

Synthesis of Ion-Exchange Catalysts by Introduction of Fluorinated Ponytails into Novel Mesoporous Polymers

 , ,
, ,
Abstract
1. Introduction
2. Experimental
2.1. Synthesis of Mesoporous Polydivinylbenzene
2.2. Acylation with Perfluorobutyryl Chloride
2.3. Sulfonation with Concentrated Sulfuric Acid
2.4. Esterification of Stearic Acid
2.5. Solid-State NMR
2.6. DFT Calculations
3. Results and Discussion







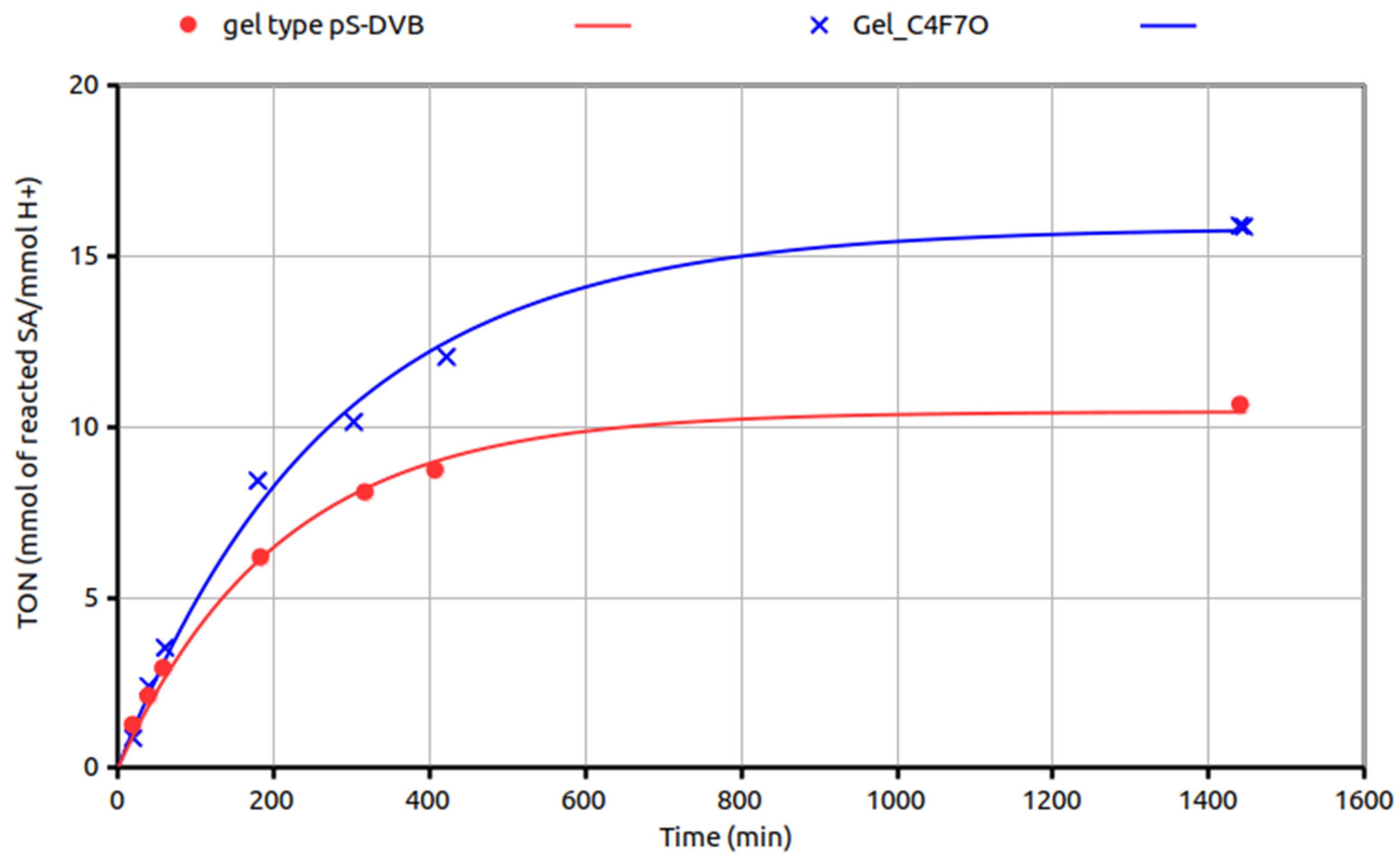
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Dorfner, K. Ion Exchangers; De Gruyter: Berlin, Germany; New York, NY, USA, 2011; ISBN 978-3-11-086243-0. [Google Scholar]
- Sherrington, D.C.; Hodge, P. Syntheses and Separations Using Functional Polymers; J. Wiley & Sons: Chichester, UK, 1988; ISBN 0-471-91848-2. [Google Scholar]
- Maul, J.; Frushour, B.G.; Kontoff, J.R.; Eichenauer, H.; Ott, K.-H.; Schade, C. Polystyrene and Styrene Copolymers. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; ISBN 978-3-527-30673-2. [Google Scholar]
- de Dardel, F.; Arden, T.V. Ion Exchangers. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2008; ISBN 978-3-527-30673-2. [Google Scholar]
- Corain, B.; Zecca, M.; Canton, P.; Centomo, P. Synthesis and Catalytic Activity of Metal Nanoclusters inside Functional Resins: An Endeavour Lasting 15 Years. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2010, 368, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Marco, Z.; Paolo, C.; Benedetto, C. CHAPTER 10—Metal Nanoclusters Supported on Cross-Linked Functional Polymers: A Class of Emerging Metal Catalysts. In Metal Nanoclusters in Catalysis and Materials Science; Corain, B., Schmid, G., Toshima, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 201–232. ISBN 978-0-444-53057-8. [Google Scholar]
- De Zan, L.; Gasparovicova, D.; Kralik, M.; Centomo, P.; Carraro, M.; Campestrini, S.; Jerabek, K.; Corain, B. Nanoclustered Palladium(0) Supported on a Gel-Type Poly-Acrylonitrile–N,N-Dimethylacrylamide–Ethylenedimethacrylate Resin: Nanostructural Aspects and Catalytic Behaviour. J. Mol. Catal. Chem. 2007, 265, 1–8. [Google Scholar] [CrossRef]
- Centomo, P.; Zecca, M.; Corain, B. Template Controlled Synthesis (TCS) of Size-Controlled Metal Nanoclusters: Preparation of Nanostructured Metals Supported by Inorganic Supports. J. Clust. Sci. 2007, 18, 947–962. [Google Scholar] [CrossRef]
- Centomo, P.; Jeřábek, K.; Canova, D.; Zoleo, A.; Maniero, A.L.; Sassi, A.; Canton, P.; Corain, B.; Zecca, M. Highly Hydrophilic Copolymers of N,N-Dimethylacrylamide, Acrylamido-2-Methylpropanesulfonic Acid, and Ethylenedimethacrylate: Nanoscale Morphology in the Swollen State and Use as Exotemplates for Synthesis of Nanostructured Ferric Oxide. Chem.—Eur. J. 2012, 18, 6632–6643. [Google Scholar] [CrossRef]
- Mbaraka, I.K.; Shanks, B.H. Design of Multifunctionalized Mesoporous Silicas for Esterification of Fatty Acid. J. Catal. 2005, 229, 365–373. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q. Solid Acid Catalysis Using Ion-Exchange Resins. Appl. Catal. Gen. 2001, 221, 45–62. [Google Scholar] [CrossRef]
- Jerabek, K.; Hankova, L.; Holub, L.; Corain, B.; Zecca, M.; Centomo, P.; Bonato, I. Strongly Acidic Ion Exchanger Catalyst and Method of Preparing the Same. WO2012127414A1, 27 September 2012. [Google Scholar]
- Centomo, P.; Bonato, I.; Hanková, L.; Holub, L.; Jeřábek, K.; Zecca, M. Novel Ion-Exchange Catalysts for Reactions Involving Lipophilic Reagents: Perspectives in the Reaction of Esterifications of Fatty Acids with Methanol. Top. Catal. 2013, 56, 611–617. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of Biomass to Selected Chemical Products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef]
- Soltani, S.; Rashid, U.; Al-Resayes, S.I.; Nehdi, I.A. Recent Progress in Synthesis and Surface Functionalization of Mesoporous Acidic Heterogeneous Catalysts for Esterification of Free Fatty Acid Feedstocks: A Review. Energy Convers. Manag. 2017, 141, 183–205. [Google Scholar] [CrossRef]
- Riess, J.G.; Blanc, M.L. Solubility and Transport Phenomena in Perfluorochemicals Relevant to Blood Substitution and Other Biomedical Applications. Pure Appl. Chem. 1982, 54, 2383–2406. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, S.; Liu, F.; Du, Y.; Liu, S.; Ji, Y.; Yokoi, T.; Tatsumi, T.; Xiao, F.-S. Superhydrophobic Nanoporous Polymers as Efficient Adsorbents for Organic Compounds. Nano Today 2009, 4, 135–142. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bagno, A.; Rastrelli, F.; Saielli, G. Predicting 13C NMR Spectra by DFT Calculations. J. Phys. Chem. A 2003, 107, 9964–9973. [Google Scholar] [CrossRef]
- Bagno, A.; Rastrelli, F.; Saielli, G. Toward the Complete Prediction of the 1H and 13C NMR Spectra of Complex Organic Molecules by DFT Methods: Application to Natural Substances. Chem.—Eur. J. 2006, 12, 5514–5525. [Google Scholar] [CrossRef] [PubMed]
- Jerabek, K. Characterization of Swollen Polymer Gels Using Size Exclusion Chromatography. Anal. Chem. 1985, 57, 1598–1602. [Google Scholar] [CrossRef]
- Jeřábek, K.; Hanková, L.; Holub, L. Working-State Morphologies of Ion Exchange Catalysts and Their Influence on Reaction Kinetics. J. Mol. Catal. Chem. 2010, 333, 109–113. [Google Scholar] [CrossRef]
- Jeřábek, K.; Zecca, M.; Centomo, P.; Marchionda, F.; Peruzzo, L.; Canton, P.; Negro, E.; Di Noto, V.; Corain, B. Synthesis of Nanocomposites from Pd0 and a Hyper-Cross-Linked Functional Resin Obtained from a Conventional Gel-Type Precursor. Chem.—Eur. J. 2013, 19, 9381–9387. [Google Scholar] [CrossRef]
- Liu, F.; Meng, X.; Zhang, Y.; Ren, L.; Nawaz, F.; Xiao, F.-S. Efficient and Stable Solid Acid Catalysts Synthesized from Sulfonation of Swelling Mesoporous Polydivinylbenzenes. J. Catal. 2010, 271, 52–58. [Google Scholar] [CrossRef]
- Hanková, L.; Holub, L.; Jeřábek, K. Formation of Porous Polymer Morphology by Microsyneresis during Divinylbenzene Polymerization. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 774–781. [Google Scholar] [CrossRef]
- Law, R.V.; Sherrington, D.C.; Snape, C.E.; Ando, I.; Korosu, H. Solid State 13C MAS NMR Studies of Anion Exchange Resins and Their Precursors. Ind. Eng. Chem. Res. 1995, 34, 2740–2749. [Google Scholar] [CrossRef]
- Ford, W.T.; Periyasamy, M.; Mohanraj, S.; McEnroe, F.J. Peak Area Measurements of Cross-Polarization Magic-Angle-Spinning 13C-NMR Spectra of Crosslinked Polystyrenes. J. Polym. Sci. Part Polym. Chem. 1989, 27, 2345–2355. [Google Scholar] [CrossRef]
- Law, R.V.; Sherrington, D.C.; Snape, C.E. Quantitative Solid State 13C NMR Studies of Highly Cross-Linked Poly(Divinylbenzene) Resins. Macromolecules 1997, 30, 2868–2875. [Google Scholar] [CrossRef]
- Tanaka, S.; Matsumoto, M.; Goseki, R.; Ishizone, T.; Hirao, A. Living Anionic Polymerization of 1,4-Divinylbenzene and Its Isomers. Macromolecules 2013, 46, 146–154. [Google Scholar] [CrossRef]
- Zhao, C.; Zhou, R.; Pan, H.; Jin, X.; Qu, Y.; Wu, C.; Jiang, X. Thermal Decomposition of Some Perfluoro- and Polyfluorodiacyl Peroxides. J. Org. Chem. 1982, 47, 2009–2013. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Macro | Meso | Micro | Vp | |
|---|---|---|---|---|
| Pd-SpDVB | 10% | 81% | 9% | 0.85 |
| pDVB_C4F7O | 39% | 54% | 7% | 0.72 |
| C (%, w/w) | H (%, w/w) | Fraction of Acylated Rings (%) | ||
|---|---|---|---|---|
| b | a | |||
| Gel_C4F7O | 76.36 | 6.05 | 19 | 21 |
| pDVB_C4F7O | 78.69 | 7.05 | 10 | 13 |
| Ca | Cb | Cc | Cd,e | |
|---|---|---|---|---|
| Gel_C4F7O | 43.03 ppm | 28.33 ppm | 143.42 ppm | 128.29 ppm |
| pDVB_C4F7O | 39.02 ppm | 25.52 ppm | 141.09 ppm | 124.47 ppm |
| Resin-CO-CF2-CF2-CF3 | 182.67 ppm |
| Resin-CO-CF2-CF2-CF3 | 129.82 ppm |
| Resin-CO-CF2-CF2-CF3 | 118.47 ppm |
| Resin-CO-CF2-CF2-CF3 | Resin-CO-CF2-CF2-CF3 | Resin-CO-CF2-CF2-CF3 | |
|---|---|---|---|
| FBMPB a | −130.42 | −139.54 | −93.94 |
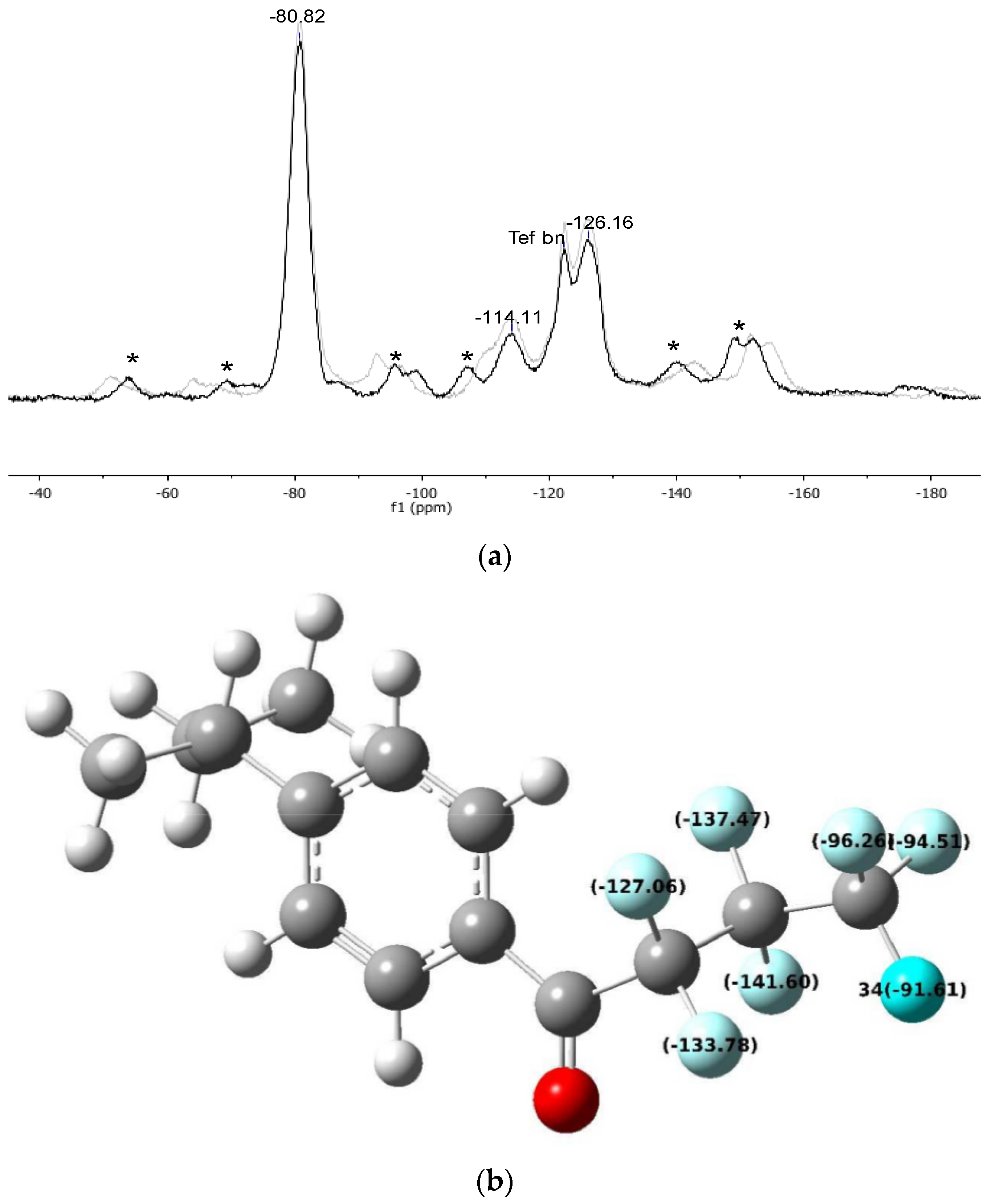
| Gel_C4F7O | −114.11 | −126.16 | −80.82 |
| pDVB_C4F7O | −117.26 | −125.36 | −80 |
| Δ (Gel_C4F7O) | 16.31 | 13.38 | 13.12 |
| Δ (pDVB_C4F7O) | 13.16 | 14.18 | 13.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalla Valle, C.; Sandri, F.; Zecca, M.; Rastrelli, F.; Campestrini, S.; Centomo, P. Synthesis of Ion-Exchange Catalysts by Introduction of Fluorinated Ponytails into Novel Mesoporous Polymers. Materials 2023, 16, 3808. https://doi.org/10.3390/ma16103808
Dalla Valle C, Sandri F, Zecca M, Rastrelli F, Campestrini S, Centomo P. Synthesis of Ion-Exchange Catalysts by Introduction of Fluorinated Ponytails into Novel Mesoporous Polymers. Materials. 2023; 16(10):3808. https://doi.org/10.3390/ma16103808
Chicago/Turabian StyleDalla Valle, Chiara, Francesco Sandri, Marco Zecca, Federico Rastrelli, Sandro Campestrini, and Paolo Centomo. 2023. "Synthesis of Ion-Exchange Catalysts by Introduction of Fluorinated Ponytails into Novel Mesoporous Polymers" Materials 16, no. 10: 3808. https://doi.org/10.3390/ma16103808
APA StyleDalla Valle, C., Sandri, F., Zecca, M., Rastrelli, F., Campestrini, S., & Centomo, P. (2023). Synthesis of Ion-Exchange Catalysts by Introduction of Fluorinated Ponytails into Novel Mesoporous Polymers. Materials, 16(10), 3808. https://doi.org/10.3390/ma16103808