
Polar GaN Surfaces under Gallium Rich Conditions: Revised Thermodynamic Insights from Ab Initio Calculations


Abstract
1. Introduction
2. Calculation Methods
2.1. Computational Model
2.2. Thermodynamic Model
3. Results and Discussion
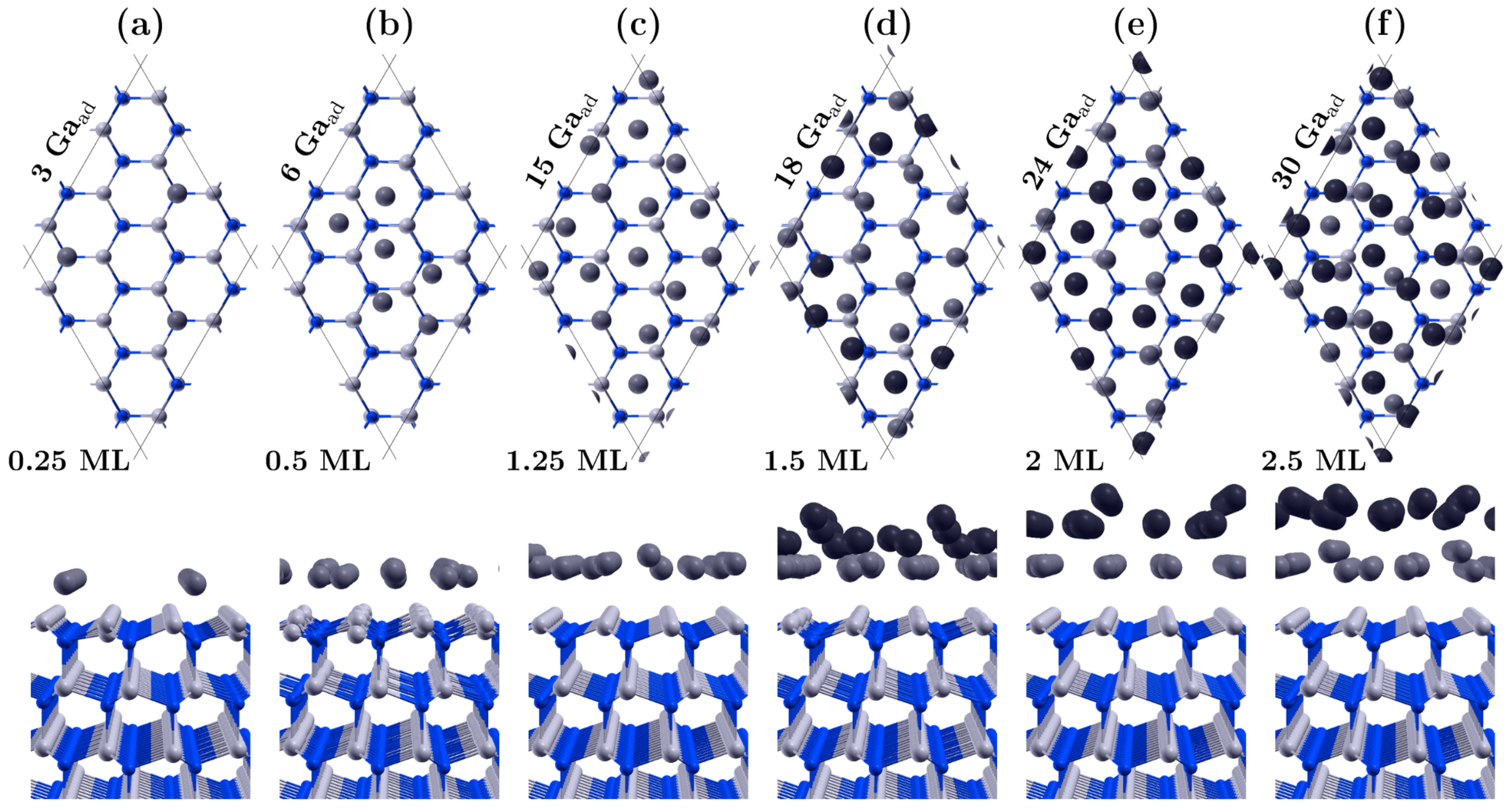
3.1. Covering the Polar GaN Surface by Gallium Atoms
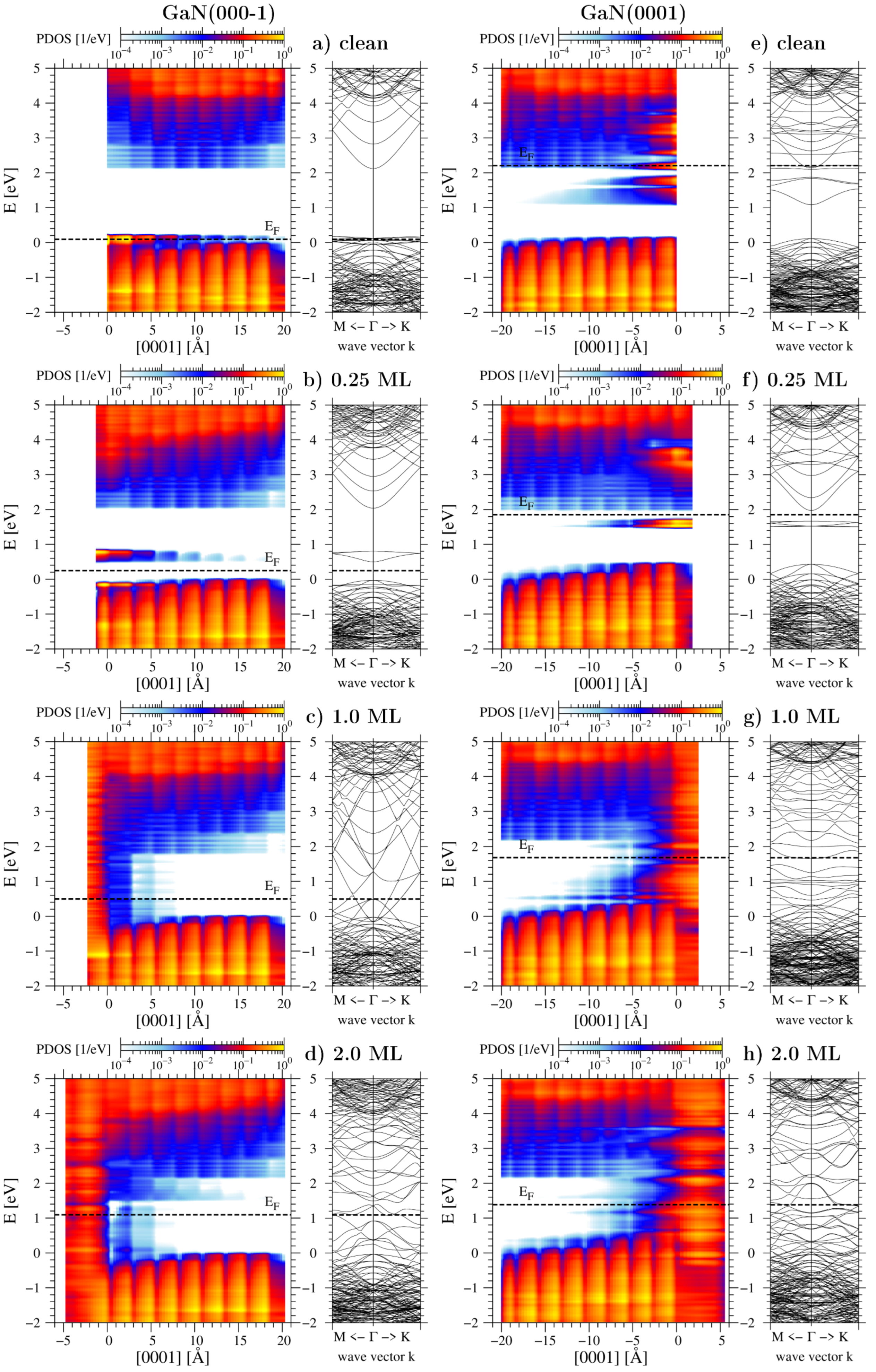
3.2. Chemical Potential of Gallium Adatoms and Electronic Properties of Surfaces
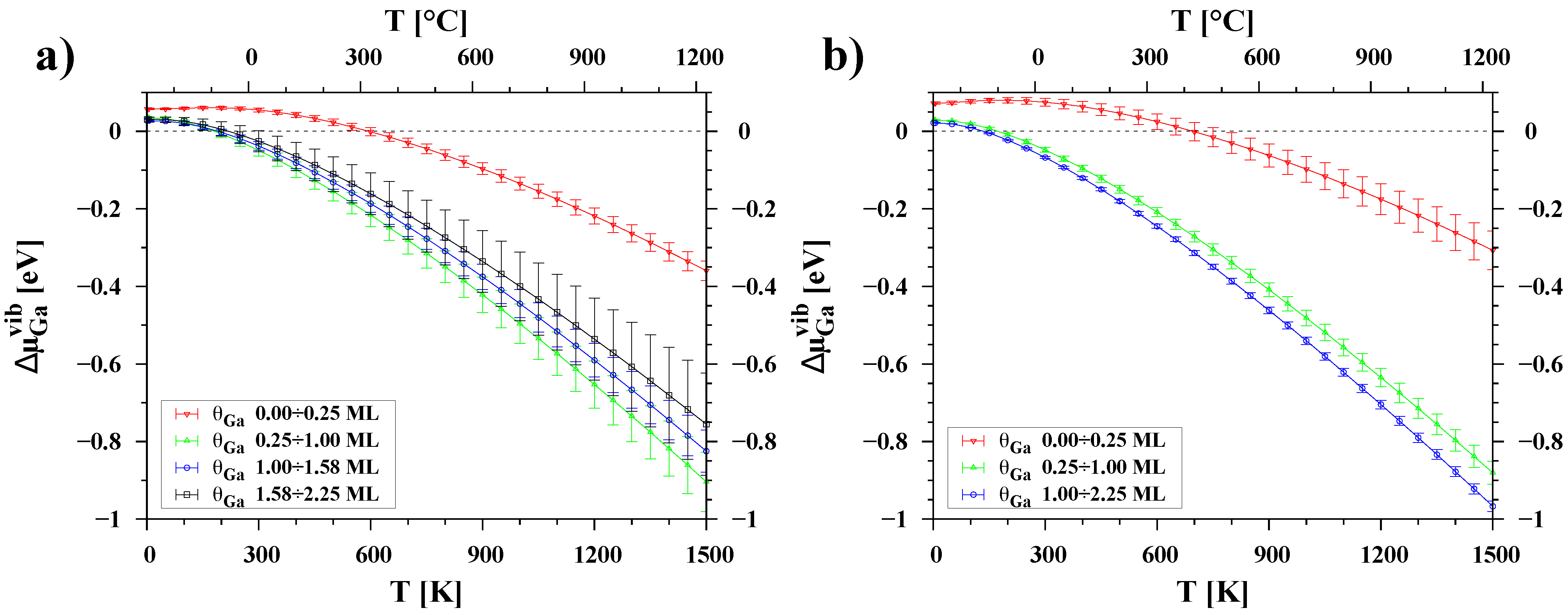
3.3. Temperature Dependence of Chemical Potential
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Yoshikawa, A. Molecular beam epitaxy growth of GaN, AlN and InN. Prog. Cryst. Growth Charact. Mater. 2004, 48–49, 42–103. [Google Scholar] [CrossRef]
- Heying, B.; Averbeck, R.; Chen, L.F.; Haus, E.; Riechert, H.; Speck, J.S. Control of GaN surface morphologies using plasma-assisted molecular beam epitaxy. J. Appl. Phys. 2000, 88, 1855–1860. [Google Scholar] [CrossRef]
- Heying, B.; Smorchkova, I.; Poblenz, C.; Elsass, C.; Fini, P.; Den Baars, S.; Mishra, U.; Speck, J.S. Optimization of the surface morphologies and electron mobilities in GaN grown by plasma-assisted molecular beam epitaxy. Appl. Phys. Lett. 2000, 77, 2885–2887. [Google Scholar] [CrossRef]
- Monroy, E.; Sarigiannidou, E.; Fossard, F.; Gogneau, N.; Bellet-Amalric, E.; Rouvière, J.L.; Monnoye, S.; Mank, H.; Daudin, B. Growth kinetics of N-face polarity GaN by plasma-assisted molecular-beam epitaxy. Appl. Phys. Lett. 2004, 84, 3684–3686. [Google Scholar] [CrossRef]
- Grandjean, N.; Massies, J.; Semond, F.; Karpov, S.Y.; Talalaev, R.A. GaN evaporation in molecular-beam epitaxy environment. Appl. Phys. Lett. 1999, 74, 1854–1856. [Google Scholar] [CrossRef]
- Smith, A.R.; Feenstra, R.M.; Greve, D.W.; Shin, M.S.; Skowronski, M.; Neugebauer, J.; Northrup, J.E. Reconstructions of GaN(0001) and (000-1) surfaces: Ga-rich metallic structures. J. Vac. Sci. Technol. B 1998, 16, 2242–2249. [Google Scholar] [CrossRef]
- Adelmann, C.; Brault, J.; Mula, G.; Daudin, B.; Lymperakis, L.; Neugebauer, J. Gallium adsorption on (0001) GaN surfaces. Phys. Rev. B 2003, 67, 165419. [Google Scholar] [CrossRef]
- Northrup, J.E.; Neugebauer, J.; Feenstra, R.M.; Smith, A.R. Structure of GaN(0001): The laterally contracted Ga bilayer model. Phys. Rev. B 2000, 61, 9932–9935. [Google Scholar] [CrossRef]
- Neugebauer, J.; Zywietz, T.K.; Scheffler, M.; Northrup, J.E.; Chen, H.; Feenstra, R.M. Adatom Kinetics On and Below the Surface: The Existence of a New Diffusion Channel. Phys. Rev. Lett. 2003, 90, 056101. [Google Scholar] [CrossRef]
- Hacke, P.; Feuillet, G.; Okumura, H.; Yoshida, S. Monitoring surface stoichiometry with the (2 × 2) reconstruction during growth of hexagonal-phase GaN by molecular beam epitaxy. Appl. Phys. Lett. 1996, 69, 2507–2509. [Google Scholar] [CrossRef]
- Koblmuller, G.; Averbeck, R.; Riechert, H.; Pongratz, P. Direct observation of different equilibrium Ga adlayer coverages and their desorption kinetics on GaN (0001) and (000-1) surfaces. Phys. Rev. B 2004, 69, 035325. [Google Scholar] [CrossRef]
- Koblmuller, G.; Brown, J.; Averbeck, R.; Riechert, H.; Pongratz, P.; Speck, J.S. Ga Adlayer Governed Surface Defect Evolution of (0001)GaN Films Grown by Plasma-Assisted Molecular Beam Epitaxy. Jpn. J. Appl. Phys. 2005, 44, L906–L908. [Google Scholar] [CrossRef]
- Koblmuller, G.; Brown, J.; Averbeck, R.; Riechert, H.; Pongratz, P.; Speck, J.S. Continuous evolution of Ga adlayer coverages during plasma-assisted molecular-beam epitaxy of (0001) GaN. Appl. Phys. Lett. 2005, 86, 041908. [Google Scholar] [CrossRef]
- Brown, J.S.; Koblmuller, G.; Wu, F.; Averbeck, R.; Riechert, H.; Speck, J.S. Ga adsorbate on (0001) GaN: In situ characterization with quadrupole mass spectrometry and reflection high-energy electron diffraction. J. Appl. Phys. 2006, 99, 074902. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [Google Scholar] [CrossRef]
- Inatomi, Y.; Kangawa, Y. Theoretical study of adatom stability on polar GaN surfaces during MBE and MOVPE. Appl. Surf. Sci. 2020, 502, 144205. [Google Scholar] [CrossRef]
- Kempisty, P.; Kangawa, Y. Evolution of the free energy of the GaN(0001) surface based on first-principles phonon calculations. Phys. Rev. B 2019, 100, 085304. [Google Scholar] [CrossRef]
- Pedroza, L.S.; da Silva, A.J.R.; Capelle, K. Gradient-dependent density functionals of the Perdew-Burke-Ernzerhof type for atoms, molecules, and solids. Phys. Rev. B 2009, 79, 201106. [Google Scholar] [CrossRef]
- Odashima, M.M.; Capelle, K.; Trickey, S.B. Tightened Lieb-Oxford Bound for Systems of Fixed Particle Number. J. Chem. Theory Comput. 2009, 5, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Soler, J.M.; Artacho, E.; Gale, J.D.; Garcia, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA method for ab initio order-N materials simulation. J. Phys. Condens. Matter 2002, 14, 2745–2779. [Google Scholar] [CrossRef]
- SIESTA. Available online: http://departments.icmab.es/leem/siesta (accessed on 20 July 2023).
- Leszczynski, M.; Teisseyre, H.; Suski, T.; Grzegory, I.; Bockowski, M.; Jun, J.; Porowski, S.; Pakula, K.; Baranowski, J.M.; Foxon, C.T.; et al. Lattice parameters of gallium nitride. Appl. Phys. Lett. 1996, 69, 73–75. [Google Scholar] [CrossRef]
- Shiraishi, K. A New Slab Model Approach for Electronic Structure Calculation of Polar Semiconductor Surface. J. Phys. Soc. Jpn. 1990, 59, 3455–3458. [Google Scholar] [CrossRef]
- Bengtsson, L. Dipole correction for surface supercell calculations. Phys. Rev. B 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Bitzek, E.; Koskinen, P.; Gahler, F.; Moseler, M.; Gumbsch, P. Structural Relaxation Made Simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef]
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Phys. Rev. Lett. 1997, 78, 4063–4066. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J.; Hafner, J. Ab initio Force Constant Approach to Phonon Dispersion Relations of Diamond and Graphite. Europhys. Lett. 1995, 32, 729–734. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Campbell, C.T.; Sprowl, L.H.; Arnadóttir, L. Equilibrium Constants and Rate Constants for Adsorbates: Two-Dimensional (2D) Ideal Gas, 2D Ideal Lattice Gas, and Ideal Hindered Translator Models. J. Phys. Chem. C 2016, 120, 10283–10297. [Google Scholar] [CrossRef]
- Elyukhin, V.; Pena-Sierra, R.; Garcia-Salgado, G. Thermodynamic models of molecular beams. Superf. Vacio 2004, 17, 25–28. [Google Scholar]
- Niu, H.; Bonati, L.; Piaggi, P.M.; Parrinello, M. Ab initio phase diagram and nucleation of gallium. Nat. Commun. 2020, 11, 2654. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, S.; Kempisty, P.; Strak, P.; Sakowski, K. Fermi level pinning and the charge transfer contribution to the energy of adsorption at semiconducting surfaces. J. Appl. Phys. 2014, 115, 043529. [Google Scholar] [CrossRef]
- Bui, K.; Boero, M.; Shiraishi, K.; Oshiyama, A. A two-dimensional liquid-like phase on Ga-rich GaN(0001) surfaces evidenced by first principles molecular dynamics. Jpn. J. Appl. Phys. 2020, 59, SGGK04. [Google Scholar] [CrossRef]
- Held, R.; Nowak, G.; Ishaug, B.E.; Seutter, S.M.; Parkhomovsky, A.; Dabiran, A.M.; Cohen, P.I.; Grzegory, I.; Porowski, S. Structure and composition of GaN(0001) A and B surfaces. J. Appl. Phys. 1999, 85, 7697–7704. [Google Scholar] [CrossRef]
- Fireman, M.; Li, H.; Keller, S.; Mishra, U.K.; Speck, J.S. Growth of N-polar GaN by ammonia molecular beam epitaxy. J. Cryst. Growth 2018, 481, 65–70. [Google Scholar] [CrossRef]
- Okumura, H.; McSkimming, B.M.; Huault, T.; Chaix, C.; Speck, J.S. Growth diagram of N-face GaN (000-1) grown at high rate by plasma-assisted molecular beam epitaxy. Appl. Phys. Lett. 2014, 104, 012111. [Google Scholar] [CrossRef]
- Kempisty, P. Gallium Adatoms on GaN Polar Surfaces—Thermodynamic Data from DFT Calculations. 2023. Available online: https://doi.org/10.18150/KOCW7R (accessed on 24 July 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Ga Coverage [ML] | Chemical Potential [eV] |
|---|---|---|
| GaN(000-1) | 0.00–0.25 | |
| 0.25–1.00 | ||
| 1.00–1.58 | ||
| 1.58–2.25 | ||
| GaN(0001) | 0.00–0.25 | |
| 0.25–1.00 | ||
| 1.00–2.50 |
| Surface | Ga Coverage | Fitting Parameters | ||
|---|---|---|---|---|
| [ML] | [eV] | a | b [eV] | |
| GaN(000-1) | 0.00–0.25 | −3.137 | 3.455 | 0.07304 |
| 0.25–1.00 | −0.768 | 2.811 | 0.01013 | |
| 1.00–1.58 | −0.157 | 3.020 | 0.02087 | |
| 1.58–2.25 | 0.061 | 1.784 | 0.00204 | |
| GaN(0001) | 0.00–0.25 | −0.851 | 3.167 | 0.06327 |
| 0.25–1.00 | −0.256 | 2.924 | 0.01168 | |
| 1.00–2.25 | −0.119 | 3.001 | 0.01088 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kempisty, P.; Kawka, K.; Kusaba, A.; Kangawa, Y. Polar GaN Surfaces under Gallium Rich Conditions: Revised Thermodynamic Insights from Ab Initio Calculations. Materials 2023, 16, 5982. https://doi.org/10.3390/ma16175982
Kempisty P, Kawka K, Kusaba A, Kangawa Y. Polar GaN Surfaces under Gallium Rich Conditions: Revised Thermodynamic Insights from Ab Initio Calculations. Materials. 2023; 16(17):5982. https://doi.org/10.3390/ma16175982
Chicago/Turabian StyleKempisty, Pawel, Karol Kawka, Akira Kusaba, and Yoshihiro Kangawa. 2023. "Polar GaN Surfaces under Gallium Rich Conditions: Revised Thermodynamic Insights from Ab Initio Calculations" Materials 16, no. 17: 5982. https://doi.org/10.3390/ma16175982
APA StyleKempisty, P., Kawka, K., Kusaba, A., & Kangawa, Y. (2023). Polar GaN Surfaces under Gallium Rich Conditions: Revised Thermodynamic Insights from Ab Initio Calculations. Materials, 16(17), 5982. https://doi.org/10.3390/ma16175982