
Long-Term Oxidation of Zirconium Alloy in Simulated Nuclear Reactor Primary Coolant—Experiments and Modeling


Abstract
1. Introduction
2. Materials and Methods
3. Results
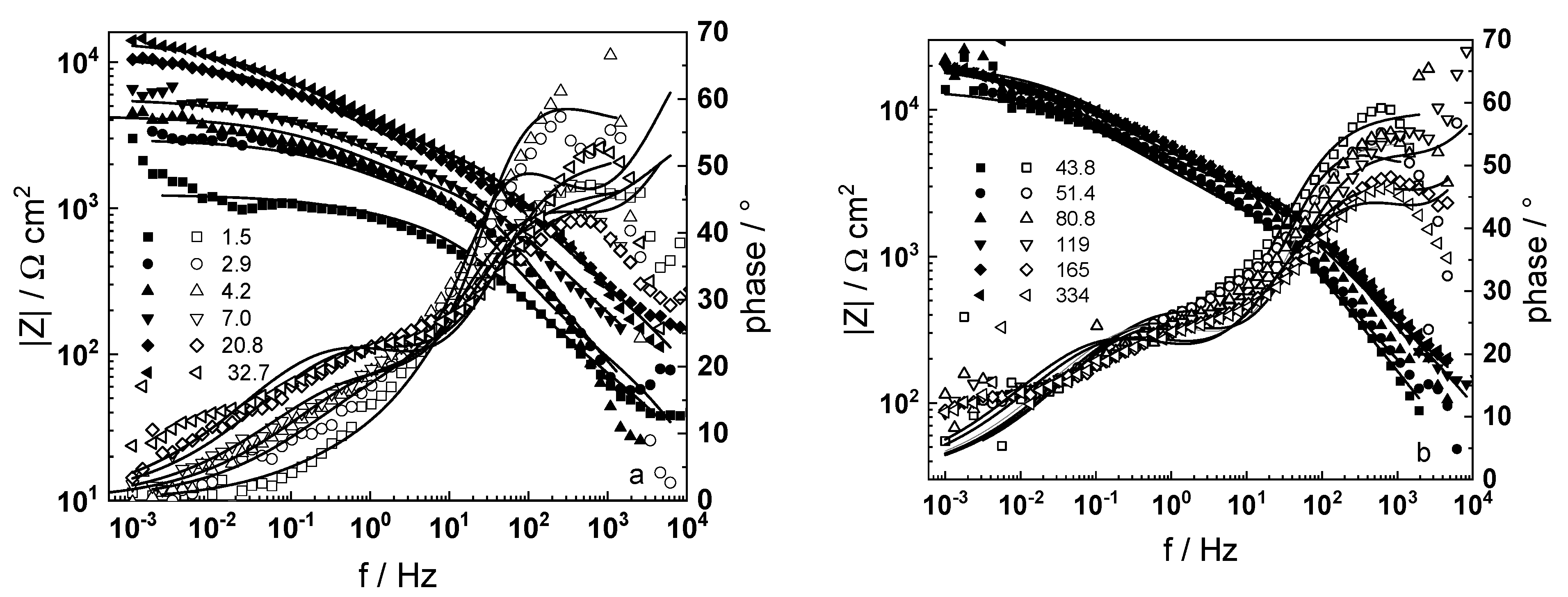
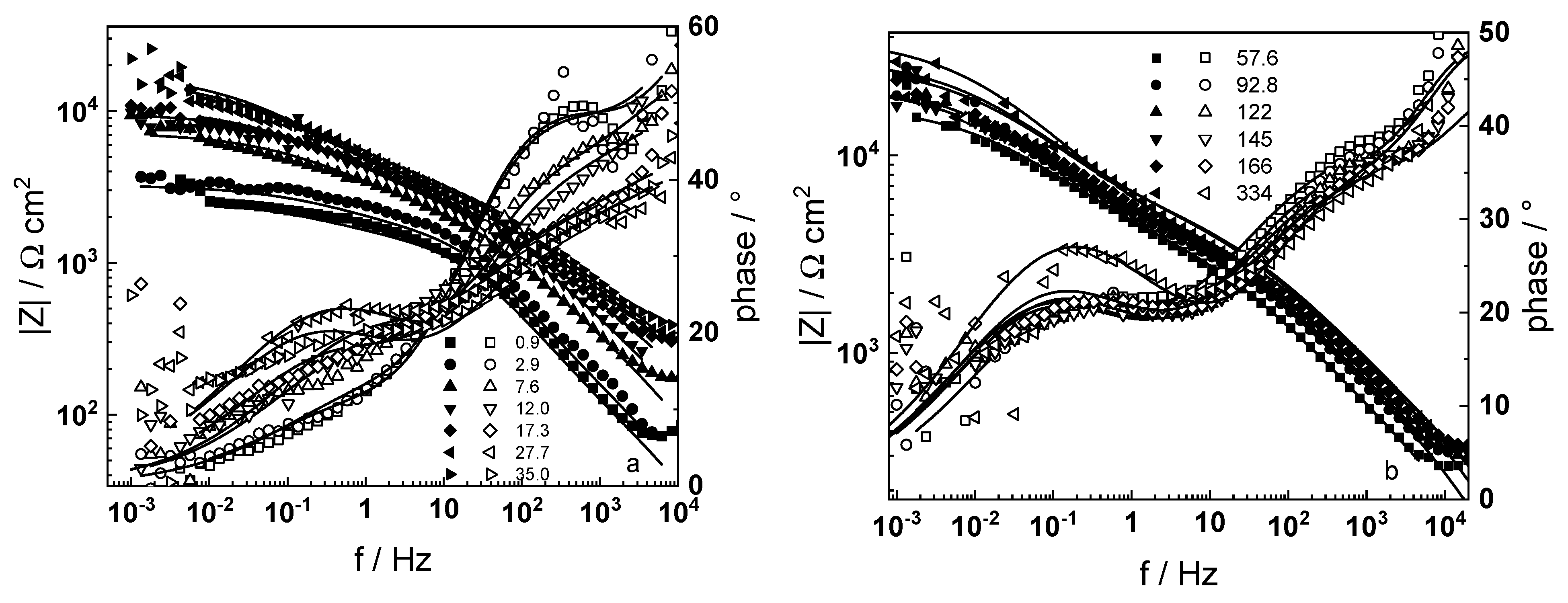
3.1. Electrochemical Impedance Spectroscopy
3.2. Chemical Composition and Thickness
4. Discussion
4.1. Physical Model and Basic Equations
4.2. Parameter Estimation
- The rate constant of metal oxidation at the alloy/oxide interface decreases significantly with time indicating that the corrosion rate decreases with increasing film thickness. Nearly constant values are reached after ca. 100–200 h of oxidation, i.e., a quasi-steady state is achieved. On the other hand, the rate of incorporation of oxygen at the oxide/coolant interface is almost independent on time, suggesting that this process is of secondary significance when compared to metal oxidation at the inner interface;
- The charge transfer resistance at the oxide/coolant interface increases with oxidation time, which means that the respective rate of water reduction decreases. This is in line with the decrease of metal oxidation rate leading to a smaller electron supply rate for the coupled cathodic reaction, taking into account the fact that the diffusion coefficient of electrons does not exhibit any dependence on oxidation time.
4.3. Influence of Internal Stresses on Oxygen Transport by Vacancy Mechanism
4.4. Influence of Space Charge of Substitutional Ions on the Field Strength in the Oxide
4.5. Kinetics of Barrier Oxide Growth
4.6. Model Validation Based on Computational Results
5. Conclusions
- Electrochemical impedance measurements allow to discernment of the contribution of barrier and outer layer conductivities, kinetics of interfacial reactions, and ionic defect transport in the overall oxidation process;
- Layer thicknesses estimated from EIS are in good agreement with those determined by ex-situ analysis techniques (GDOES and SEM). The thickness of the barrier protective layer is much larger than that of the outer layer except for oxidation of ex-situ pre-hydrogenated samples;
- The proposed kinetic model is able to reproduce quantitatively the impedance spectra as depending on oxidation time in a variety of experimental conditions;
- Taking into account the influence of compressive stress and space charge in the oxide allows rationalization of the dependences of the diffusion coefficient of oxygen via vacancy mechanism and the field strength in the oxide on film thickness, or equivalent oxidation time. Barrier thickness vs. time data are successfully interpreted with the same set of kinetic parameters furnishing further credibility to the model.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tupin, M. Understanding the Corrosion Processes of Fuel Cladding in Pressurized Water Reactors. In Nuclear Corrosion, European Federation of Corrosion; Elsevier: Amsterdam, The Netherlands, 2020; pp. 252–299. [Google Scholar]
- Yagnik, S.; Garde, A. Zirconium Alloys for LWR Fuel Cladding and Core Internals. In Structural Alloys for Nuclear Energy Applications; Elsevier: Amsterdam, The Netherlands, 2019; pp. 247–291. [Google Scholar]
- Fromhold, A.T. Theory of Metal Oxidation-Vol. I: Fundamentals; North-Holland: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Cox, B. Some thoughts on the mechanisms of in-reactor corrosion of zirconium alloys. J. Nucl. Mater. 2005, 336, 331–368. [Google Scholar] [CrossRef]
- Motta, A.T. Mechanistic Understanding of Zirconium Alloy Fuel Cladding Performance. In Zirconium in the Nuclear Industry, Proceedings of the 18th International Symposium; Comstock, R.J., Motta, A.T., Eds.; ASTM International: West Conshohocken, PA, USA, 2018; pp. 19–51. [Google Scholar]
- Actis-Dato, L.O.; Aldave de Las Heras, L.; Betti, M.; Toscano, E.H.; Miserque, F.; Gouder, T. Investigation of mechanisms of corrosion due to diffusion of impurities by direct current glow discharge mass spectrometry depth profiling. J. Anal. Atom. Spectrom. 2000, 15, 1479–1484. [Google Scholar] [CrossRef]
- Moya, J.S.; Diaz, M.; Bartolomeé, J.F.; Roman, E.; Sacedon, J.L.; Izquierdo, J. Zirconium oxide film formation on zircaloy by water corrosion. Acta Mater. 2000, 48, 4749–4754. [Google Scholar] [CrossRef]
- Sawicki, J. Evidence of Ni2FeBO5 and m-ZrO2 precipitates in fuel rod deposits in AOA-affected high boiling duty PWR core. J. Nucl. Mater. 2008, 374, 248–269. [Google Scholar] [CrossRef]
- Henshaw, J.; McGurk, J.C.; Sims, H.E.; Tuson, A.; Dickinson, S.; Deshon, J. A model of chemistry and thermal hydraulics in PWR fuel crud deposits. J. Nucl. Mater. 2006, 353, 1–11. [Google Scholar] [CrossRef]
- Nagy, G.; Kerner, Z.; Pajkossy, T. In situ electrochemical impedance spectroscopy of Zr–1%Nb under VVER primary circuit conditions. J. Nucl. Mater. 2002, 300, 230–236. [Google Scholar] [CrossRef]
- Vermoyal, J.J.; Hammou, A.; Dessemond, L.; Frichet, A. Electrical characterization of waterside corrosion films formed on ZrNb(1%)O(0.13%). Electrochim. Acta 2002, 47, 2679–2695. [Google Scholar] [CrossRef]
- Schefold, J.; Lincot, D.; Ambard, A.; Kerrec, O. The Cyclic Nature of Corrosion of Zr and Zr-Sn in High-Temperature Water at 633 K. A Long-Term In Situ Impedance Spectroscopic Study. J. Electrochem. Soc. 2003, 150, B451–B461. [Google Scholar] [CrossRef]
- Chen, Y.; Urquidi-Macdonald, M.; Macdonald, D.D. The electrochemistry of zirconium in aqueous solutions at elevated temperatures and pressures. J. Nucl. Mater. 2006, 348, 133–147. [Google Scholar] [CrossRef]
- Ai, J.Y.; Chen, M.; Urquidi-Macdonald, M.; Macdonald, D.D. Electrochemical Impedance Spectroscopic Study of Passive Zirconium. I. High-Temperature, Deaerated Aqueous Solutions. J. Electrochem. Soc. 2007, 154, C43. [Google Scholar] [CrossRef]
- Ai, J.Y.; Chen, M.; Urquidi-Macdonald, M.; Macdonald, D.D. Electrochemical Impedance Spectroscopic Study of Passive Zirconium. II. High-Temperature, Hydrogenated Aqueous Solutions. J. Electrochem. Soc. 2007, 154, C52. [Google Scholar] [CrossRef]
- Couet, A.; Motta, A.T.; Ambard, A.; Livigni, D. In-situ electrochemical impedance spectroscopy measurements of zirconium alloy oxide conductivity: Relationship to hydrogen pickup. Corros. Sci. 2017, 119, 1–13. [Google Scholar] [CrossRef]
- Kim, T.; Couet, A.; Kim, S.; Lee, Y.; Yoo, S.C.; Kim, J.H. In-situ electrochemical study of zirconium alloy in high temperature hydrogenated water conditions. Corros. Sci. 2020, 173, 108745. [Google Scholar] [CrossRef]
- Renciukova, V.; Macak, J.; Sajdl, P.; Novotný, R.; Krausova, A. Corrosion of zirconium alloys demonstrated by using impedance spectroscopy. J. Nucl. Mater. 2018, 510, 312–321. [Google Scholar] [CrossRef]
- Bojinov, M.; Cai, W.; Kinnunen, P.; Saario, T. Kinetic parameters of the oxidation of zirconium alloys in simulated WWER water—Effect of KOH content. J. Nucl. Mater. 2008, 378, 45–54. [Google Scholar] [CrossRef]
- Bojinov, M.; Karastoyanov, V.; Kinnunen, P.; Saario, T. Influence of water chemistry on the corrosion mechanism of a zirconium-niobium alloy in simulated light water reactor coolant conditions. Corros. Sci. 2010, 52, 54–67. [Google Scholar] [CrossRef]
- Beverskog, B.; Bojinov, M.; Kinnunen, P.; Laitinen, T.; Makela, K.; Saario, T. A mixed-conduction model for oxide films on. Fe, Cr and Fe–Cr alloys in high-temperature aqueous electrolytes—II. Adaptation and justification of the model. Corros. Sci. 2002, 44, 1923–1940. [Google Scholar] [CrossRef]
- Boukamp, B. Practical application of the Kramers-Kronig transformation on impedance measurements in solid state electrochemistry. Solid State Ion. 1993, 62, 131–141. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane analysis of α-dispersions in some polymer systems. J. Polym. Sci. C 1966, 14, 99–117. [Google Scholar]
- Yamamoto, Y.; Morishita, K.; Iwakiri, H.; Kaneta, Y. Stress dependence of oxygen diffusion in ZrO2 film. Nucl. Instrum. Methods Phys. Res. 2013, B303, 42–45. [Google Scholar] [CrossRef]
- Swan, H.; Blackmur, M.S.; Hyde, J.M.; Laferrere, A.; Ortner, S.R.; Styman, P.D.; Staines, C.; Gass, M.; Hulme, H.; Cole-Baker, A.; et al. The measurement of stress and phase fraction distributions in pre and post-transition Zircaloy oxides using nano-beam synchrotron X-ray diffraction. J. Nucl. Mater. 2016, 479, 559–575. [Google Scholar] [CrossRef]
- Moorehead, M.; Yu, Z.; Borrel, L.; Hu, J.; Cai, Z.; Couet, A. Comprehensive investigation of the role of Nb on the oxidation kinetics of Zr-Nb alloys. Corros. Sci. 2019, 155, 173–181. [Google Scholar] [CrossRef]
- Couet, A.; Borrel, L.; Liu, J.; Hu, J.; Grovenor, C. An integrated modeling and experimental approach to study hydrogen pickup mechanism in zirconium alloys. Corros. Sci. 2019, 159, 108134. [Google Scholar] [CrossRef]
- Fromhold, A., Jr. Single Carrier Steady-State Theory for Formation of Anodic Films Under Conditions of High Space Charge in Very Large Electric Fields. J. Electrochem. Soc. 1977, 124, 538–549. [Google Scholar] [CrossRef]
- Moorehead, M.; Hu, J.; Couet, A.; Cai, Z. Progressing Zirconium-Alloy Corrosion Models Using Synchrotron XANES. In Proceedings of the 18th International Conference on Environmental Degradation of Materials in Nuclear Power Systems—Water Reactors, Portland, OR, USA, 13–17 August 2017; Jackson, J., Paraventi, D., Wright, M., Eds.; The Minerals, Metals & Materials Series. Springer: Cham, Switzerland, 2018; pp. 565–576. [Google Scholar]
- Hu, J.; Garner, A.; Frankel, P.; Li, M.; Kirk, M.A.; Lozano-Perez, S.; Preuss, M.; Grovenor, C. Effect of neutron and ion irradiation on the metal matrix and oxide corrosion layer on Zr-1.0Nb cladding alloys. Acta Mater. 2019, 173, 313–326. [Google Scholar] [CrossRef]
- Likhanskii, V.V.; Evdokimov, I.A.; Aliev, T.N.; Kon’kov, V.F.; Markelov, V.A.; Novikov, V.V.; Khokhunova, T.N. Corrosion model for zirconium-niobium alloys in pressurized water reactors. At. Energy 2014, 116, 186–193. [Google Scholar] [CrossRef]
- Ramezani, L.; Mansouri, M.; Rahgoshay, M. Modeling the Water Side Corrosion and Hydrogen Pickup of the WWER fuel clad. Nucl. Technol. Radiat. Prot. 2018, 33, 334–340. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Zr | Nb | Sn | Fe | N | C | O |
|---|---|---|---|---|---|---|---|
| Content/wt.% | Base | 1.05 | 0.04 | 0.05 | <0.01 | 0.02 | 0.10 |
| Experiment | Oxide Thickness (GDOES) µm/2 Points | Oxide Thickness (SEM) µm/22 Points |
|---|---|---|
| w/o LiOH, 720 h | 0.44 ± 0.01 | 0.40 ± 0.04 |
| with LiOH, 720 h | 0.31 ± 0.01 | 0.27 ± 0.04 |
| w/o LiOH, ex-situ charged, 360 h | 0.32 ± 0.01 | 0.33 ± 0.04 |
| Oxide Structure | Isotropic Compression | Compression in Direction a | Compression in Direction b | Compression in Direction c |
|---|---|---|---|---|
| Ef m-ZrO2(O1)/eV | 0.010 | 0.012 | 0.011 | 0.032 |
| Ef m-ZrO2(O2)/eV | 0.012 | 0.016 | 0.010 | 0.022 |
| Ef t-ZrO2/eV | 0.015 | 0.011 | 0.006 | |
| EV t-ZrO2/eV | 0.034 | 0.070 | 0.008 | 0.030 |
| Ratio of Diffusion Coefficients | Isotropic Compression | Compression in Direction a | Compression in Direction b | Compression in Direction c |
|---|---|---|---|---|
| 0.37 | 0.19 | 0.68 | 0.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betova, I.; Bojinov, M.; Karastoyanov, V. Long-Term Oxidation of Zirconium Alloy in Simulated Nuclear Reactor Primary Coolant—Experiments and Modeling. Materials 2023, 16, 2577. https://doi.org/10.3390/ma16072577
Betova I, Bojinov M, Karastoyanov V. Long-Term Oxidation of Zirconium Alloy in Simulated Nuclear Reactor Primary Coolant—Experiments and Modeling. Materials. 2023; 16(7):2577. https://doi.org/10.3390/ma16072577
Chicago/Turabian StyleBetova, Iva, Martin Bojinov, and Vasil Karastoyanov. 2023. "Long-Term Oxidation of Zirconium Alloy in Simulated Nuclear Reactor Primary Coolant—Experiments and Modeling" Materials 16, no. 7: 2577. https://doi.org/10.3390/ma16072577
APA StyleBetova, I., Bojinov, M., & Karastoyanov, V. (2023). Long-Term Oxidation of Zirconium Alloy in Simulated Nuclear Reactor Primary Coolant—Experiments and Modeling. Materials, 16(7), 2577. https://doi.org/10.3390/ma16072577