

First-Principles Study of Nitrogen Adsorption and Dissociation on ZrMnFe(110) Surface


Abstract
1. Introduction
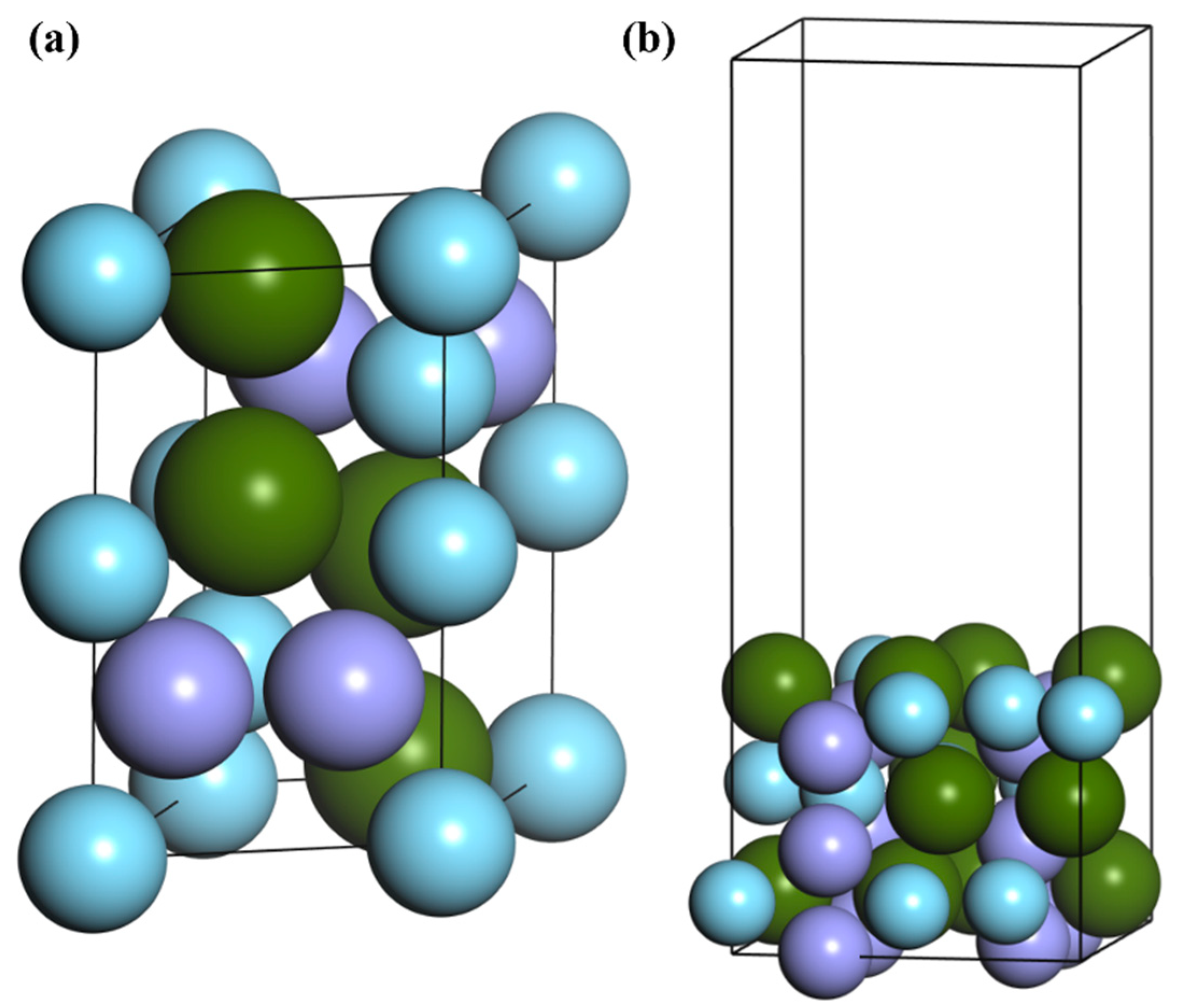
2. Calculation Model and Method
3. Calculation Results and Discussion
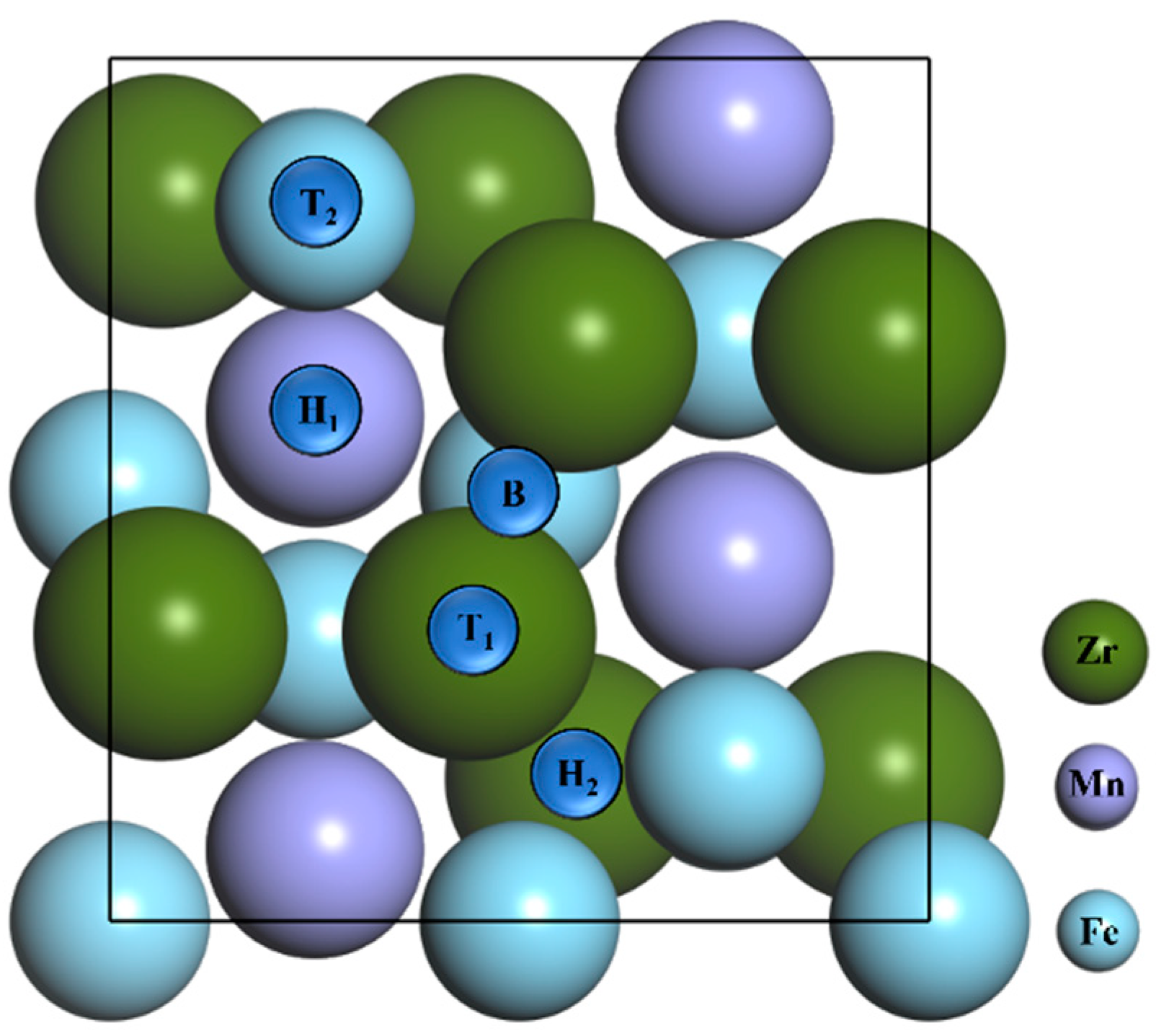
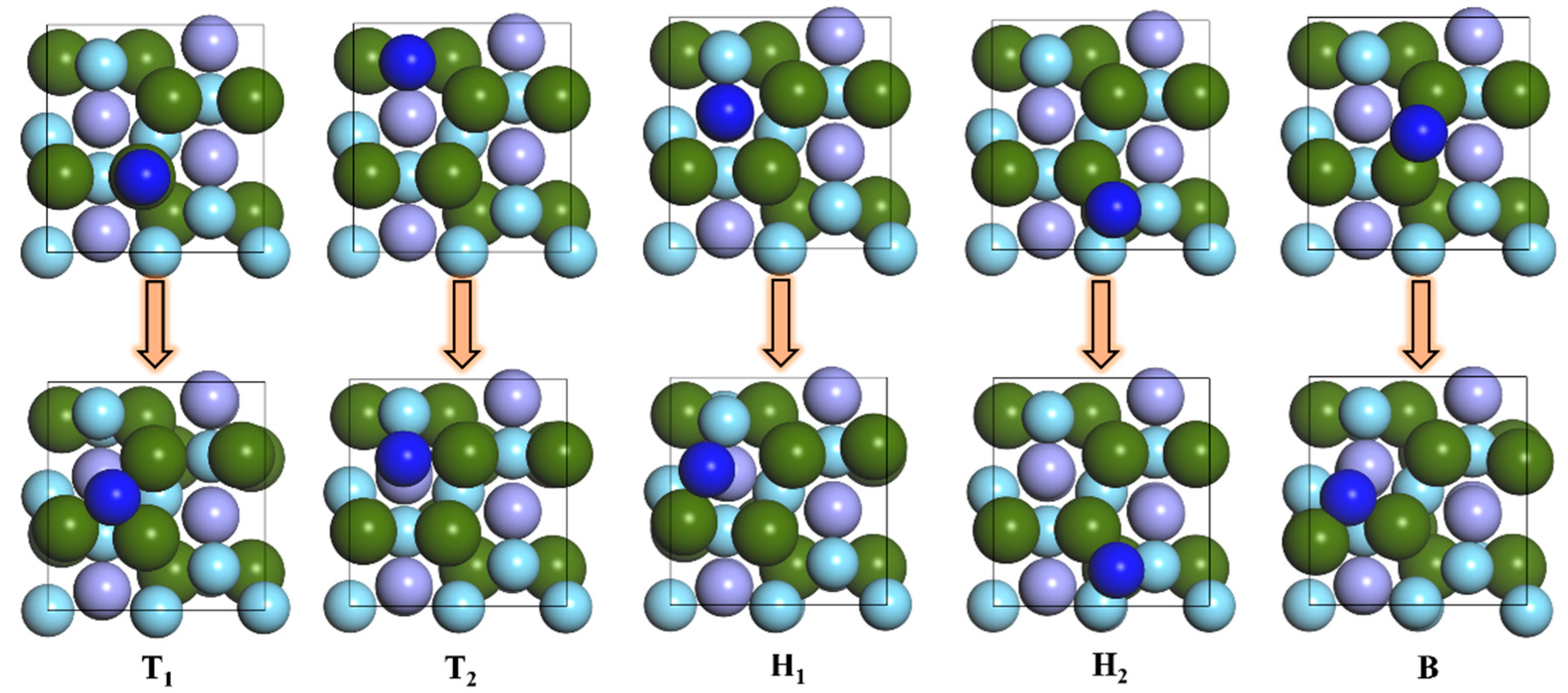
3.1. Adsorption of N Atoms on ZrMnFe(110) Surface
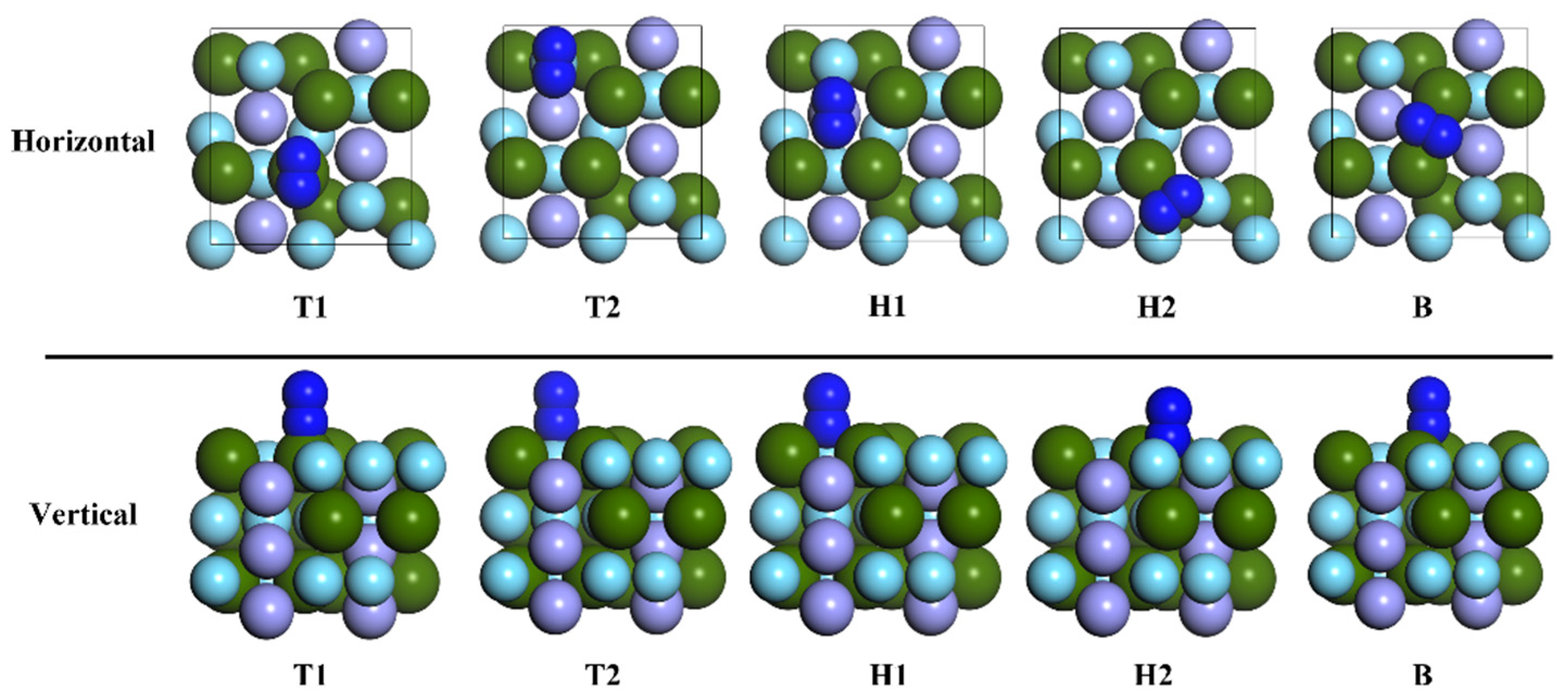
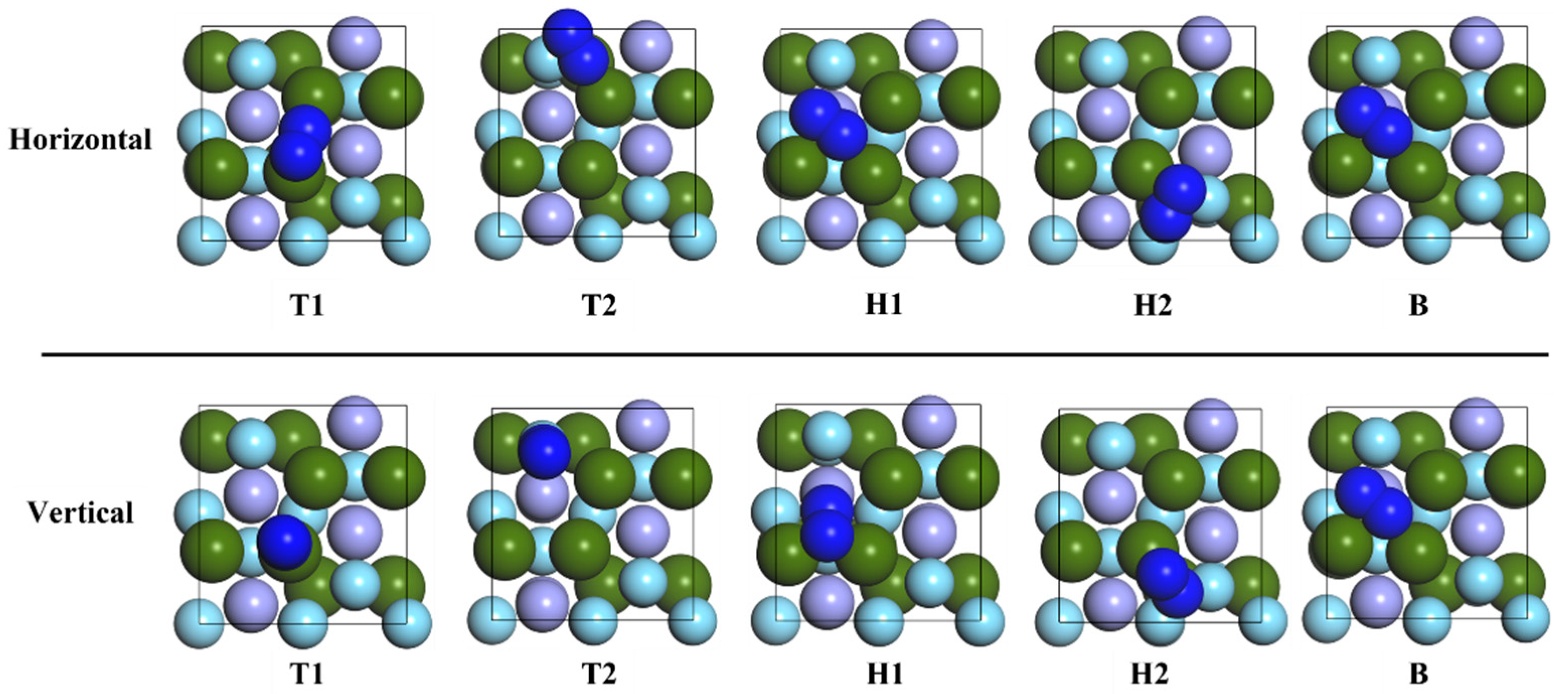
3.2. Adsorption of N2 Molecule on ZrMnFe(110) Surface
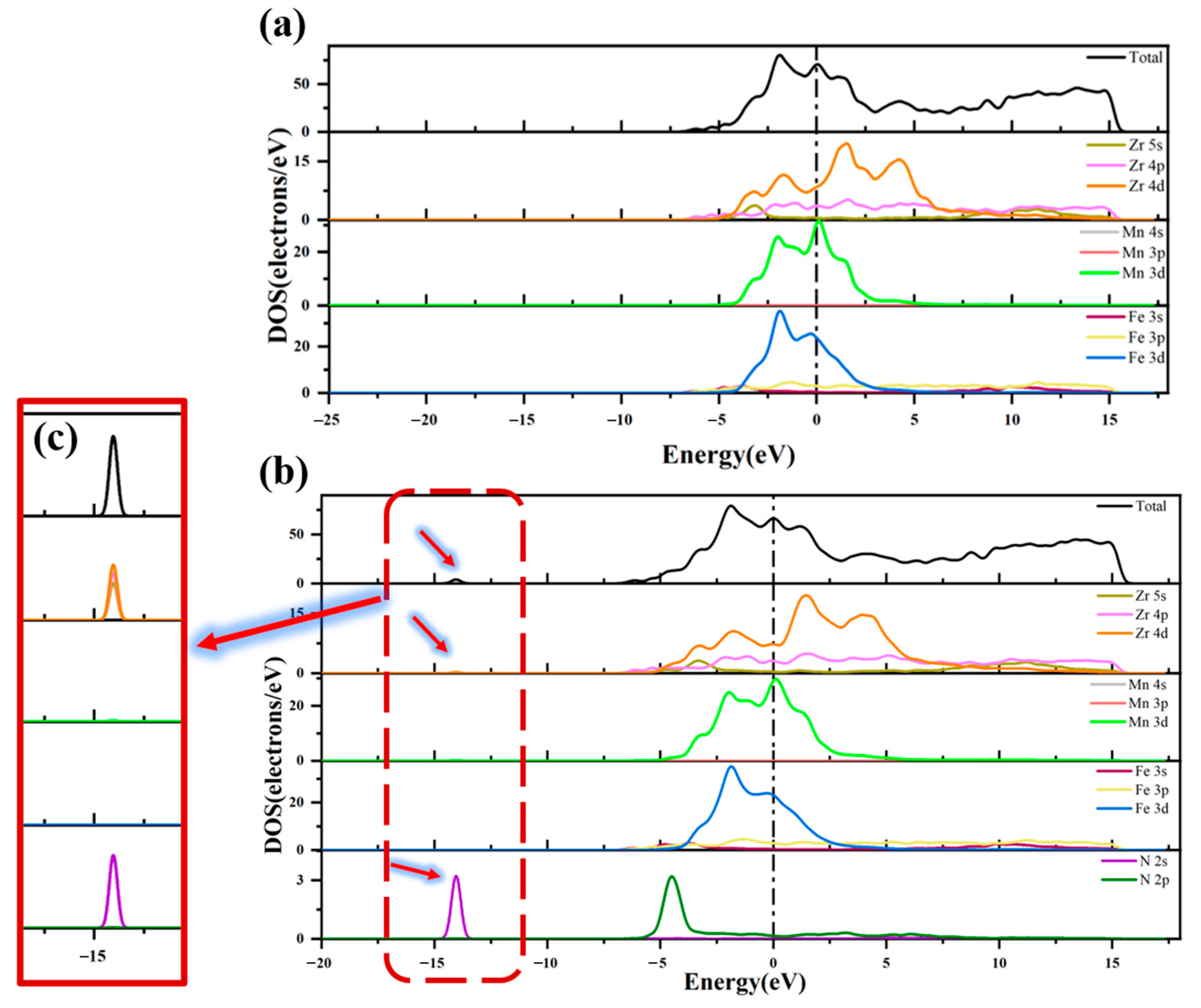
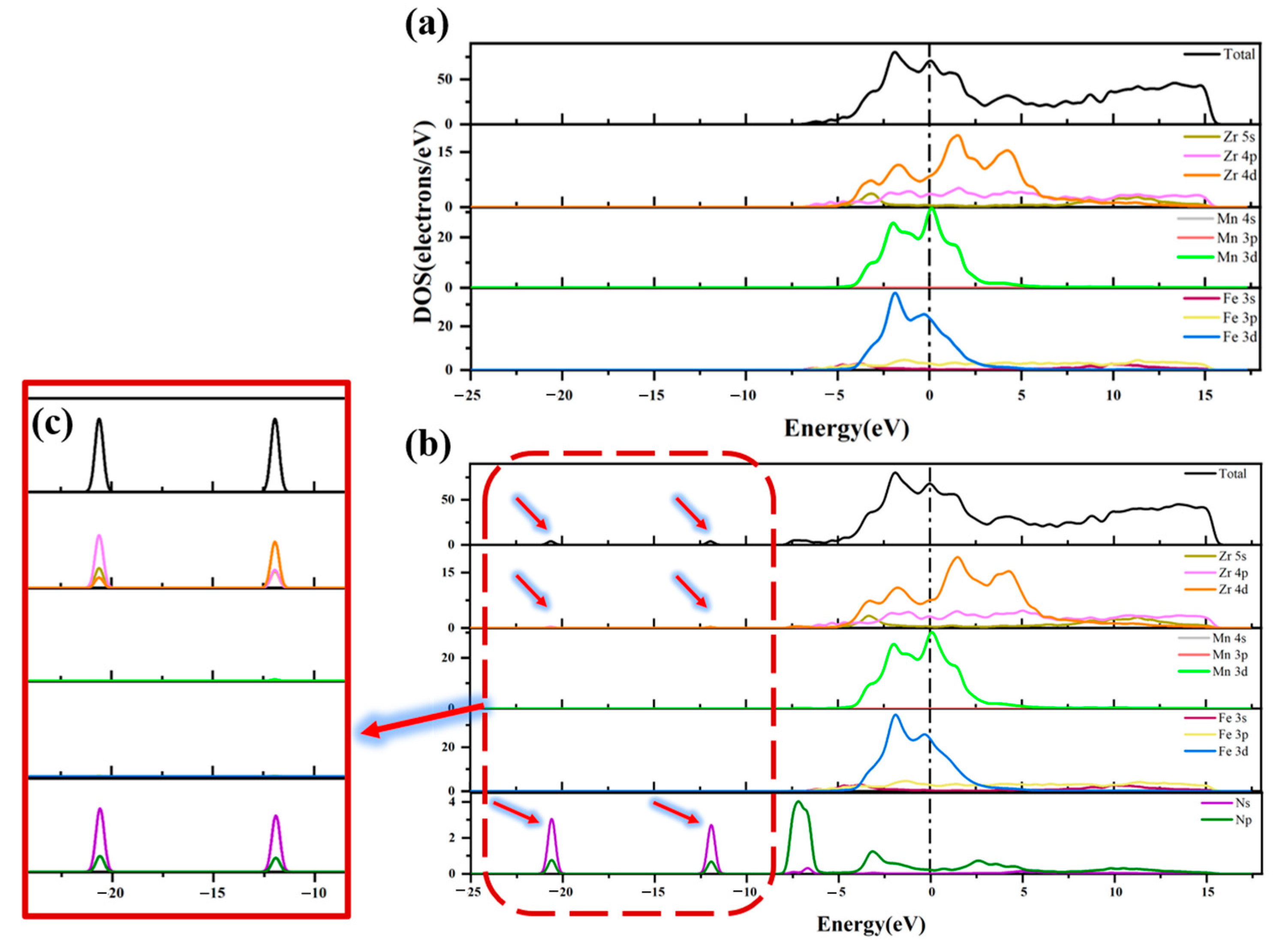
3.3. Analysis of Electronic Structure
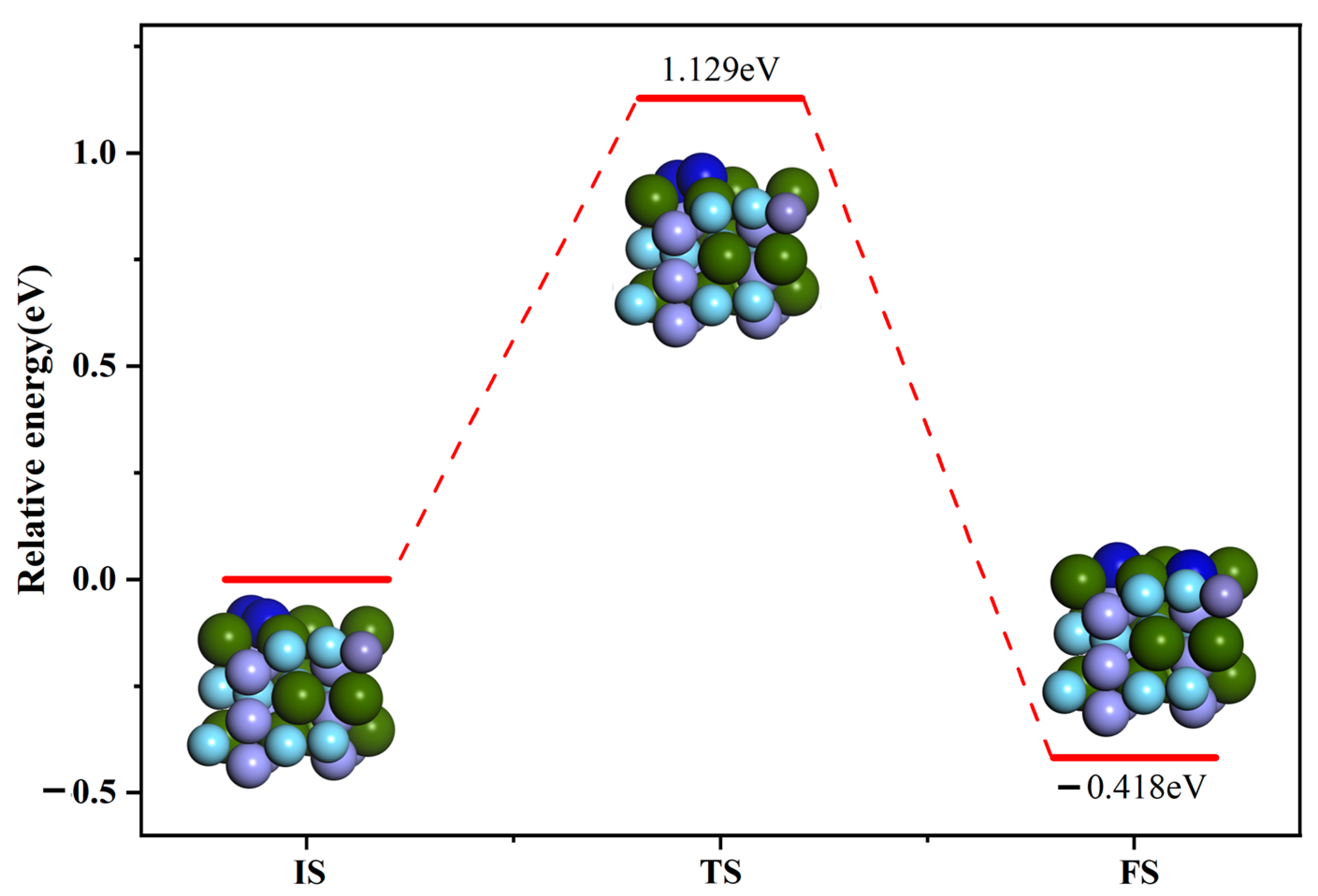
3.4. Interface Reaction
4. Conclusions
- (1)
- The adsorption behavior of the N atom and N2 molecule on the ZrMnFe(110) surface was investigated theoretically by using the first-principles calculation method. The results show that vacancies Hollow 1 and H1-Horis are the most stable adsorption configurations of the N atom and N2 molecule on the ZrMnFe(110) surface, with adsorption energy of 6.057 eV and 10.215 eV, respectively. The N2 molecular bond is activated after adsorption, with its length relaxing from 1.099 to 1.350 Å.
- (2)
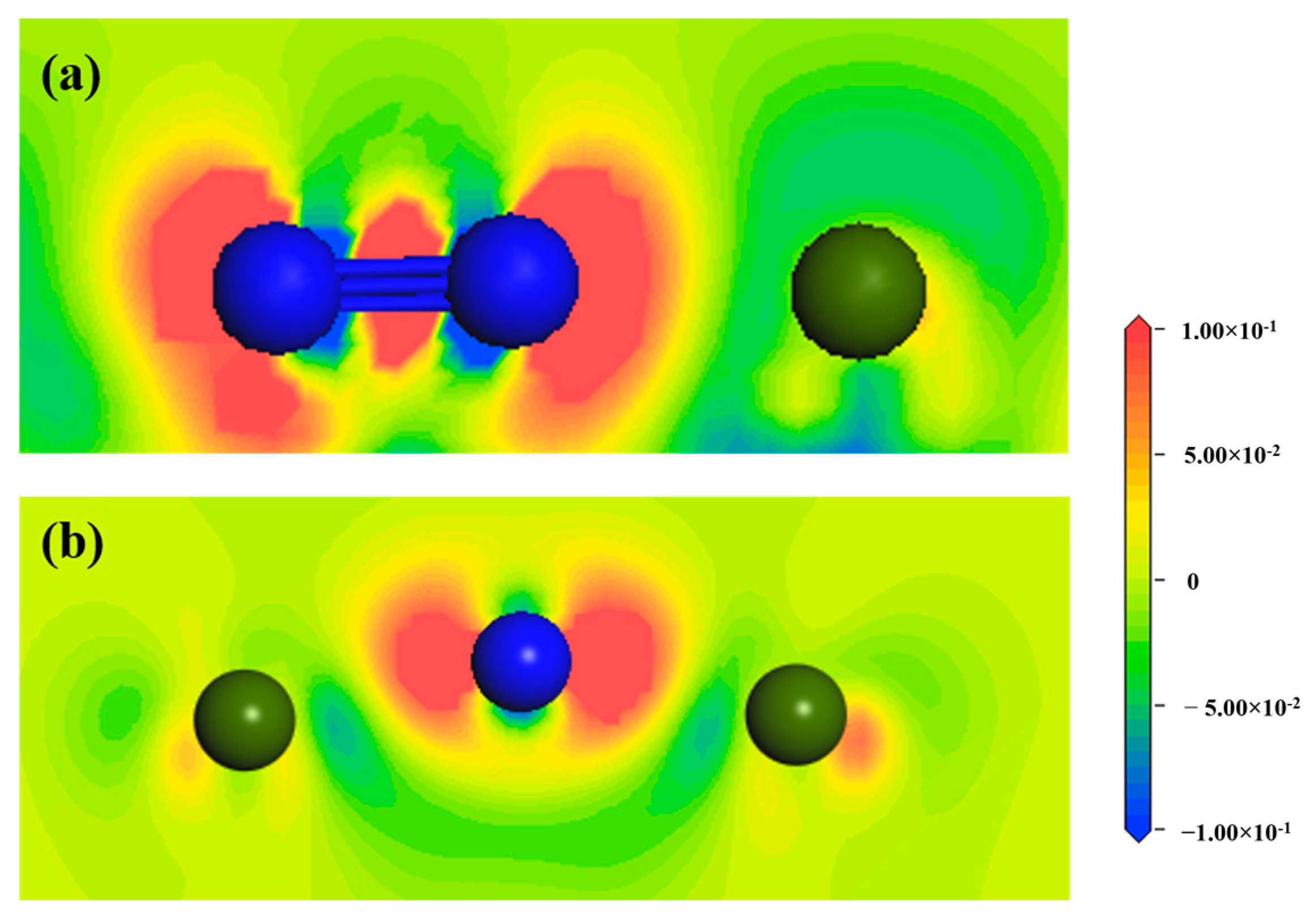
- Differential charge density analysis shows that both N atoms and N2 molecules gain electrons after adsorption, while Zr atoms on the ZrMnFe(110) surface lose electrons, forming a N-Zr ionic bond, but N2 molecules do not completely dissociate with the N-N bond, which is attributed to the hybridization of the 5s, 4p and 4d orbitals of Zr and the 2s orbitals of N after adsorption according to the density analysis of the bound states.
- (3)
- The calculated transition states show that the maximum energy barrier to be overcome during the dissociation of the N2 molecule on the surface of ZrMnFe(110) is 1.129 eV, while that during the permeation of the N atom from the surface of ZrMnFe(110) is lower at 0.766 eV. Moreover, N2 molecules are more likely to undergo osmotic reaction after dissociation reaction on the ZrMnFe(110) surface. All these results can provide theoretical guidance for exploring the adsorption mechanism of the N2 molecule on the ZrMnFe(110) surface.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beckner, M.; Dailly, A. Hydrogen and methane storage in adsorbent materials for automotive applications. Int. J. Energy Res. 2016, 40, 91–99. [Google Scholar] [CrossRef]
- Ma, S.; Zhou, H.-C. Gas storage in porous metal-organic frameworks for clean energy applications. Chem. Commun. 2010, 46, 44–53. [Google Scholar] [CrossRef]
- Banerjee, D.; Cairns, A.J.; Liu, J.; Motkuri, R.K.; Nune, S.K.; Fernandez, C.A.; Krishna, R.; Strachan, D.M.; Thallapally, P.K. Potential of Metal–Organic Frameworks for Separation of Xenon and Krypton. Acc. Chem. Res. 2015, 48, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.E.; Hoelder, J.S. Screening Tests for Improved Methane Cracking Materials. Fusion Sci. Technol. 2008, 54, 611–614. [Google Scholar] [CrossRef]
- Panasenko, A.E.; Shichalin, O.O.; Yarusova, S.B.; Ivanets, A.I.; Belov, A.A.; Dran, A.N.; Azon, S.A.; Fedorets, A.N.; Buravlev, I.Y.; Mayorov, V.Y.; et al. A novel approach for rice straw agricultural waste utilization: Synthesis of solid aluminosilicate matrices for cesium immobilization. Nucl. Eng. Technol. 2022, 54, 3250–3259. [Google Scholar] [CrossRef]
- Shichalin, O.O.; Yarusova, S.B.; Ivanets, A.I.; Papynov, E.K.; Belov, A.A.; Azon, S.A.; Buravlev, I.Y.; Panasenko, A.E.; Zadorozhny, P.A.; Mayorov, V.Y.; et al. Synthesis and spark plasma sintering of solid-state matrices based on calcium silicate for 60Co immobilization. J. Alloy. Compd. 2022, 912, 165233. [Google Scholar] [CrossRef]
- Shichalin, O.O.; Papynov, E.K.; Nepomnyushchaya, V.A.; Ivanets, A.I.; Belov, A.A.; Dran'Kov, A.N.; Yarusova, S.B.; Buravlev, I.Y.; Tarabanova, A.E.; Fedorets, A.N. Hydrothermal synthesis and spark plasma sintering of NaY zeolite as solid-state matrices for cesium-137 immobilization. J. Eur. Ceram. Soc. 2022, 42, 3004–3014. [Google Scholar] [CrossRef]
- Dran, A.; Shichalin, O.; Papynov, E.; Nomerovskii, A.; Mayorov, V.; Pechnikov, V.; Ivanets, A.; Buravlev, I.; Yarusova, S.; Zavjalov, A.; et al. Hydrothermal synthesis, structure and sorption performance to cesium and strontium ions of nanostructured magnetic zeolite composites. Nucl. Eng. Technol. 2022, 54, 1991–2003. [Google Scholar] [CrossRef]
- Yarusova, S.B.; Shichalin, O.O.; Belov, A.A.; Azon, S.A.; Buravlev, I.Y.; Golub, A.V.; Mayorov, V.Y.; Gerasimenko, A.V.; Papynov, E.K.; Ivanets, A.I.; et al. Synthesis of amorphous KAlSi3O8 for cesium radionuclide immobilization into solid matrices using spark plasma sintering technique. Ceram. Int. 2022, 48, 3808–3817. [Google Scholar] [CrossRef]
- Li, B.; Wen, H.M.; Wei, Z.; Chen, B. Porous Metal-Organic Frameworks for Gas Storage and Separation: What, How, and Why? J. Phys. Chem. Lett. 2014, 5, 3468–3479. [Google Scholar] [CrossRef]
- Chuntonov, K.; Setina, J.; Douglass, G. The Newest Getter Technologies: Materials, Processes, Equipment. J. Mater. Sci. Chem. Eng. 2015, 3, 57–67. [Google Scholar] [CrossRef]
- Zhao, Y.; Seredych, M.; Zhong, Q.; Bandosz, T.J. Aminated graphite oxides and their composites with copper-based metal–organic framework: In search for efficient media for CO2 sequestration. Rsc Adv. 2013, 3, 9932–9941. [Google Scholar] [CrossRef]
- Dassanayake, R.S.; Gunathilake, C.; Abidi, N.; Jaroniec, M. Activated carbon derived from chitin aerogels: Preparation and CO2 adsorption. Cellulose 2018, 25, 1911–1920. [Google Scholar] [CrossRef]
- Sladekova, K.; Campbell, C.; Grant, C.; Fletcher, A.J.; Gomes, J.R.B.; Jorge, M. The effect of atomic point charges on adsorption isotherms of CO2 and water in metal organic frameworks. Adsorption 2020, 26, 663–685. [Google Scholar] [CrossRef]
- Papynov, E.K.; Portnyagin, A.S.; Modin, E.B.; Yu, M.V.; Shichalin, O.O.; Golikov, A.P.; Pechnikov, V.S.; Gridasova, E.A.; Tananaev, I.G.; Avramenko, V.A. A complex approach to assessing porous structure of structured ceramics obtained by SPS technique. Mater. Charact. 2018, 145, 294–302. [Google Scholar] [CrossRef]
- James, D.W.; Morgan, G.A. Evaluation of the Effects of Impurities on SAES ST198 Hydrogen Gettering. Fusion Sci. Technol. 2017, 71, 321–325. [Google Scholar] [CrossRef]
- Larson, E.J.; Cook, K.J.; Wermer, J.R.; Tuggle, D.G. Nitriding reactions with a Zr–Mn–Fe metal getter. J. Alloy. Compd. 2002, 330, 897–901. [Google Scholar] [CrossRef]
- James, D.W.; Morgan, G.A. Evaluation of getters for methane and ammonia decomposition. Fusion Eng. Des. 2018, 133, 1–5. [Google Scholar] [CrossRef]
- Hsu, R.H.; Hoelder, J.S. Testing of a Prototype SAES St909 Getter Bed for Conditioning Gas to a Tritium Stripper System. Fusion Sci. Technol. 2005, 48, 171–174. [Google Scholar] [CrossRef]
- Staff, A. SAESST909 Bench Scale Methane Cracking Tests. Nucl. Sci. Eng. 2002, 41, 998–1003. [Google Scholar]
- Stein, F.; Leineweber, A. Laves phases: A review of their functional and structural applications and an improved fundamental understanding of stability and properties. J. Mater. Sci. 2021, 56, 5321–5427. [Google Scholar] [CrossRef]
- Wan, C.B.; Jiang, X.P.; Yin, X.H.; Ju, X. High-capacity Zr-based AB2-type alloys as metal hydride battery anodes. J. Alloy. Compd. 2020, 828, 154402. [Google Scholar] [CrossRef]
- Prigent, J.; Latroche, M.; Leoni, E.; Rohr, V. Hydrogen trapping properties of Zr-based intermetallic compounds in the presence of CO contaminant gas. J. Alloy. Compd. 2011, 509, S801–S803. [Google Scholar] [CrossRef]
- Kumar, L.; Ramanujan, R.V.; Tewari, R.; Mukhopadhyay, P.; Banerjee, S. Active eutectoid decomposition in Zr-3 wt.% Fe. Scr. Mater. 1999, 40, 723–728. [Google Scholar] [CrossRef]
- Stein, F.; Sauthoff, G.; Palm, M. Experimental Determination Phase Equilibria, and Temperatures in the Fe-Zr System of Intermetallic Phases, Invariant Reaction. Scand. J. Immunol. 1985, 21, 267–273. [Google Scholar] [CrossRef]
- Li, L.; Zeng, F.; Li, W.; Wang, Z.; Liu, W. Nitrogen absorption behavior and mechanism of TiZrMnFe getter alloy. Vacuum 2020, 183, 109814. [Google Scholar] [CrossRef]
- Jahnke, M.C.; Hahn, E. Crystal Structure and Microstructure Analysis of Alloys Zr(Mn 1 x M x ) 2 H y with M = V, Fe, Co, Ni, Al and their Hydrides*. Z. Für Phys. Chem. 1992, 1, 199–210. [Google Scholar]
- Baerends, E.J. Perspective on "Self-consistent equations including exchange and correlation effects"—Kohn W, Sham LJ (1965) Phys Rev A 140:133-1138. Theor. Chem. Acc. 2000, 103, 265–269. [Google Scholar] [CrossRef]
- Adamo, M.; Scuseria, G.E. The meta-GGA functional: Thermochemistry with a kinetic energy density dependent exchange-correlation functional. J. Chem. Phys. 2000, 112, 2643–2649. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.J.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Blochl, P.; Blöchl, E.; Blöchl, P. Projected augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Am. Phys. Soc. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys Rev B Condens Matter 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Huda, M.N. Hydrogen Molecule Adsorption and Dissociation on Plutonium (111) Surface. Am. Phys. Soc. 2005, 72, 085101. [Google Scholar]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Bell, S.; Crighton, J.S. Locating transition states. J. Chem. Phys. 1984, 80, 2464–2475. [Google Scholar] [CrossRef]
- Fischer, S.; Karplus, M. Conjugate peak refinement: An algorithm for finding reaction paths and accurate transition states in systems with many degrees of freedom. Chem. Phys. Lett. 1992, 194, 252–261. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-Adsorption Site | Site after Adsorption | Adsorption Energy Ea/eV | Distance/Å |
|---|---|---|---|
| Top 1 | Hollow 1 | / | / |
| Top 2 | Hollow 1 | / | / |
| Hollow 1 | Hollow 1 | / | / |
| Hollow 2 | Hollow 2 | 5.592 | 3.357 |
| Bridge | Hollow 1 | 6.057 | 0.659 |
| Pre-Adsorption Site | Site after Adsorption | Adsorption Energy Ea/eV | N-N Distance/Å |
|---|---|---|---|
| T1-Hor | T1-B | 10.215 | 1.210 |
| T2-Hor | T2-B | 9.772 | 1.285 |
| H1-Hor | H1 | 10.215 | 1.350 |
| H2-Hor | H2 | 8.389 | 1.255 |
| B-Hor | H1 | 10.214 | 1.350 |
| T1-Ver | T1 | 7.807 | 1.127 |
| T2-Ver | T2 | 8.068 | 1.139 |
| H1-Ver | H1-B | 9.398 | 1.296 |
| H2-Ver | H2 | 8.389 | 1.238 |
| B-Ver | H1 | 10.215 | 1.350 |
| Free N2 | / | / | 1.099 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Zeng, F.; Chen, M.; Dai, Y.; Gao, Y.; Huang, R.; Gu, Y.; Song, J. First-Principles Study of Nitrogen Adsorption and Dissociation on ZrMnFe(110) Surface. Materials 2023, 16, 3323. https://doi.org/10.3390/ma16093323
Yang Q, Zeng F, Chen M, Dai Y, Gao Y, Huang R, Gu Y, Song J. First-Principles Study of Nitrogen Adsorption and Dissociation on ZrMnFe(110) Surface. Materials. 2023; 16(9):3323. https://doi.org/10.3390/ma16093323
Chicago/Turabian StyleYang, Qiaobin, Fanhao Zeng, Meiyan Chen, Yu Dai, Yafang Gao, Rui Huang, Yi Gu, and Jiangfeng Song. 2023. "First-Principles Study of Nitrogen Adsorption and Dissociation on ZrMnFe(110) Surface" Materials 16, no. 9: 3323. https://doi.org/10.3390/ma16093323
APA StyleYang, Q., Zeng, F., Chen, M., Dai, Y., Gao, Y., Huang, R., Gu, Y., & Song, J. (2023). First-Principles Study of Nitrogen Adsorption and Dissociation on ZrMnFe(110) Surface. Materials, 16(9), 3323. https://doi.org/10.3390/ma16093323