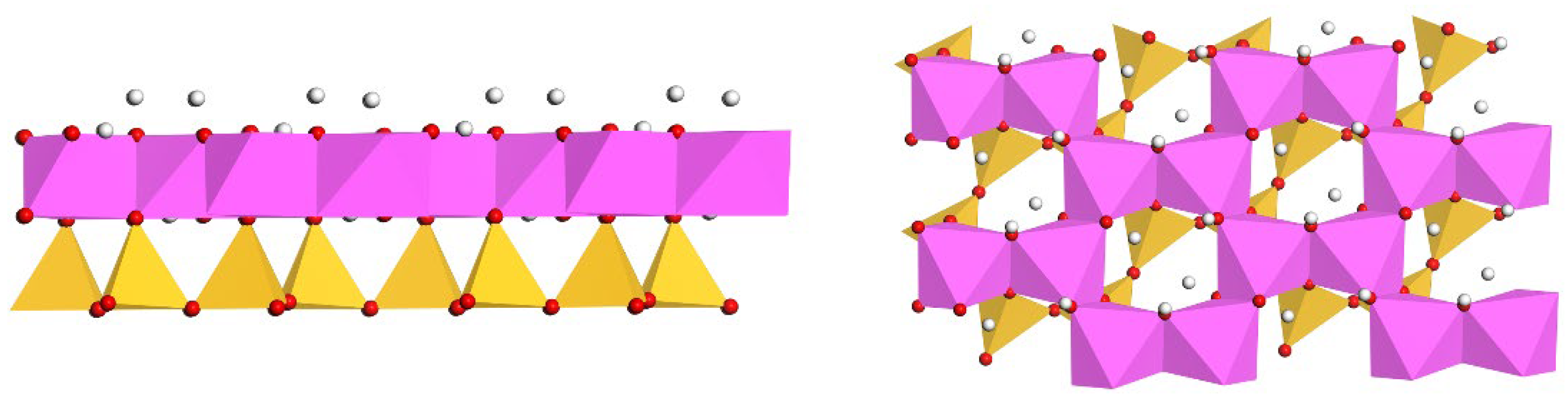
2.2. Model Construction
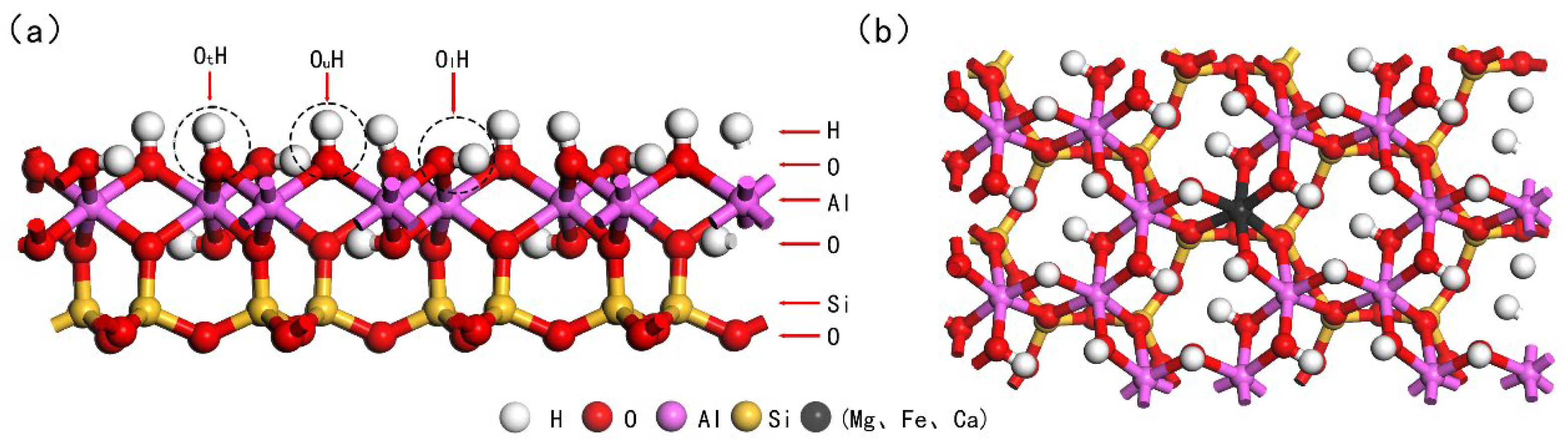
3. Results and Discussion
3.1. Structure and Properties of the Adsorption Model
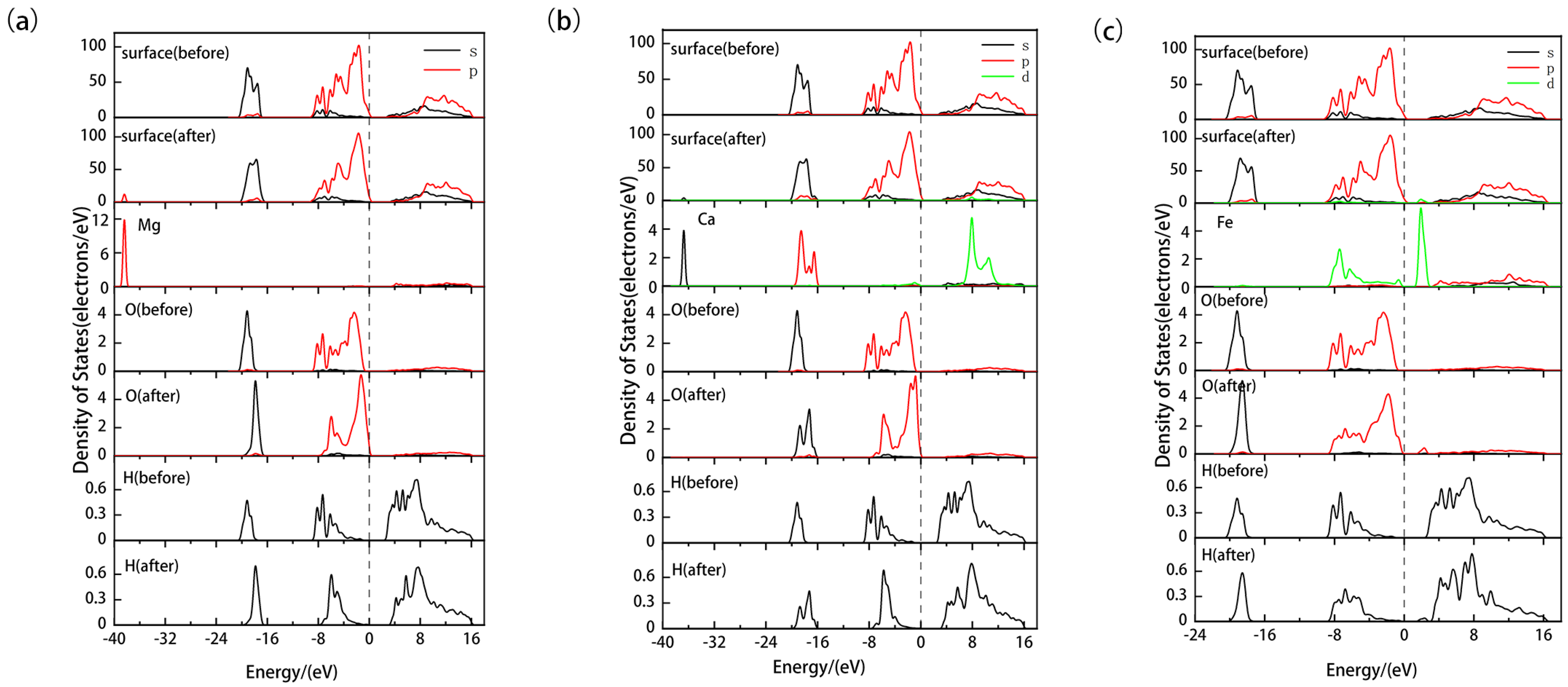
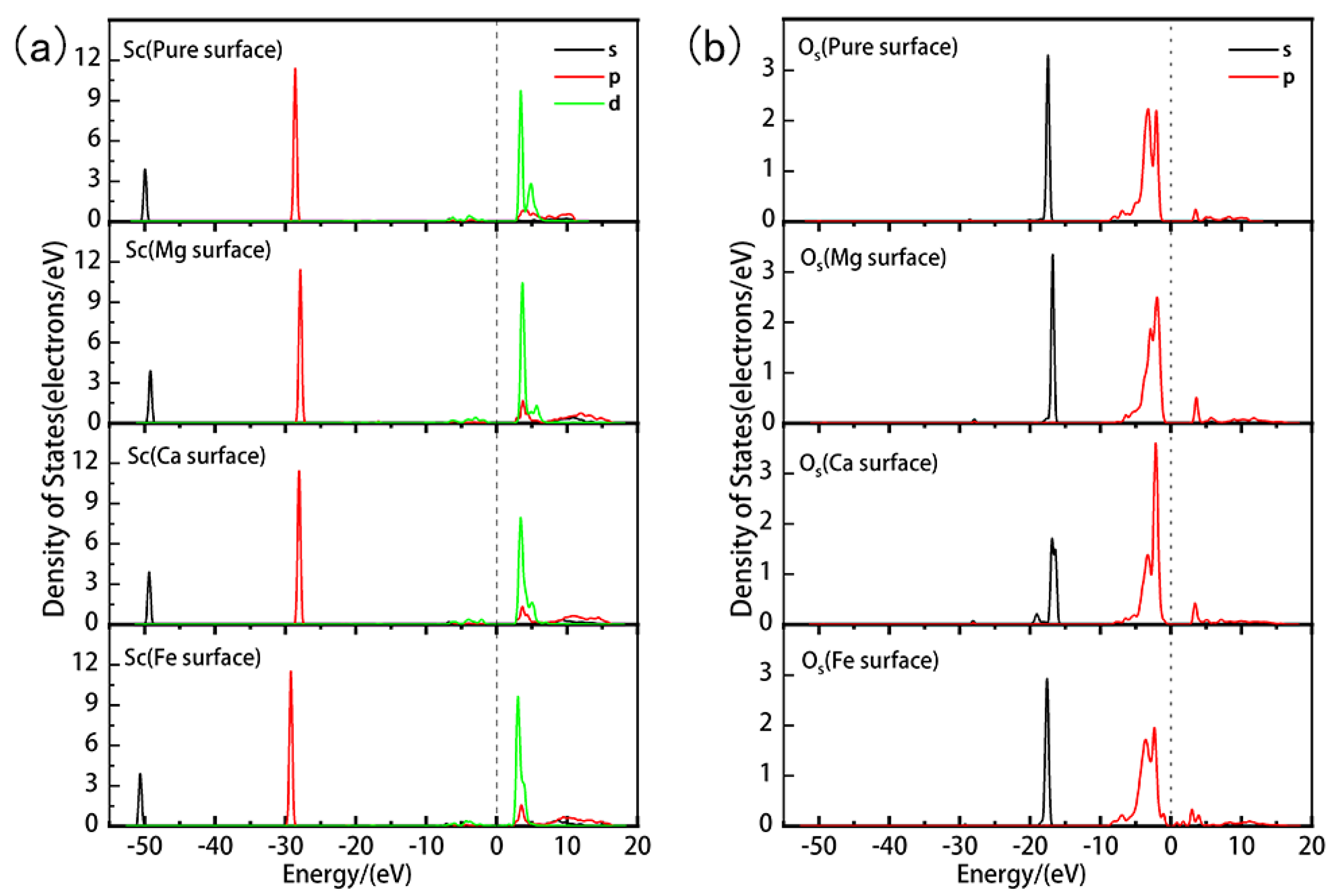
3.2. Properties of Ideal and Doped Kaolinite (001) Al-OH Surfaces
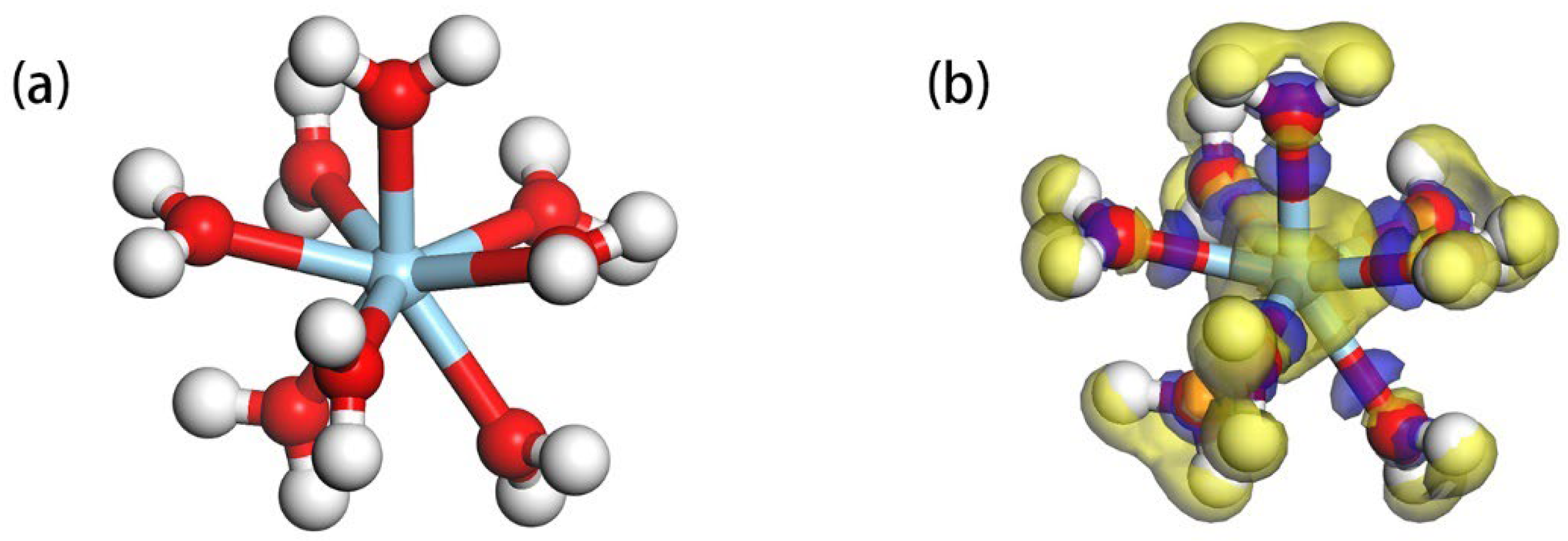
3.3. Adsorption of Hydrated on the Ideal Kaolinite (001) Al-OH Surface
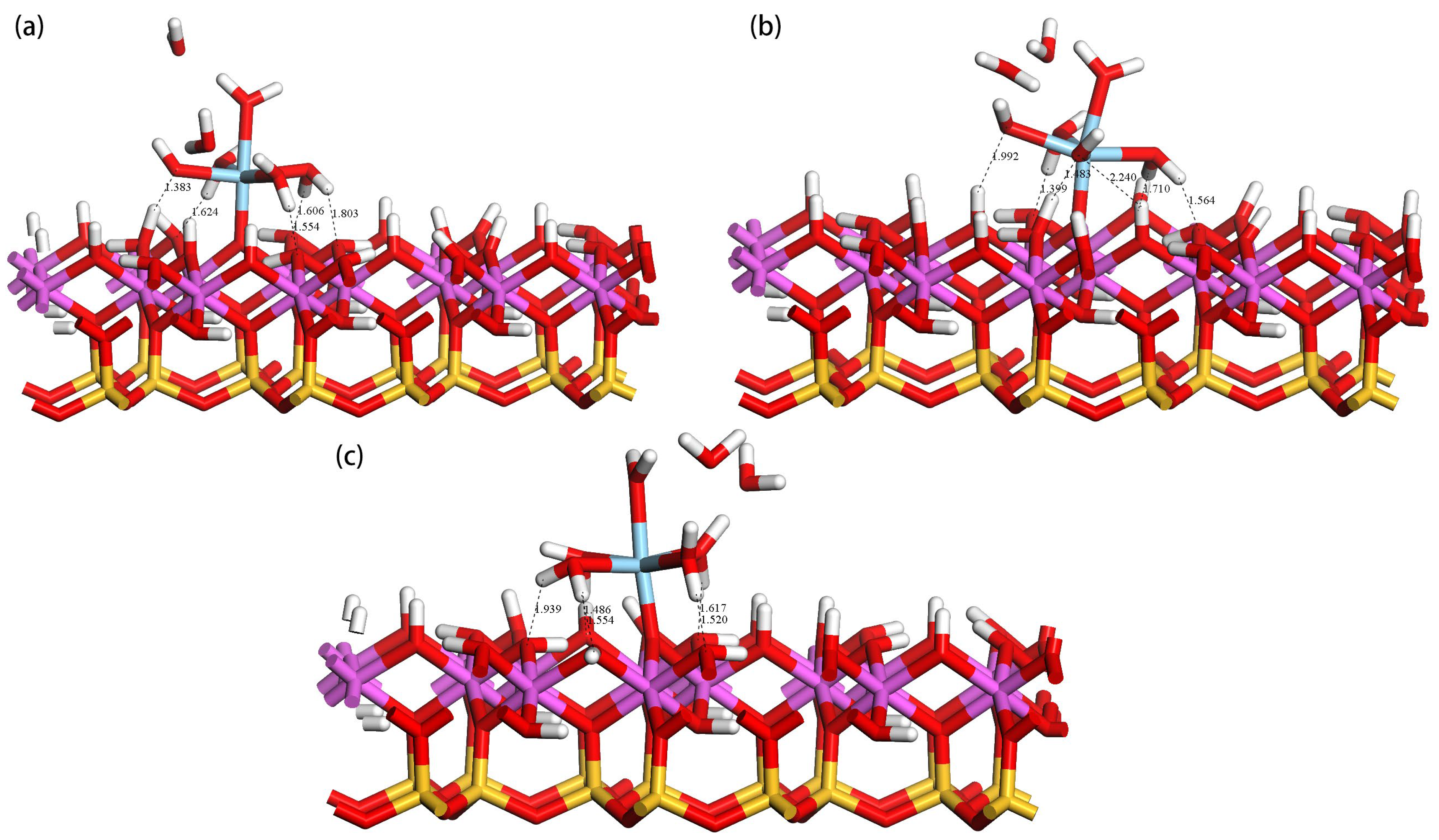
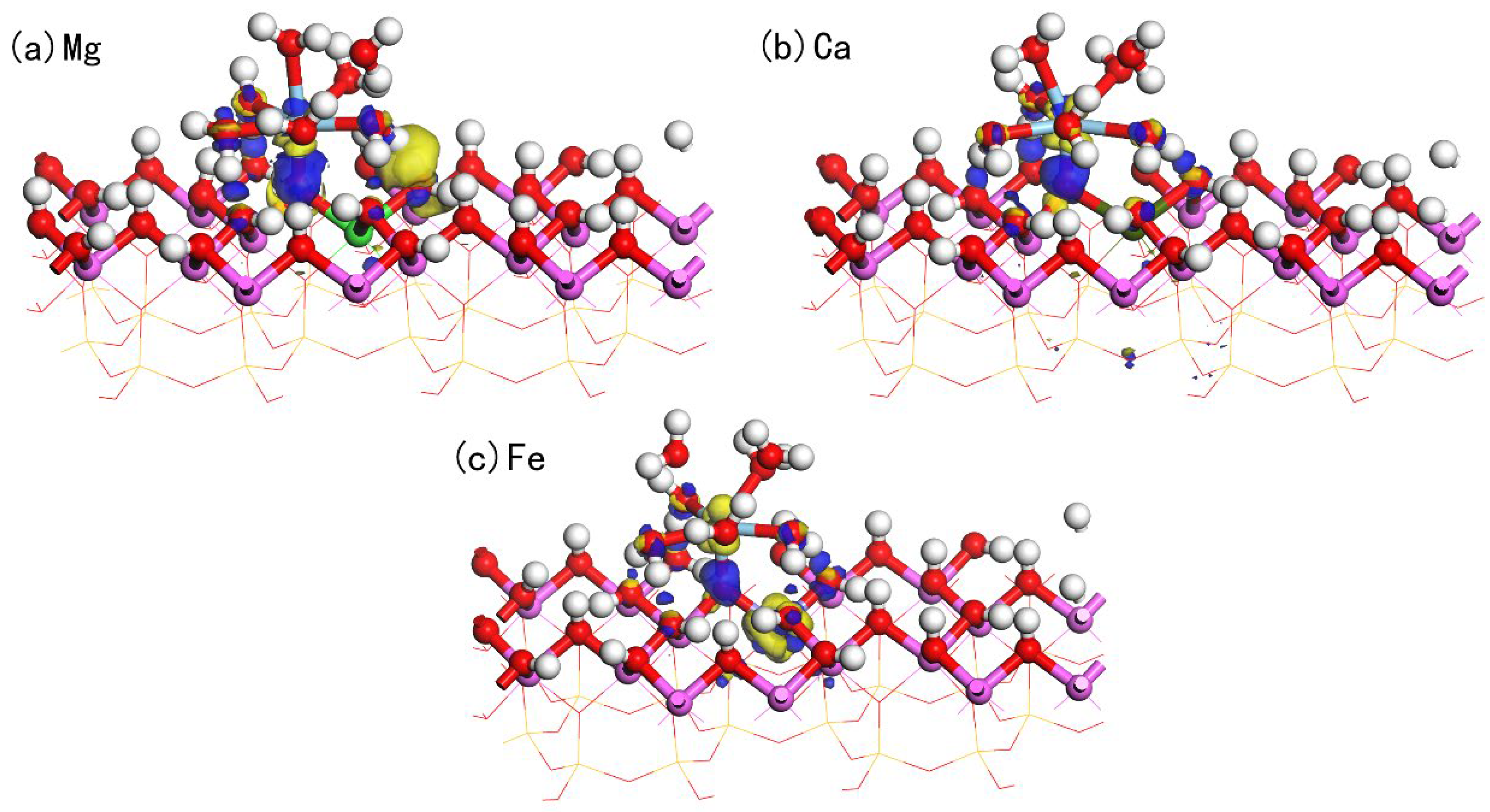
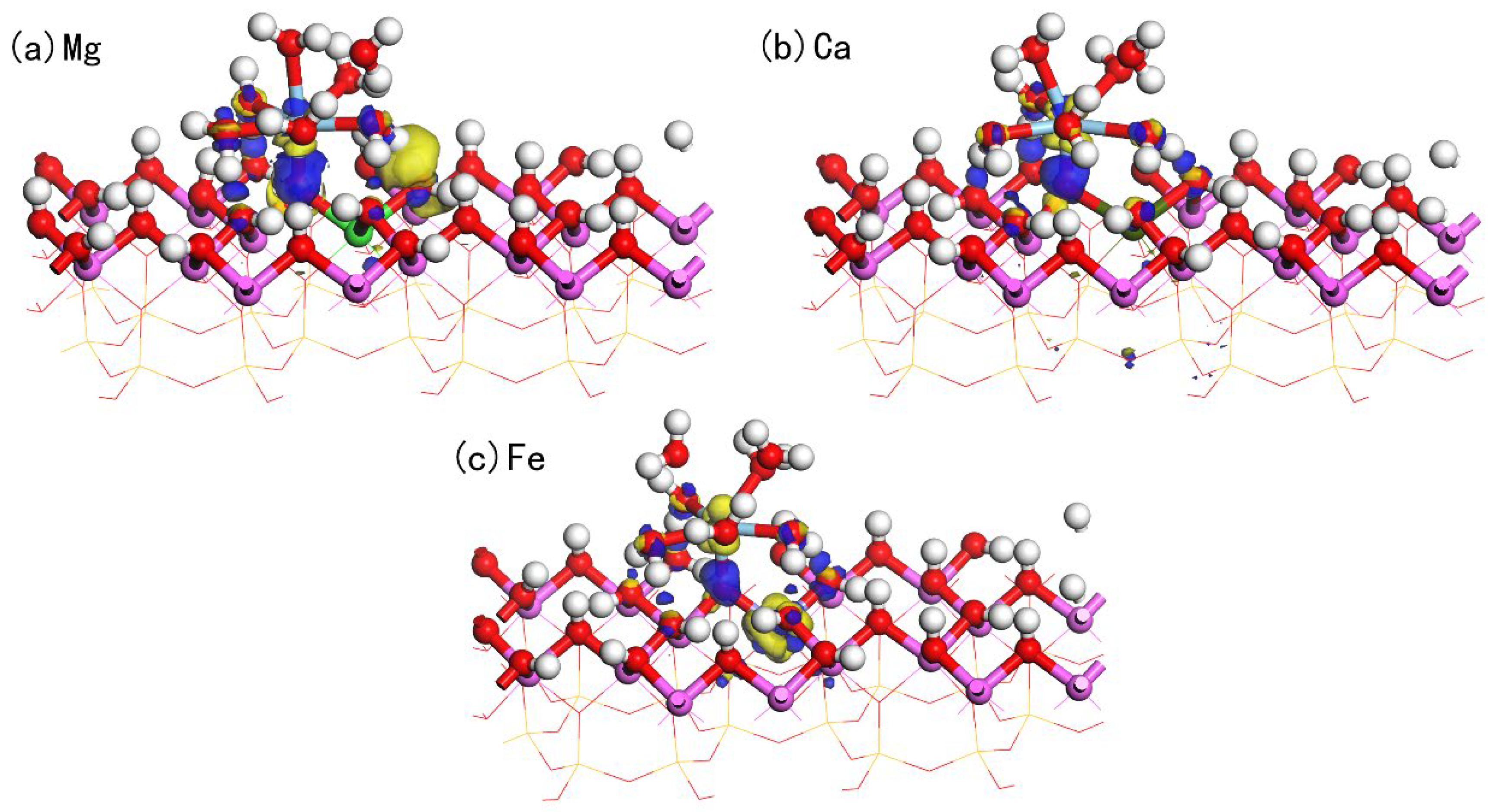
3.4. Adsorption of Hydrated on Doped Kaolinite (001) Al-OH Surface
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hou, Y.; Nichele, F.; Chi, H.; Lodesani, A.; Wu, Y.; Ritter, M.F.; Haxell, D.Z.; Davydova, M.; Ilić, S.; Glezakou-Elbert, O.; et al. Ubiquitous Superconducting Diode Effect in Superconductor Thin Films. Phys. Rev. Lett. 2023, 131, 027001. [Google Scholar] [CrossRef]
- Meier, W.R.; Madhogaria, R.P.; Mozaffari, S.; Marshall, M.; Graf, D.E.; McGuire, M.A.; Arachchige, H.W.S.; Allen, C.L.; Driver, J.; Cao, H.; et al. Tiny Sc Allows the Chains to Rattle: Impact of Lu and Y Doping on the Charge-Density Wave in ScV6Sn6. J. Am. Chem. Soc. 2023, 145, 20943–20950. [Google Scholar] [CrossRef]
- Cheisson, T.; Schelter, E.J. Rare earth elements: Mendeleev’s bane, modern marvels. Science 2019, 363, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, K.N.; Sinha, K.P. Magnetic superconductors: Model theories and experimental properties of rare-earth compounds. Phys. Rep. 1984, 115, 93–149. [Google Scholar] [CrossRef]
- Chen, J.; Qiu, J.; Huang, L.; Chen, X.; Yang, Y.; Xiao, Y. Coordination–reduction leaching process of ion-adsorption type rare earth ore with ascorbic acid. J. Rare Earths 2023, 41, 1225–1233. [Google Scholar] [CrossRef]
- Xiao, Y.-F.; Feng, Z.-Y.; Hu, G.-H.; Huang, L.; Huang, X.-W.; Chen, Y.-Y.; Li, M.-L. Leaching and mass transfer characteristics of elements from ion-adsorption type rare earth ore. Rare Met. 2015, 34, 357–365. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, J. Recovery of rare earth from ion-adsorption rare earth ores with a compound lixiviant. Sep. Purif. Technol. 2015, 142, 203–208. [Google Scholar] [CrossRef]
- Zhang, Q.; Ren, F.; Li, F.; Chen, G.; Yang, G.; Wang, J.; Du, K.; Liu, S.; Li, Z. Ammonia nitrogen sources and pollution along soil profiles in an in-situ leaching rare earth ore. Environ. Pollut. 2020, 267, 115449. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-F.; Wu, H.; Li, X.-D.; Ou, J.-C.; Huang, X.-L. Anti-adsorption mechanism of ion-adsorption type rare earth tailings. Rare Met. 2023, 42, 1420–1426. [Google Scholar] [CrossRef]
- Li, M.Y.H.; Zhou, M.-F. The role of clay minerals in formation of the regolith-hosted heavy rare earth element deposits. Am. Mineral. 2020, 105, 92–108. [Google Scholar] [CrossRef]
- Kameda, J.; Yamagishi, A.; Kogure, T. Morphological characteristics of ordered kaolinite: Investigation using electron back-scattered diffraction. Am. Mineral. 2005, 90, 1462–1465. [Google Scholar] [CrossRef]
- Chen, J.; Ling, Y.; Min, F.; Cheng, Y.; Chu, X.; Shang, H. Mechanism research on surface hydration of Fe(II)-doped kaolinite, insights from molecular dynamics simulations. J. Mol. Liq. 2023, 386, 122469. [Google Scholar] [CrossRef]
- Borst, A.M.; Smith, M.P.; Finch, A.A.; Estrade, G.; Villanova-de-Benavent, C.; Nason, P.; Marquis, E.; Horsburgh, N.J.; Goodenough, K.M.; Xu, C.; et al. Adsorption of rare earth elements in regolith-hosted clay deposits. Nat. Commun. 2020, 11, 4386. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.; Rendtorff, N.M. Local Environments in Iron-Bearing Clay Minerals by DFT Approaches: The Case of Structural Fe in Kaolinite. Appl. Clay Sci. 2021, 213, 106251. [Google Scholar] [CrossRef]
- Malden, P.J.; Meads, R.E. Substitution by Iron in Kaolinite. Nature 1967, 215, 844–846. [Google Scholar] [CrossRef]
- Jafaar Ghafil, A.; Mazloom, G.; Abdi, J.; Tamtaji, M.; Banisharif, F. Ti3C2Tx/ZIF-67 hybrid nanocomposite as a highly effective adsorbent for Pb (II) removal from water: Synthesis and DFT calculations. Appl. Surf. Sci. 2024, 643, 158642. [Google Scholar] [CrossRef]
- Santana, E.; Possa, R.D.; Novais, A.L.F.; Manzoni, V.; Novais, E.R.P.; Martins, T.C.; Gester, R.; Andrade-Filho, T. Adsorption study of 4-nitrophenol onto kaolinite (001) surface: A van der Waals density functional study. Mater. Chem. Phys. 2021, 271, 124887. [Google Scholar] [CrossRef]
- El Hassani, A.A.; Tanji, K.; El Mrabet, I.; Fahoul, Y.; El Gaidoumi, A.; Benjelloun, A.T.; Sfaira, M.; Zaitan, H.; Kherbeche, A. A combined molecular dynamics simulation, DFT calculations, and experimental study of the adsorption of Rhodamine B dye on kaolinite and hydroxyapatite in aqueous solutions. Surf. Interfaces 2023, 36, 102647. [Google Scholar] [CrossRef]
- Kremleva, A.; Krüger, S.; Rösch, N. Density Functional Model Studies of Uranyl Adsorption on (001) Surfaces of Kaolinite. Langmuir 2008, 24, 9515–9524. [Google Scholar] [CrossRef]
- Kasprzhitskii, A.; Lazorenko, G.; Kharytonau, D.S.; Osipenko, M.A.; Kasach, A.A.; Kurilo, I.I. Adsorption mechanism of aliphatic amino acids on kaolinite surfaces. Appl. Clay Sci. 2022, 226, 106566. [Google Scholar] [CrossRef]
- Lainé, J.; Foucaud, Y.; Bonilla-Petriciolet, A.; Badawi, M. Molecular picture of the adsorption of phenol, toluene, carbon dioxide and water on kaolinite basal surfaces. Appl. Surf. Sci. 2022, 585, 152699. [Google Scholar] [CrossRef]
- García, K.I.; Quezada, G.R.; Arumí, J.L.; Urrutia, R.; Toledo, P.G. Adsorption of Phosphate Ions on the Basal and Edge Surfaces of Kaolinite in Low Salt Aqueous Solutions Using Molecular Dynamics Simulations. J. Phys. Chem. C 2021, 125, 21179–21190. [Google Scholar] [CrossRef]
- Peng, C.; Zhong, Y.; Wang, G.; Min, F.; Qin, L. Atomic-level insights into the adsorption of rare earth Y(OH)3−nn+ (n = 1–3) ions on kaolinite surface. Appl. Surf. Sci. 2019, 469, 357–367. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Z.; Gao, W.; Wang, Y.-F.; Huang, B.-W. Theoretical Investigation on Rare Earth Elements of Y, Nd and La Atoms’ Adsorption on the Kaolinite (001) and (001−) Surfaces. Minerals 2021, 11, 856. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Für Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Segall, M.D.; Philip, J.D.L.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, K.; Wu, G.; Jiang, D.; Lan, Y. Adsorption of Sc on the Surface of Kaolinite (001): A Density Functional Theory Study. Materials 2023, 16, 5349. [Google Scholar] [CrossRef] [PubMed]
- Bish, D.L. Rietveld Refinement of the Kaolinite Structure at 1.5 K. Clays Clay Miner. 1993, 41, 738–744. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.I.; Zaanen, J.; Andersen, O.K. Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 1991, 44, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yang, B.; Zhou, X.; Qiu, X.; Zhu, D.; Wu, H.; Li, M.; Long, Q.; Xia, Y.; Chen, J.; et al. Adsorption mechanism of hydrated Lu(OH)2+ and Al(OH)2+ ions on the surface of kaolinite. Powder Technol. 2022, 407, 117611. [Google Scholar] [CrossRef]
- Frost, R.L.; Horváth, E.; Makó, É.; Kristóf, J. Modification of low- and high-defect kaolinite surfaces: Implications for kaolinite mineral processing. J. Colloid Interface Sci. 2004, 270, 337–346. [Google Scholar] [CrossRef]
- Chen, J.; Min, F.; Liu, L.; Cai, C. Systematic exploration of the interactions between Fe-doped kaolinite and coal based on DFT calculations. Fuel 2020, 266, 117082. [Google Scholar] [CrossRef]
- Zhu, B.-L.; Qi, C.-L.; Zhang, Y.-H.; Bisson, T.; Xu, Z.; Fan, Y.-J.; Sun, Z.-X. Synthesis, characterization and acid-base properties of kaolinite and metal (Fe, Mn, Co) doped kaolinite. Appl. Clay Sci. 2019, 179, 105138. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom | s | p | d | Total | Charge/e | Sc-O Bond | Bond Length(Å) |
|---|---|---|---|---|---|---|---|
| O1 | 1.84 | 5.10 | 0 | 6.94 | −0.94 | 0.13 | 2.191 |
| O2 | 1.84 | 5.10 | 0 | 6.94 | −0.94 | 0.13 | 2.211 |
| O3 | 1.86 | 5.10 | 0 | 6.96 | −0.96 | 0.13 | 2.282 |
| O4 | 1.86 | 5.12 | 0 | 6.98 | −0.98 | 0.14 | 2.322 |
| O5 | 1.82 | 5.12 | 0 | 6.94 | −0.94 | 0.14 | 2.357 |
| O6 | 1.84 | 5.12 | 0 | 6.96 | −0.96 | 0.13 | 2.357 |
| O7 | 1.84 | 5.10 | 0 | 6.94 | −0.94 | 0.13 | 2.181 |
| O8 | 1.86 | 5.11 | 0 | 6.97 | −0.97 | 0.14 | 2.344 |
| Sc | 2.20 | 5.88 | 0.88 | 8.96 | 2.04 | \ | \ |
| Surface | (eV/Atom) | Atom Energy (eV) | ||
|---|---|---|---|---|
| Mg-doped | −4.311 | H | O | Al |
| Ca-doped | −4.286 | |||
| Fe-doped | −4.179 | −12.5 | −434.36 | −52.79 |
| Site | State | N a | R(Sc-Ow)avg b/Å | R(Sc-Os) c/Å | Hs-Ow d/Å | Hw-Os e/ Å | Eads/kJ·mol−1 |
|---|---|---|---|---|---|---|---|
| OuH | before | 7 | 2.266 | 1.965 | 1.383 | 1.554, 1.606, 1.624, 1.803 | −986.56 |
| After | 5 | 2.175 | |||||
| OtH | before | 7 | 2.270 | 1.966 | 1.483, 2.240, 1.992 | 1.399, 1.564, 1.710 | −922.24 |
| After | 5 | 2.187 | |||||
| OlH | before | 7 | 2.270 | 1.968 | \ | 1.486, 1.520, 1.554, 1.617, 1.939 | −915.04 |
| After | 5 | 2.182 |
| Atomic | s | p | d | Total | Charge (e) | Sc-Os Bond |
|---|---|---|---|---|---|---|
| Sc before | 2.18 | 5.88 | 0.90 | 8.96 | 2.04 | 0.45 |
| Sc after | 2.18 | 5.90 | 1.02 | 9.10 | 1.90 | |
| Os before | 1.87 | 5.34 | 0 | 7.21 | −1.21 | |
| Os after | 1.86 | 5.14 | 0 | 7.00 | −1.00 |
| Site | N | R(Sc-Ow)avg /Å | R(Sc-Os) /Å | Hs-Ow /Å | Hw-Os /Å | Eads /kJ·mol−1 |
|---|---|---|---|---|---|---|
| Pure surface | 5 | 2.175 | 1.965 | 1.383 | 1.554, 1.606, 1.624, 1.803 | −986.56 |
| Mg surface | 6 | 2.319 | 1.873 | 2.030, 2.230, 2.375, 2.429 | 1.442, 1.503, 1.704 | −1104.16 |
| Ca surface | 6 | 2.310 | 1.869 | \ | 1.485, 1.523, 1.560, 1.689, 1.759, 2.298 | −1088.16 |
| Fe surface | 5 | 2.262 | 1.971 | \ | 1.470, 1.565, 1.576, 1.653, 1.682 | −853.92 |
| State | Sc | Os | Sc-Os | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| s | p | d | Total | Charge | s | p | Total | Charge | ||
| Pure surface | 2.18 | 5.90 | 1.02 | 9.10 | 1.90 | 1.86 | 5.14 | 7.00 | −1.00 | 0.45 |
| Mg surface | 2.20 | 5.88 | 1.22 | 9.30 | 1.70 | 1.87 | 5.09 | 6.96 | −0.96 | 0.60 |
| Ca surface | 2.18 | 5.90 | 1.21 | 9.29 | 1.71 | 1.85 | 5.06 | 6.91 | −0.91 | 0.58 |
| Fe surface | 2.20 | 5.94 | 1.02 | 9.16 | 1.84 | 1.85 | 5.05 | 6.91 | −0.91 | 0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Zhao, Z.; Wu, G.; Jiang, D.; Lan, Y. Investigating the Influence of Impurity Defects on the Adsorption Behavior of Hydrated Sc3+ on the Kaolinite (001) Surface Using Density Functional Theory. Materials 2024, 17, 610. https://doi.org/10.3390/ma17030610
Wang K, Zhao Z, Wu G, Jiang D, Lan Y. Investigating the Influence of Impurity Defects on the Adsorption Behavior of Hydrated Sc3+ on the Kaolinite (001) Surface Using Density Functional Theory. Materials. 2024; 17(3):610. https://doi.org/10.3390/ma17030610
Chicago/Turabian StyleWang, Kaiyu, Zilong Zhao, Guoyuan Wu, Dengbang Jiang, and Yaozhong Lan. 2024. "Investigating the Influence of Impurity Defects on the Adsorption Behavior of Hydrated Sc3+ on the Kaolinite (001) Surface Using Density Functional Theory" Materials 17, no. 3: 610. https://doi.org/10.3390/ma17030610
APA StyleWang, K., Zhao, Z., Wu, G., Jiang, D., & Lan, Y. (2024). Investigating the Influence of Impurity Defects on the Adsorption Behavior of Hydrated Sc3+ on the Kaolinite (001) Surface Using Density Functional Theory. Materials, 17(3), 610. https://doi.org/10.3390/ma17030610