
Surface Functionalization of Ureteral Stents-Based Polyurethane: Engineering Antibacterial Coatings

 ,
,  ,
,  , , ,
, , , 

Abstract
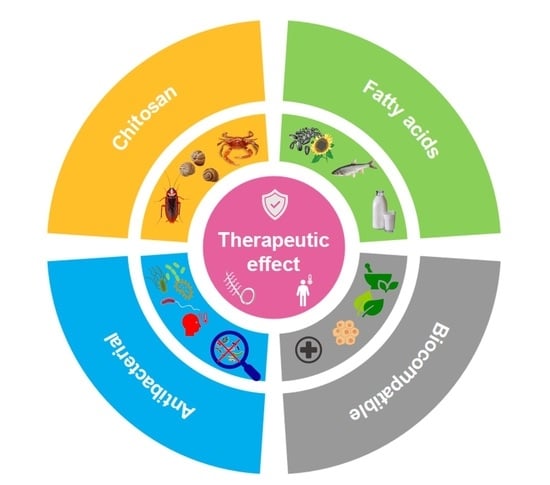
1. Introduction
2. Materials and Methods
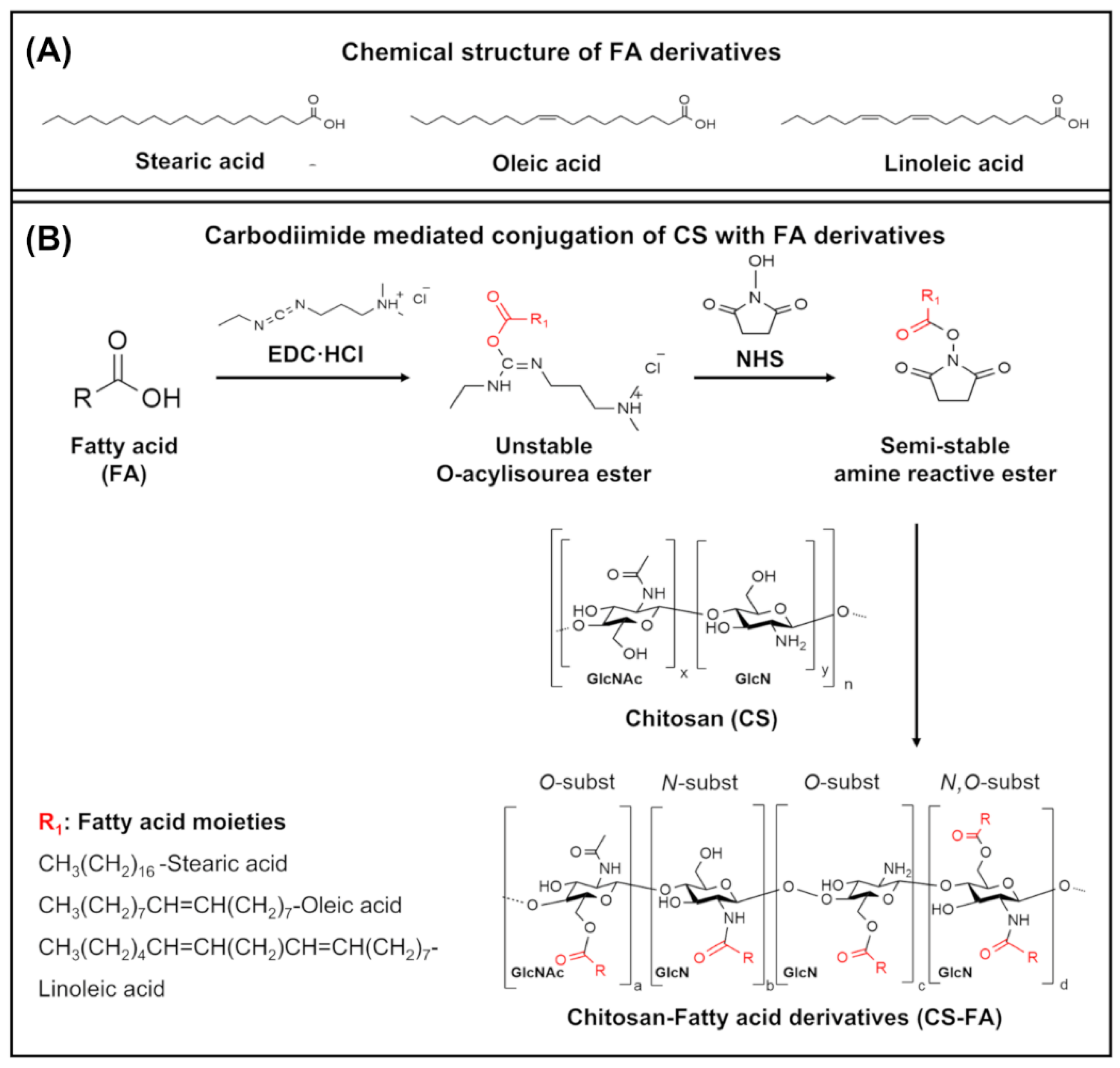
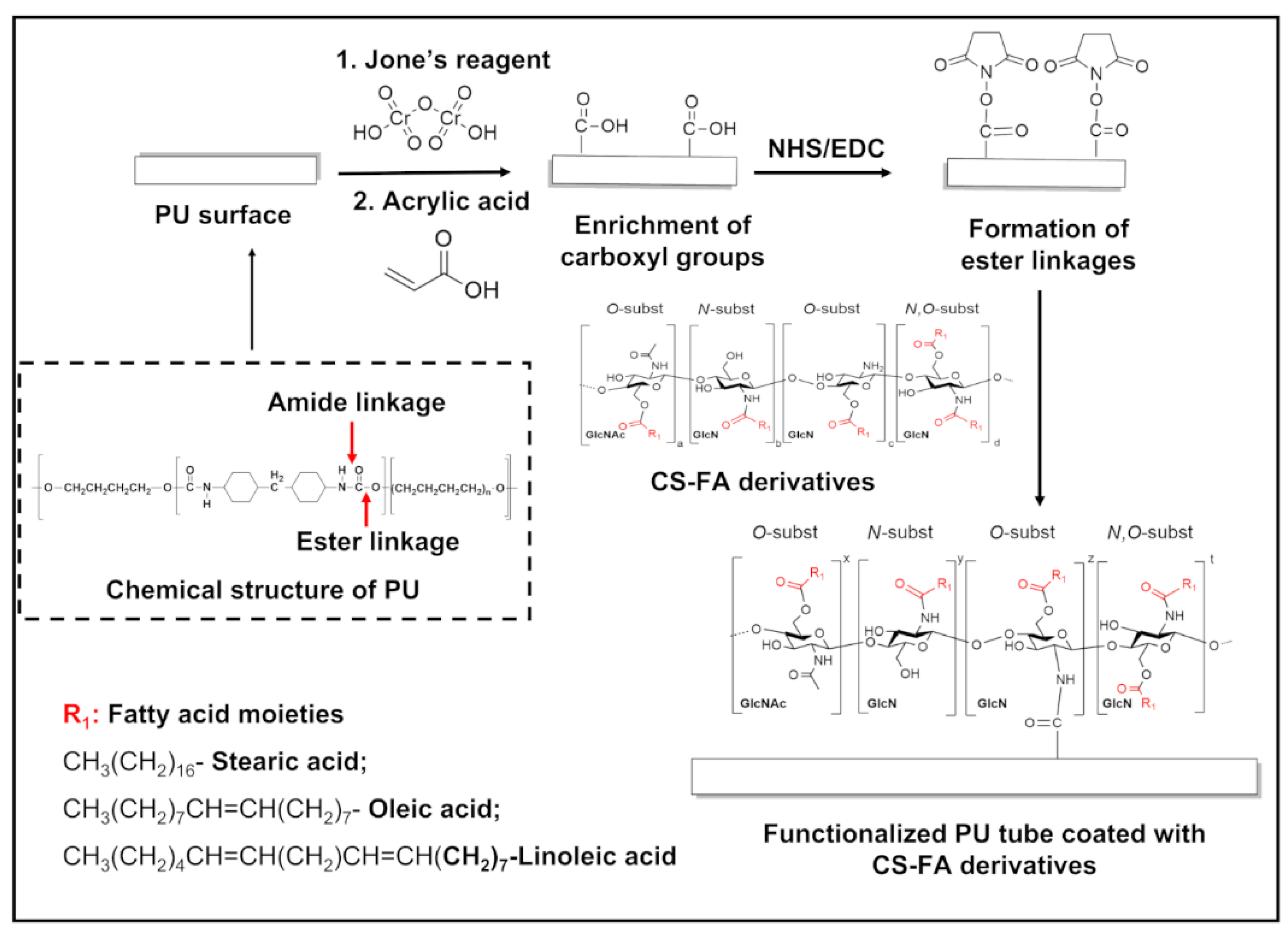
2.1. Grafting of FA Derivatives on CS
2.2. Fabrication of CS-FA-Coated PU Stents
2.3. Physicochemical Characterization
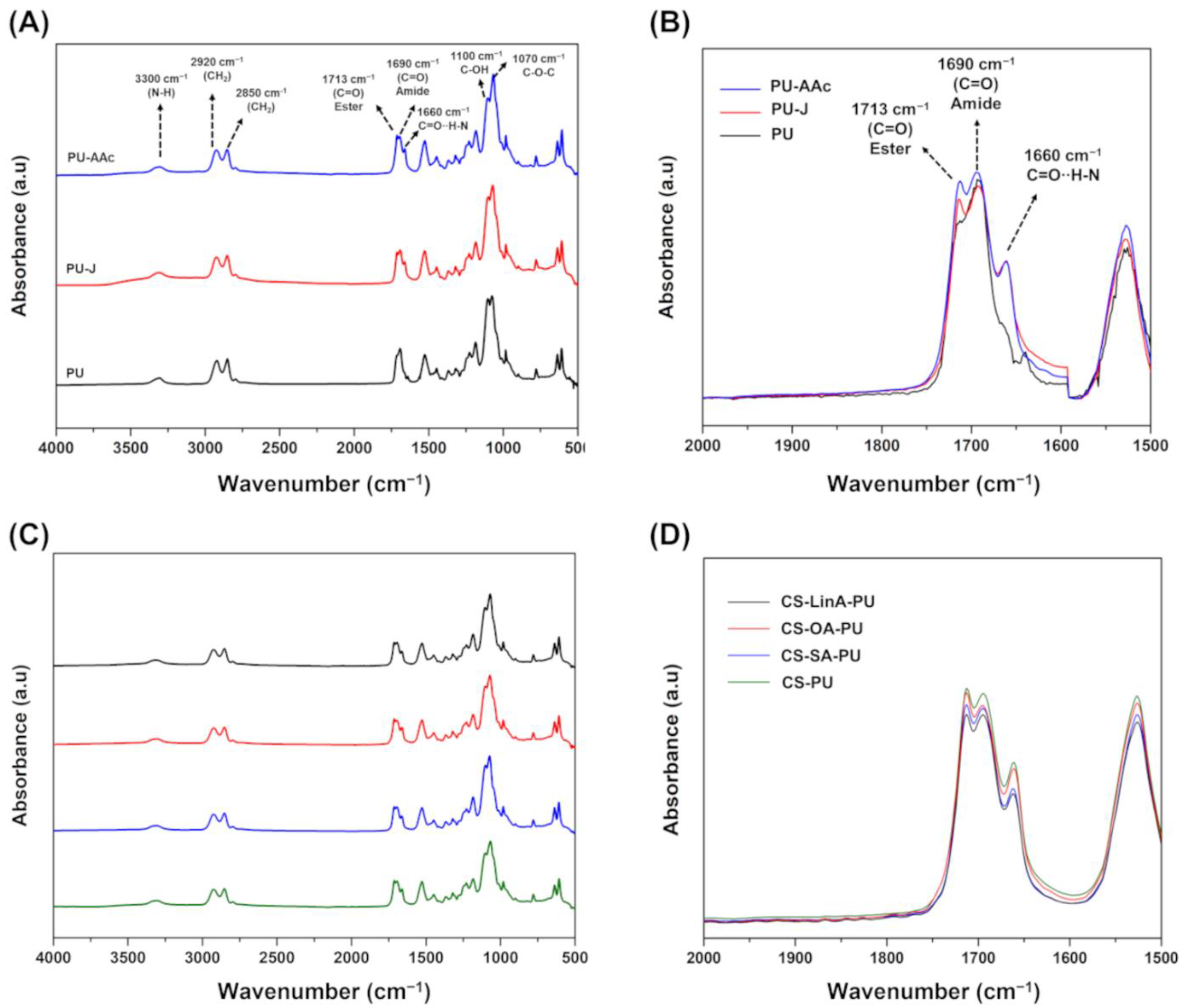
2.3.1. Fourier Transform Infra-Red in Attenuated Total Reflection Mode–(FTIR-ATR)
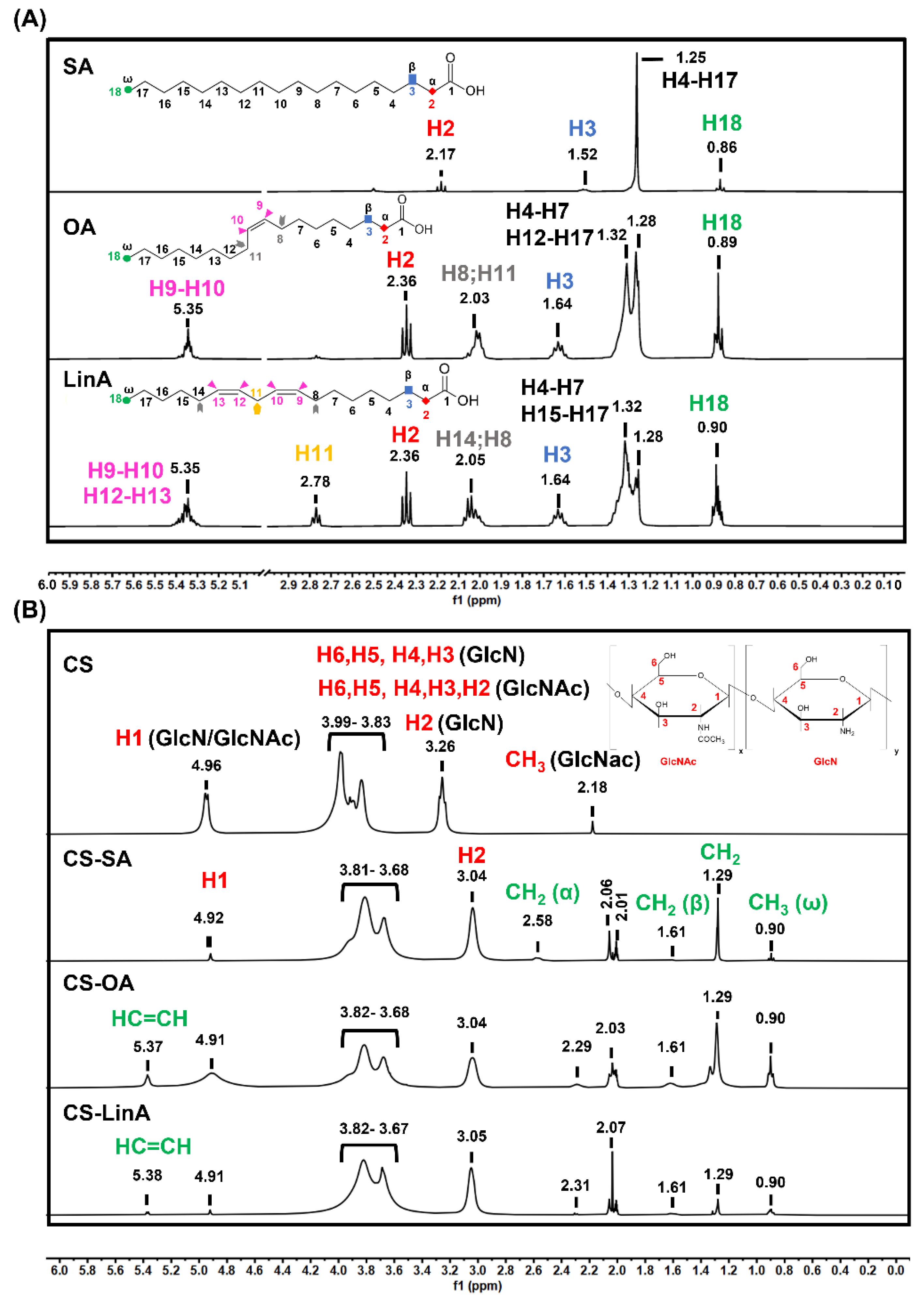
2.3.2. Proton Nuclear Magnetic Resonance (1H-NMR)
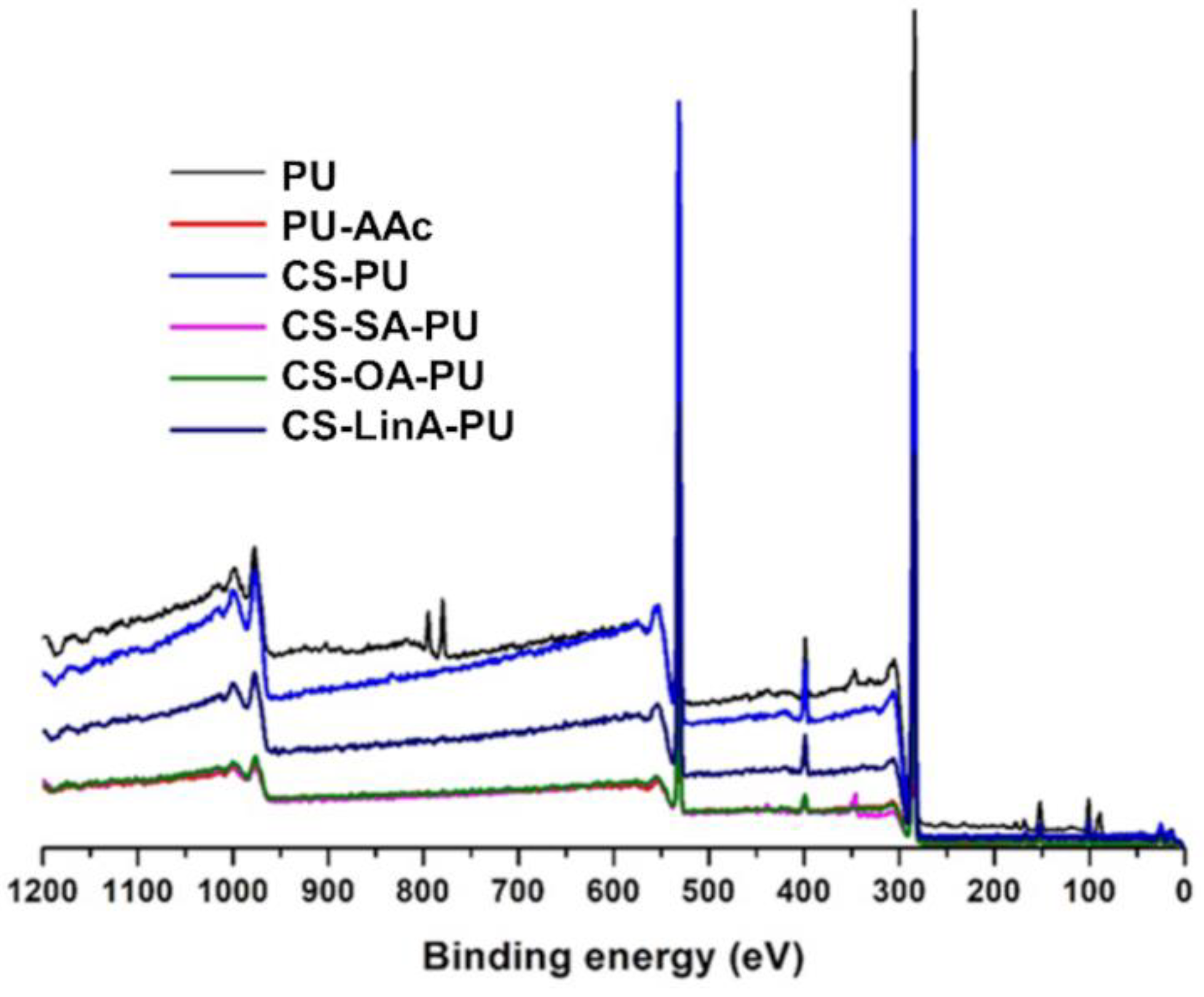
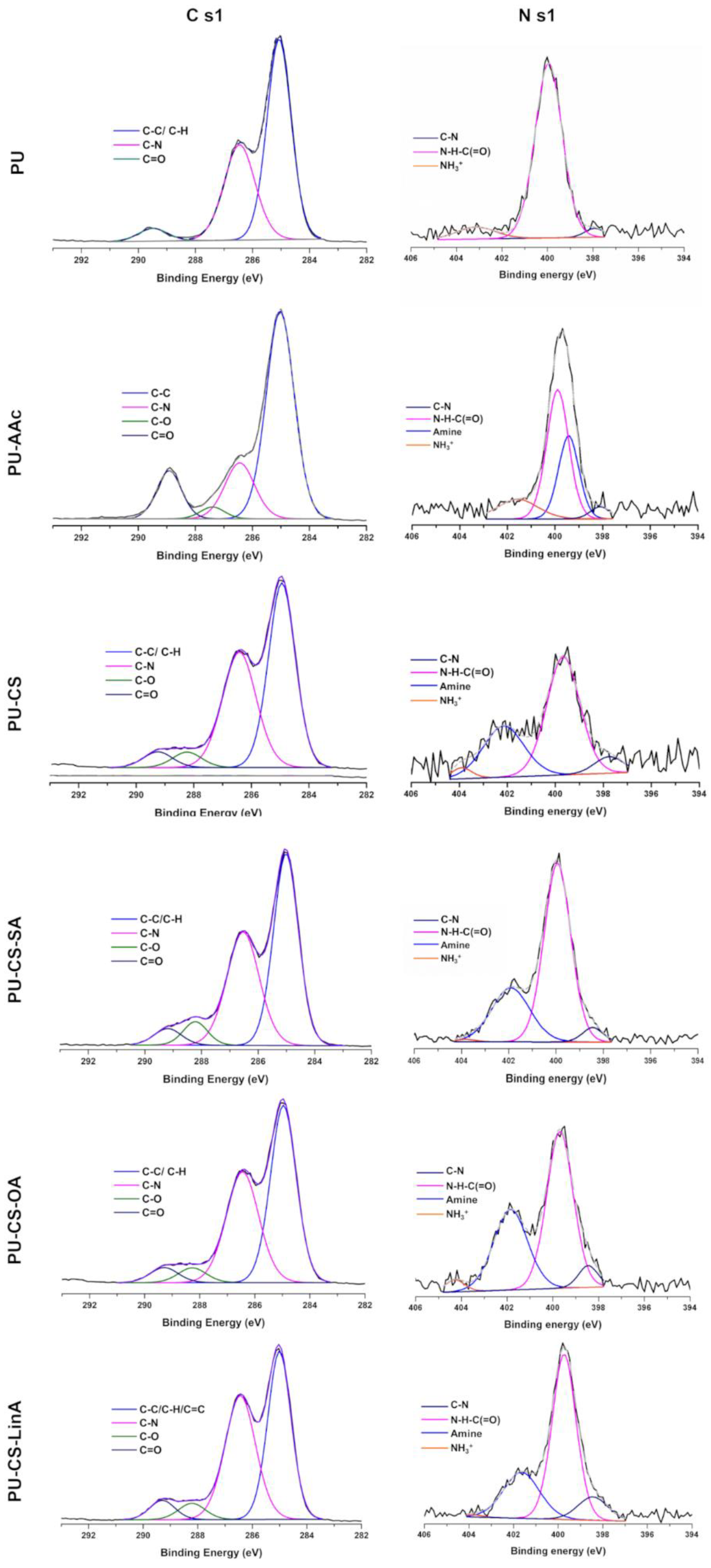
2.3.3. X-rays Photoelectron Spectroscopy (XPS)
2.3.4. Differential Scanning Calorimetry (DSC)
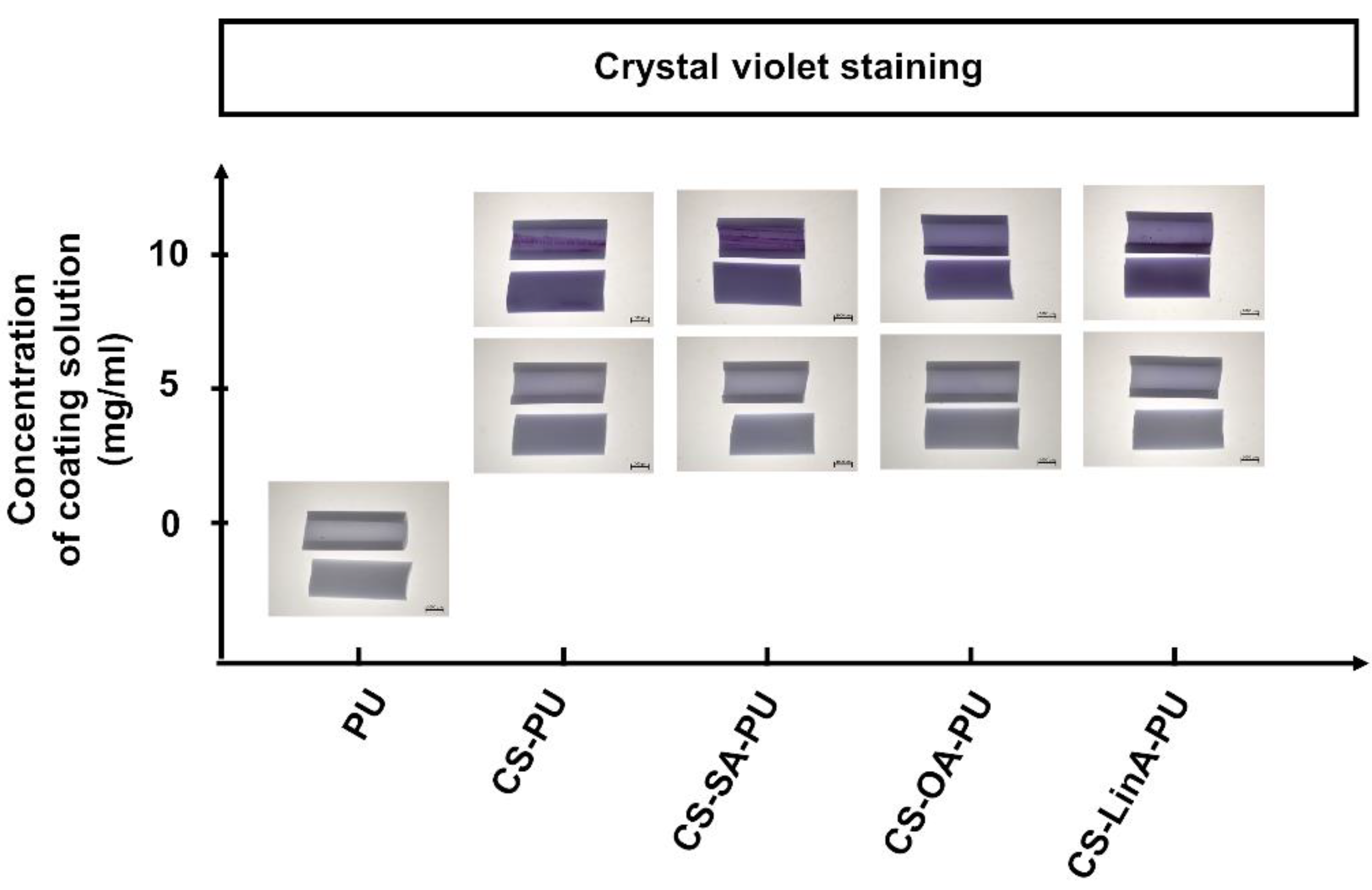
2.3.5. Crystal Violet Staining
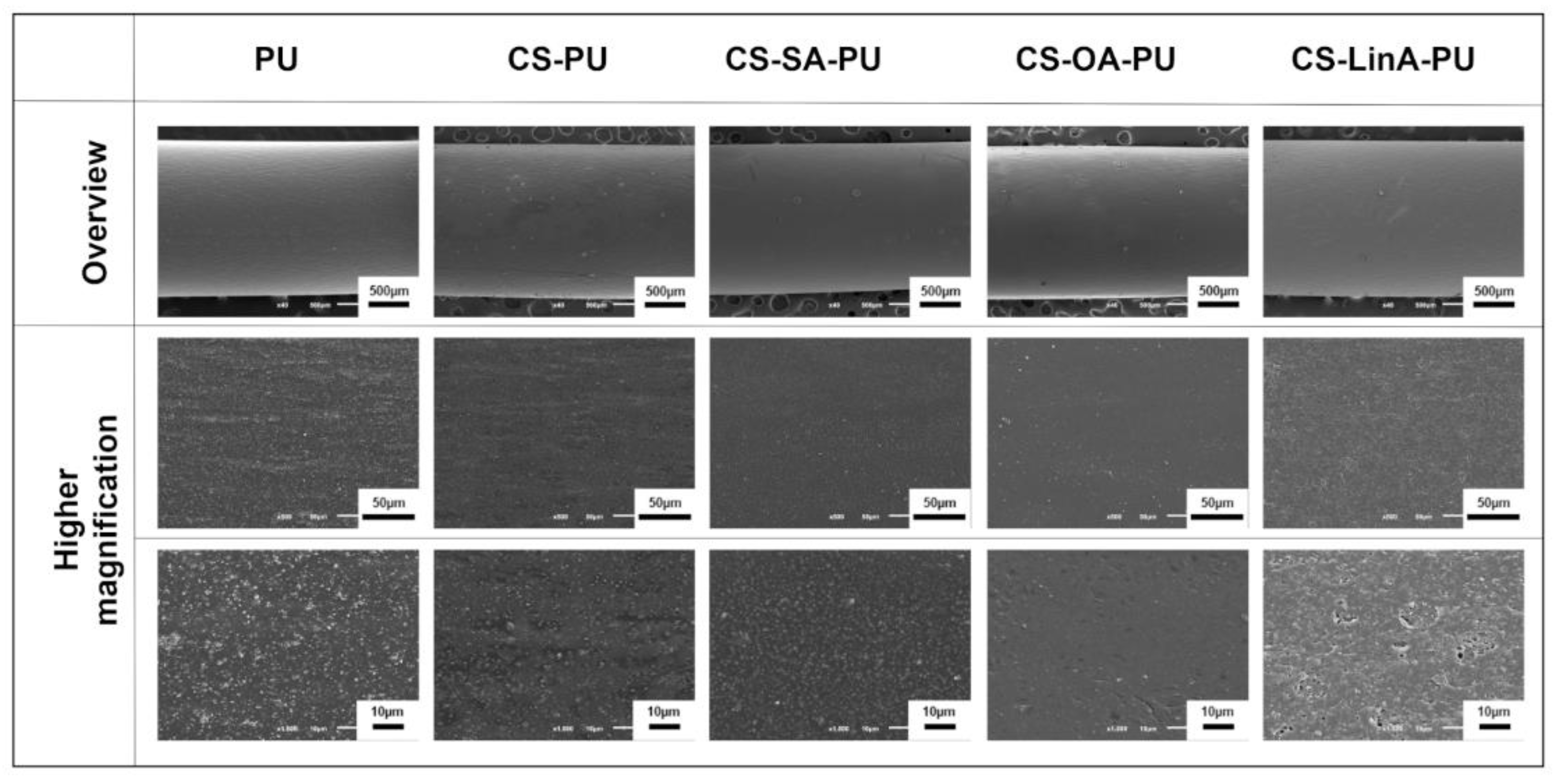
2.3.6. Scanning Electron Microscopy (SEM)
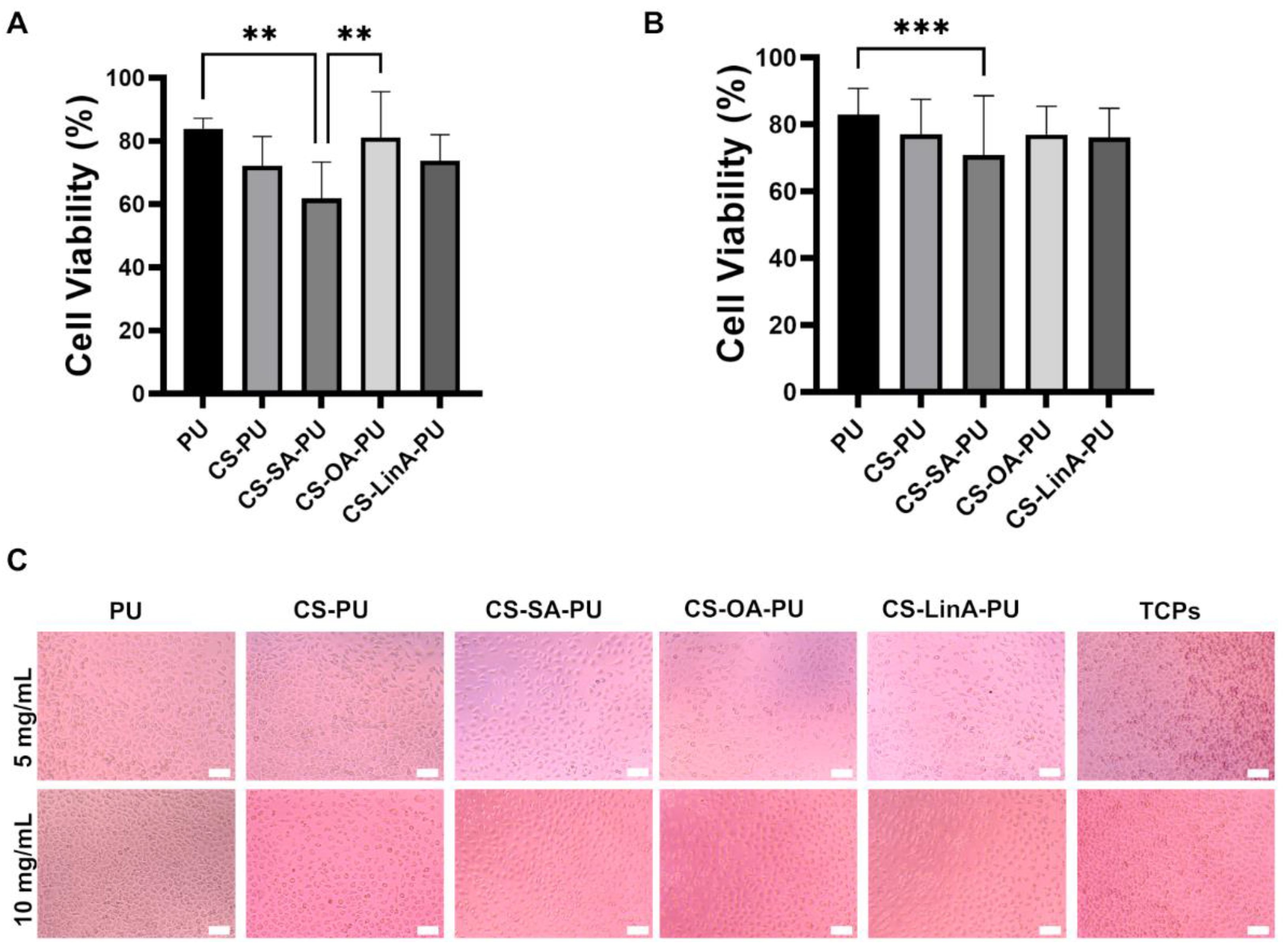
2.4. Cytotoxicity Studies against L929 Murine Fibroblasts
2.5. In Vitro Antibacterial Activity
2.5.1. Bacterial Attachment Assay
2.5.2. Live/Dead Fluorescence Assay
2.6. Statistical Analysis
3. Results and Discussion
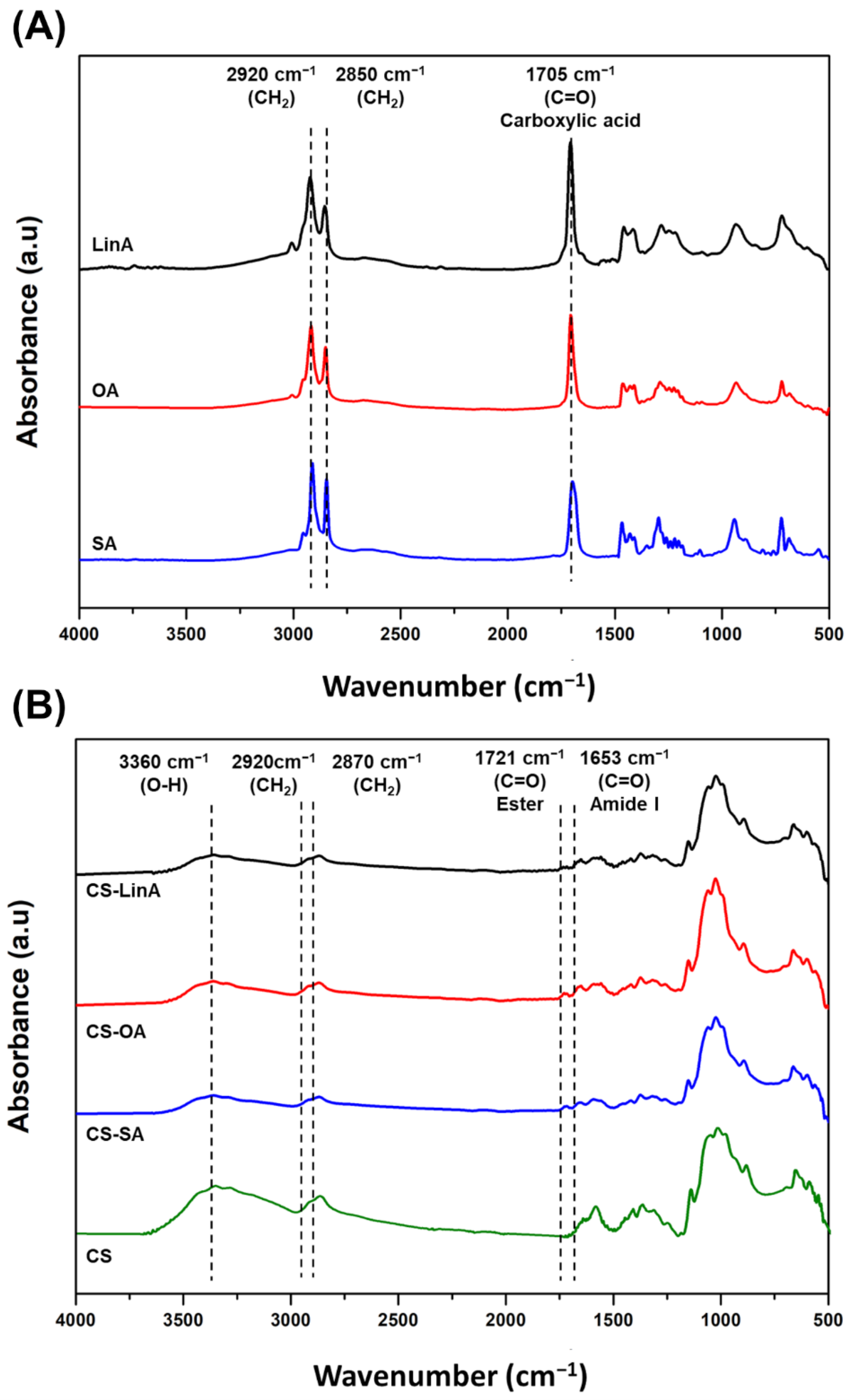
3.1. Structural Characterization of CS-FA Derivatives
3.2. Functionalized PU Stents with CS-FA Derivatives: Physicochemical Characterization
3.3. Biological Performance
3.3.1. Cytotoxicity Studies against L929 Murine Fibroblasts
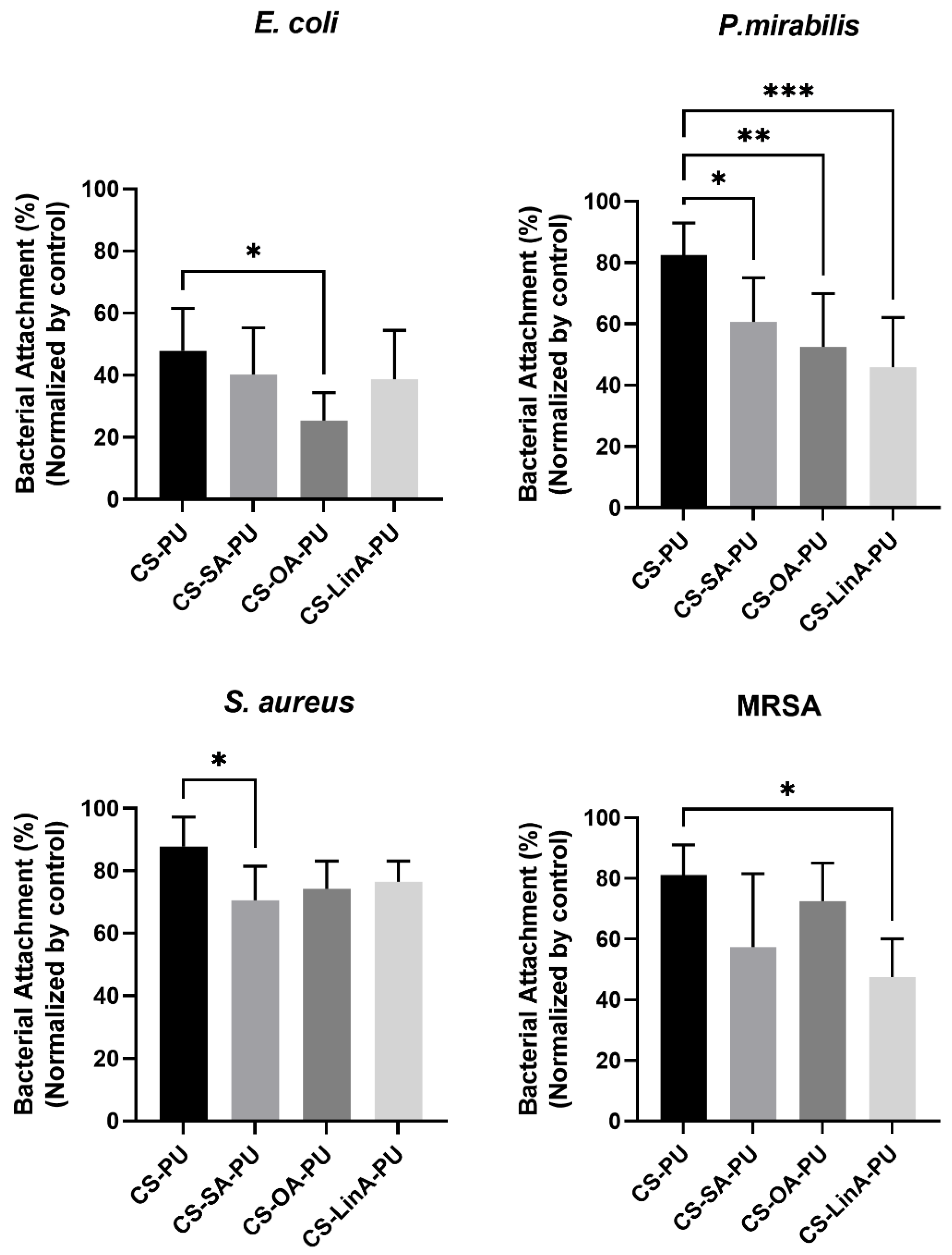
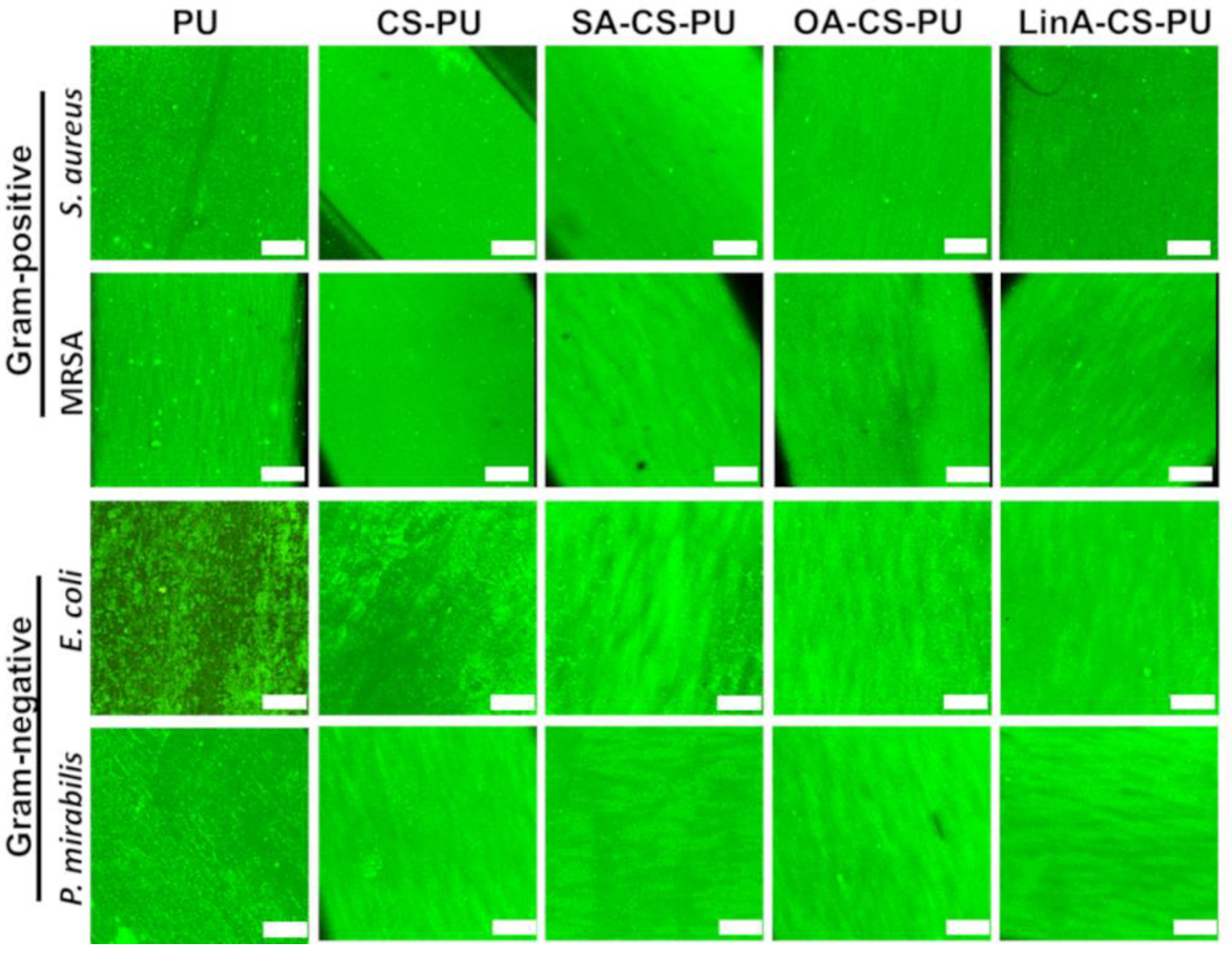
3.3.2. Antibacterial Assessments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andersen, M.J.; Flores-Mireles, A.L. Urinary Catheter Coating Modifications: The Race against Catheter-Associated Infections. Coatings 2020, 10, 23. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Chai, T.C. The Bladder Is Not Sterile: An Update on the Urinary Microbiome. Curr. Bladder. Dysfunct. Rep. 2019, 14, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Chua, R.R.Y.; Ho, B.; Tambyah, P.A.; Hadinoto, K.; Leong, S.S.J. Development of a Catheter Functionalized by a Polydopamine Peptide Coating with Antimicrobial and Antibiofilm Properties. Acta Biomater. 2015, 15, 127–138. [Google Scholar] [CrossRef]
- Monteiro, C.; Costa, F.; Pirttilä, A.M.; Tejesvi, M.V.; Martins, M.C.L. Prevention of Urinary Catheter-Associated Infections by Coating Antimicrobial Peptides from Crowberry Endophytes. Sci. Rep. 2019, 9, 10753. [Google Scholar] [CrossRef] [PubMed]
- Catheter-Associated Urinary Tract Infections (CAUTI)|HAI|CDC. Available online: https://www.cdc.gov/hai/ca_uti/uti.html (accessed on 12 July 2021).
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus Biofilm: An Emerging Battleground in Microbial Communities. Antimicrob. Resist. Infect. Control. 2019, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Singha, P.; Locklin, J.; Handa, H. A Review of the Recent Advances in Antimicrobial Coatings for Urinary Catheters. Acta Biomater. 2017, 50, 20–40. [Google Scholar] [CrossRef]
- Green, J.-B.D.; Fulghum, T.; Nordhaus, M.A. A Review of Immobilized Antimicrobial Agents and Methods for Testing. Biointerphases 2011, 6, MR13–MR28. [Google Scholar] [CrossRef]
- Green, J.-B.D.; Fulghum, T.; Nordhaus, M.A. Immobilized antimicrobial agents: A critical perspective. Chem. Rev. 2009, 109, 5437–5527. [Google Scholar]
- Zhu, S.; Lou, C.-W.; Zhang, S.; Wang, N.; Li, J.; Feng, Y.; He, R.; Xu, C.; Lin, J.-H. Clean Surface Additive Manufacturing of Aramid Paper-Based Electrically Heated Devices for Medical Therapy Application. Surf. Interfaces 2022, 29, 101689. [Google Scholar] [CrossRef]
- Saidin, S.; Jumat, M.A.; Mohd Amin, N.A.A.; Saleh Al-Hammadi, A.S. Organic and Inorganic Antibacterial Approaches in Combating Bacterial Infection for Biomedical Application. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 118, 111382. [Google Scholar] [CrossRef]
- Vladkova, T.G.; Staneva, A.D.; Gospodinova, D.N. Surface Engineered Biomaterials and Ureteral Stents Inhibiting Biofilm Formation and Encrustation. Surf. Coat. Technol. 2020, 404, 126424. [Google Scholar] [CrossRef]
- Guglielmi, P.; Pontecorvi, V.; Rotondi, G. Natural Compounds and Extracts as Novel Antimicrobial Agents. Expert Opin. Ther. Pat. 2020, 30, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Parfene, G.; Horincar, V.; Tyagi, A.K.; Malik, A.; Bahrim, G. Production of Medium Chain Saturated Fatty Acids with Enhanced Antimicrobial Activity from Crude Coconut Fat by Solid State Cultivation of Yarrowia Lipolytica. Food Chem. 2013, 136, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Arachidonic Acid and Other Unsaturated Fatty Acids and Some of Their Metabolites Function as Endogenous Antimicrobial Molecules: A Review. J. Adv. Res. 2018, 11, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Walvekar, P.; Gannimani, R.; Rambharose, S.; Mocktar, C.; Govender, T. Fatty Acid Conjugated Pyridinium Cationic Amphiphiles as Antibacterial Agents and Self-Assembling Nano Carriers. Chem. Phys. Lipids 2018, 214, 1–10. [Google Scholar] [CrossRef]
- Desbois, A.P.; Smith, V.J. Antibacterial Free Fatty Acids: Activities, Mechanisms of Action and Biotechnological Potential. Appl. Microbiol. Biotechnol. 2009, 85, 1629–1642. [Google Scholar] [CrossRef]
- Annu; Manzoor, K.; Ahmad, S.; Soundarajan, A.; Ikram, S.; Ahmed, S. Chapter 30-Chitosan Based Nanomaterials for Biomedical Applications. In Handbook of Nanomaterials for Industrial Applications; Mustansar Hussain, C., Ed.; Micro and Nano Technologies; Elsevier-New Jersey Institute of Technology: Newark, NJ, USA, 2018; pp. 543–562. ISBN 978-0-12-813351-4. [Google Scholar]
- Sharma, D.; Singh, J. Synthesis and Characterization of Fatty Acid Grafted Chitosan Polymer and Their Nanomicelles for Nonviral Gene Delivery Applications. Bioconjug. Chem. 2017, 28, 2772–2783. [Google Scholar] [CrossRef]
- Niemczyk, A.; Goszczyńska, A.; Gołda-Cępa, M.; Kotarba, A.; Sobolewski, P.; El Fray, M. Biofunctional Catheter Coatings Based on Chitosan-Fatty Acids Derivatives. Carbohydr. Polym. 2019, 225, 115263. [Google Scholar] [CrossRef]
- Desbois, A.P.; Lawlor, K.C. Antibacterial Activity of Long-Chain Polyunsaturated Fatty Acids against Propionibacterium Acnes and Staphylococcus aureus. Mar. Drugs 2013, 11, 4544–4557. [Google Scholar] [CrossRef]
- Huang, C.B.; George, B.; Ebersole, J.L. Antimicrobial Activity of N-6, n-7 and n-9 Fatty Acids and Their Esters for Oral Microorganisms. Arch. Oral Biol. 2010, 55, 555–560. [Google Scholar] [CrossRef]
- Huang, C.B.; Alimova, Y.; Myers, T.M.; Ebersole, J.L. Short- and Medium-Chain Fatty Acids Exhibit Antimicrobial Activity for Oral Microorganisms. Arch. Oral Biol. 2011, 56, 650–654. [Google Scholar] [CrossRef]
- Narayana, P.S.V.V.S.; Srihari, P.S.V.V. Biofilm Resistant Surfaces and Coatings on Implants: A Review. Mater. Today Proc. 2019, 18, 4847–4853. [Google Scholar] [CrossRef]
- Al-Aown, A.; Kyriazis, I.; Kallidonis, P.; Kraniotis, P.; Rigopoulos, C.; Karnabatidis, D.; Petsas, T.; Liatsikos, E. Ureteral Stents: New Ideas, New Designs. Ther. Adv. Urol. 2010, 2, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Mosayyebi, A.; Manes, C.; Carugo, D.; Somani, B.K. Advances in Ureteral Stent Design and Materials. Curr. Urol. Rep. 2018, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Barrera, C.; Herrera, A.P.; Rinaldi, C. Colloidal Dispersions of Monodisperse Magnetite Nanoparticles Modified with Poly(Ethylene Glycol). J. Colloid Interface Sci. 2009, 329, 107–113. [Google Scholar] [CrossRef]
- Yang, S.-H.; Lee, Y.-S.J.; Lin, F.-H.; Yang, J.-M.; Chen, K.-S. Chitosan/Poly(Vinyl Alcohol) Blending Hydrogel Coating Improves the Surface Characteristics of Segmented Polyurethane Urethral Catheters. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 83, 304–313. [Google Scholar] [CrossRef]
- Lin, W.-C.; Tseng, C.-H.; Yang, M.-C. In-Vitro Hemocompatibility Evaluation of a Thermoplastic Polyurethane Membrane with Surface-Immobilized Water-Soluble Chitosan and Heparin. Macromol. Biosci. 2005, 5, 1013–1021. [Google Scholar] [CrossRef]
- Le Tien, C.; Lacroix, M.; Ispas-Szabo, P.; Mateescu, M.-A. N-Acylated Chitosan: Hydrophobic Matrices for Controlled Drug Release. J. Control. Release 2003, 93, 1–13. [Google Scholar] [CrossRef]
- Layek, B.; Haldar, M.K.; Sharma, G.; Lipp, L.; Mallik, S.; Singh, J. Hexanoic Acid and Polyethylene Glycol Double Grafted Amphiphilic Chitosan for Enhanced Gene Delivery: Influence of Hydrophobic and Hydrophilic Substitution Degree. Mol. Pharm. 2014, 11, 982–994. [Google Scholar] [CrossRef]
- Namazi, H.; Fathi, F.; Dadkhah, A. Hydrophobically Modified Starch Using Long-Chain Fatty Acids for Preparation of Nanosized Starch Particles. Sci. Iran. 2011, 18, 439–445. [Google Scholar] [CrossRef]
- Pettit, R.K.; Weber, C.A.; Kean, M.J.; Hoffmann, H.; Pettit, G.R.; Tan, R.; Franks, K.S.; Horton, M.L. Microplate Alamar Blue Assay for Staphylococcus epidermidis Biofilm Susceptibility Testing. Antimicrob. Agents Chemother. 2005, 49, 2612–2617. [Google Scholar] [CrossRef] [PubMed]
- Piegat, A.; Goszczyńska, A.; Idzik, T.; Niemczyk, A. The Importance of Reaction Conditions on the Chemical Structure of N,O-Acylated Chitosan Derivatives. Molecules 2019, 24, 3047. [Google Scholar] [CrossRef] [PubMed]
- Hirai, A.; Odani, H.; Nakajima, A. Determination of Degree of Deacetylation of Chitosan by 1H NMR Spectroscopy. Polym. Bull. 1991, 26, 87–94. [Google Scholar] [CrossRef]
- Silva, D.S.; Almeida, A.; Prezotti, F.; Cury, B.; Campana-Filho, S.P.; Sarmento, B. Synthesis and Characterization of 3,6-O,O′-Dimyristoyl Chitosan Micelles for Oral Delivery of Paclitaxel. Colloids Surf. B Biointerfaces 2017, 152, 220–228. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, B.; Xia, Y. Synthesis and Characterization of O-Acetyl-Chitosan Acetic Ester. Int. J. Polym. Sci. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z.; Zhao, K. Chitosan Derivatives and Their Application in Biomedicine. Int. J. Mol. Sci. 2020, 21, 487. [Google Scholar] [CrossRef]
- Shanmugasundaram, N.; Ravichandran, P.; Neelakanta Reddy, P.; Ramamurty, N.; Pal, S.; Panduranga Rao, K. Collagen–Chitosan Polymeric Scaffolds for the in Vitro Culture of Human Epidermoid Carcinoma Cells. Biomaterials 2001, 22, 1943–1951. [Google Scholar] [CrossRef]
- Guinesi, L.S.; Cavalheiro, É.T.G. The Use of DSC Curves to Determine the Acetylation Degree of Chitin/Chitosan Samples. Thermochim. Acta 2006, 444, 128–133. [Google Scholar] [CrossRef]
- Hao, G.; Hu, Y.; Shi, L.; Chen, J.; Cui, A.; Weng, W.; Osako, K. Physicochemical Characteristics of Chitosan from Swimming Crab (Portunus Trituberculatus) Shells Prepared by Subcritical Water Pretreatment. Sci. Rep. 2021, 11, 1646. [Google Scholar] [CrossRef]
- Zhang, C.; Ping, Q.; Ding, Y.; Cheng, Y.; Shen, J. Synthesis, Characterization, and Microsphere Formation of Galactosylated Chitosan. J. Appl. Polym. Sci. 2004, 91, 659–665. [Google Scholar] [CrossRef]
- Harry-O’kuru, R.E.; Mohamed, A.; Xu, J.; Sharma, B.K. Synthesis and Characterization of Corn Oil Polyhydroxy Fatty Acids Designed as Additive Agent for Many Applications. J. Am. Oil Chem. Soc. 2011, 88, 1211–1221. [Google Scholar] [CrossRef]
- Habibizadeh, M.; Rostamizadeh, K.; Dalali, N.; Ramazani, A. Preparation and Characterization of PEGylated Multiwall Carbon Nanotubes as Covalently Conjugated and Non-Covalent Drug Carrier: A Comparative Study. Mater. Sci. Eng. C 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vithalkar, S.H.; Jugade, R.M. Adsorptive Removal of Crystal Violet from Aqueous Solution by Cross-Linked Chitosan Coated Bentonite. Mater. Today Proc. 2020, 29, 1025–1032. [Google Scholar] [CrossRef]
- Boffito, M.; Di Meglio, F.; Mozetic, P.; Giannitelli, S.M.; Carmagnola, I.; Castaldo, C.; Nurzynska, D.; Sacco, A.M.; Miraglia, R.; Montagnani, S.; et al. Surface Functionalization of Polyurethane Scaffolds Mimicking the Myocardial Microenvironment to Support Cardiac Primitive Cells. PLoS ONE 2018, 13, e0199896. [Google Scholar] [CrossRef]
- Yuan, H.; Xue, J.; Qian, B.; Chen, H.; Zhu, Y.; Lan, M. Preparation and Antifouling Property of Polyurethane Film Modified by Chondroitin Sulfate. Appl. Surf. Sci. 2017, 394, 403–413. [Google Scholar] [CrossRef]
- Mishra, A.K.; Chattopadhyay, D.K.; Sreedhar, B.; Raju, K.V.S.N. FT-IR and XPS Studies of Polyurethane-Urea-Imide Coatings. Prog. Org. Coat. 2006, 55, 231–243. [Google Scholar] [CrossRef]
- Niimura, N.; Iijima, Y.; Miyakoshi, T. Hardening Process and Surface Structure of Lacquer Films Studied by X-ray Photoelectron Spectroscopy. Surf. Interface Anal. 1996, 24, 237–242. [Google Scholar] [CrossRef]
- Tang, C.Y.; Kwon, Y.-N.; Leckie, J.O. Probing the Nano- and Micro-Scales of Reverse Osmosis Membranes—A Comprehensive Characterization of Physiochemical Properties of Uncoated and Coated Membranes by XPS, TEM, ATR-FTIR, and Streaming Potential Measurements. J. Membr. Sci. 2007, 287, 146–156. [Google Scholar] [CrossRef]
- Vaz, J.M.; Taketa, T.B.; Hernandez-Montelongo, J.; Chevallier, P.; Cotta, M.A.; Mantovani, D.; Beppu, M.M. Antibacterial Properties of Chitosan-Based Coatings Are Affected by Spacer-Length and Molecular Weight. Appl. Surf. Sci. 2018, 445, 478–487. [Google Scholar] [CrossRef]
- Wu, J.; Peng, C.; Xiao, H.; Bo, S.; Qiu, L.; Zhen, Z.; Liu, X. Donor Modification of Nonlinear Optical Chromophores: Synthesis, Characterization, and Fine-Tuning of Chromophores’ Mobility and Steric Hindrance to Achieve Ultra Large Electro-Optic Coefficients in Guest–Host Electro-Optic Materials. Dye. Pigment. 2014, 104, 15–23. [Google Scholar] [CrossRef]
- Shu, J.; Li, C.; Liu, M.; Liu, H.; Feng, X.; Tan, W.; Liu, F. Role of Counteranions in Sol–Gel-Derived Alkoxyl-Functionalized Ionic-Liquid-Based Organic–Inorganic Hybrid Coatings for SPME. Chromatographia 2012, 75, 1421–1433. [Google Scholar] [CrossRef]
- Zheng, W.T.; Cao, P.J.; Li, J.J.; Wang, X.; Jin, Z.S. Chemical Bonding of CNx Films Synthesized by Nitrogen Ion Implantation into Diamond and Graphite. Surf. Coat. Technol. 2003, 173, 213–218. [Google Scholar] [CrossRef]
- Siew, H.L.; Qiao, M.H.; Chew, C.H.; Mok, K.F.; Chan, L.; Xu, G.Q. Adsorption and Reaction of NH3 on Ti/Si(1 0 0). Appl. Surf. Sci. 2001, 173, 95–102. [Google Scholar] [CrossRef]
- Hussein, A.; Sarkar, S.; Lee, K.; Kim, B. Cryogenic Fracture Behavior of Epoxy Reinforced by a Novel Graphene Oxide/Poly(p-Phenylenediamine) Hybrid. Compos. Part B Eng. 2017, 129, 133–142. [Google Scholar] [CrossRef]
- Huang, H.-C.; Ye, D.-Q.; Huang, B.-C. Nitrogen Plasma Modification of Viscose-Based Activated Carbon Fibers. Surf. Coat. Technol. 2007, 201, 9533–9540. [Google Scholar] [CrossRef]
- Graf, N.; Yegen, E.; Gross, T.; Lippitz, A.; Weigel, W.; Krakert, S.; Terfort, A.; Unger, W.E.S. XPS and NEXAFS Studies of Aliphatic and Aromatic Amine Species on Functionalized Surfaces. Surf. Sci. 2009, 603, 2849–2860. [Google Scholar] [CrossRef]
- Popat, A.; Liu, J.; Lu, G.Q.; Qiao, S.Z. A PH-Responsive Drug Delivery System Based on Chitosan Coated Mesoporous Silica Nanoparticles. J. Mater. Chem. 2012, 22, 11173. [Google Scholar] [CrossRef]
- Bahrami, N.; Khorasani, S.N.; Mahdavi, H.; Ghiaci, M.; Mokhtari, R. Low-Pressure Plasma Surface Modification of Polyurethane Films with Chitosan and Collagen Biomolecules. J. Appl. Polym. Sci. 2019, 136, 47567. [Google Scholar] [CrossRef]
- Audifred-Aguilar, J.C.; Pino-Ramos, V.H.; Bucio, E. Synthesis and Characterization of Hydrophilically Modified Tecoflex® Polyurethane Catheters for Drug Delivery. Mater. Today Commun. 2021, 26, 101894. [Google Scholar] [CrossRef]
- Yilgör, E.; Burgaz, E.; Yurtsever, E.; Yilgör, İ. Comparison of Hydrogen Bonding in Polydimethylsiloxane and Polyether Based Urethane and Urea Copolymers. Polymer 2000, 41, 849–857. [Google Scholar] [CrossRef]
- Queiroz, D.P.; Norberta de Pinho, M. Structural Characteristics and Gas Permeation Properties of Polydimethylsiloxane/Poly(Propylene Oxide) Urethane/Urea Bi-Soft Segment Membranes. Polymer 2005, 46, 2346–2353. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, N.; Feng, M. Polyurethane Foams Derived from Liquefied Mountain Pine Beetle-Infested Barks. J. Appl. Polym. Sci. 2012, 123, 2849–2858. [Google Scholar] [CrossRef]
- Machado Centenaro, G.S.N.; Facin, B.R.; Valério, A.; de Souza, A.A.U.; da Silva, A.; de Oliveira, J.V.; de Oliveira, D. Application of Polyurethane Foam Chitosan-Coated as a Low-Cost Adsorbent in the Effluent Treatment. J. Water Process Eng. 2017, 20, 201–206. [Google Scholar] [CrossRef]
- Jiménez, A.; Fabra, M.J.; Talens, P.; Chiralt, A. Effect of Lipid Self-Association on the Microstructure and Physical Properties of Hydroxypropyl-Methylcellulose Edible Films Containing Fatty Acids. Carbohydr. Polym. 2010, 82, 585–593. [Google Scholar] [CrossRef]
- Bodas, D.; Khan-Malek, C. Hydrophilization and Hydrophobic Recovery of PDMS by Oxygen Plasma and Chemical Treatment—An SEM Investigation. Sens. Actuators B Chem. 2007, 123, 368–373. [Google Scholar] [CrossRef]
- Braem, A.; Van Mellaert, L.; Mattheys, T.; Hofmans, D.; De Waelheyns, E.; Geris, L.; Anné, J.; Schrooten, J.; Vleugels, J. Staphylococcal Biofilm Growth on Smooth and Porous Titanium Coatings for Biomedical Applications. J. Biomed. Mater. Res. Part A 2014, 102, 215–224. [Google Scholar] [CrossRef]
- Gedefie, A.; Demsis, W.; Ashagrie, M.; Kassa, Y.; Tesfaye, M.; Tilahun, M.; Bisetegn, H.; Sahle, Z. Acinetobacter baumannii Biofilm Formation and Its Role in Disease Pathogenesis: A Review. Infect. Drug Resist. 2021, 14, 3711–3719. [Google Scholar] [CrossRef]
- Lucarini, S.; Fagioli, L.; Campana, R.; Cole, H.; Duranti, A.; Baffone, W.; Vllasaliu, D.; Casettari, L. Unsaturated Fatty Acids Lactose Esters: Cytotoxicity, Permeability Enhancement and Antimicrobial Activity. Eur. J. Pharm. Biopharm. 2016, 107, 88–96. [Google Scholar] [CrossRef]
- Kara, F.; Aksoy, E.A.; Yuksekdag, Z.; Hasirci, N.; Aksoy, S. Synthesis and Surface Modification of Polyurethanes with Chitosan for Antibacterial Properties. Carbohydr. Polym. 2014, 112, 39–47. [Google Scholar] [CrossRef]
- Kizaloglu, A.; Kilicay, E.; Karahaliloglu, Z.; Hazer, B.; Denkbas, E.B. The Preparation of Chitosan Membrane Improved with Nanoparticles Based on Unsaturated Fatty Acid for Using in Cancer-Related Infections. J. Bioact. Compat. Polym. 2020, 35, 328–350. [Google Scholar] [CrossRef]
- Levison, M.E. Effect of Colon Flora and Short-Chain Fatty Acids on Growth In Vitro of Pseudomonas aeruginosa and Enterobacteriaceae. Infect. Immun. 1973, 8, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Vo, D.-T.; Lee, C.-K. Cells Capture and Antimicrobial Effect of Hydrophobically Modified Chitosan Coating on Escherichia coli. Carbohydr. Polym. 2017, 164, 109–117. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C 1s Assignment (%) | ||||||
| Components | PU | PU-AAc | CS-PU | CS-SA-PU | CS-OA-PU | CS-LinA-PU |
| C-C/C-H/C=C | 49.05 ± 1.31 (285.1 eV) | 45.63 ± 3.81 (284.9 eV) | 37.73 ± 1.64 (285.0 eV) | 39.11 ± 3.16 (284.9 eV) | 31.31 ± 5.13 (284.9 eV) | 34.69 ± 1.60 (284.9 eV) |
| C-N | 27.13 ± 1.20 (286.4 eV) | 21.54± 7.22 (286.4 eV) | 29.51 ± 6.94 (286.4 eV) | 25.85 ± 1.26 (286.4 eV) | 27.33 ± 4.02 (286.4 eV) | 30.02 ± 1.12 (286.4 eV) |
| C=O | 3.53 ± 1.13 (289.5 eV) | 7.47± 2.77 (289.0 eV) | 6.49 ± 4.30 (289.0 eV) | 3.05 ± 0.58 (289.2 eV) | 4.82 ± 0.55 (289.2 eV) | 3.37 ± 0.62 (289.2 eV) |
| C-O | - | 2.47± 0.08 (287.4 eV) | 3.22 ± 0.10 (288.2 eV) | 3.43 ± 0.88 (288.2 eV) | 5.66 ± 1.85 (288.2 eV) | 3.65 ± 0.42 (288.2 eV) |
| N 1s Assignment (%) | ||||||
| C–N | 0.13 ± 0.02 (398.5 eV) | 0.13 ± 0.01 (398.5 eV) | 0.17 ± 0.04 (398.5 eV) | 0.15 ± 0.01 (398.5 eV) | 0.2 7± 0.01 (398.5 eV) | 0.45 ± 0.04 (398.5 eV) |
| Urethane -N-C(=O) | 2.86 ± 0.05 (400.2 eV) | 1.05 ± 0.55 (399.7 eV) | 0.86 ± 0.36 (399.7 eV) | 1.16 ± 0.12 (399.7 eV) | 1.94 ± 0.31 (399.7 eV) | 0.97 ± 0.09 (399.7 eV) |
| NH3+ | 0.32 ± 0.02 (402.6 eV) | 0.25 ± 0.02 (403.8 eV) | 0.05 ± 0.01 (403.8 eV) | 0.11 ± 0.09 (403.8 eV) | 0.15 ± 0.01 (402.6 eV) | 0.03 ± 0.01 (403.8 eV) |
| Amine | - | 0.94 ± 0.01 (399.5 eV) | 1.66 ± 0.72 (401.8 eV) | 2.30 ± 0.44 (401.8 eV) | 2.05 ± 0.77 (401.8 eV) | 2.74 ± 0.16 (401.8 eV) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ecevit, K.; Silva, E.; Rodrigues, L.C.; Aroso, I.; Barros, A.A.; Silva, J.M.; Reis, R.L. Surface Functionalization of Ureteral Stents-Based Polyurethane: Engineering Antibacterial Coatings. Materials 2022, 15, 1676. https://doi.org/10.3390/ma15051676
Ecevit K, Silva E, Rodrigues LC, Aroso I, Barros AA, Silva JM, Reis RL. Surface Functionalization of Ureteral Stents-Based Polyurethane: Engineering Antibacterial Coatings. Materials. 2022; 15(5):1676. https://doi.org/10.3390/ma15051676
Chicago/Turabian StyleEcevit, Kardelen, Eduardo Silva, Luísa C. Rodrigues, Ivo Aroso, Alexandre A. Barros, Joana M. Silva, and Rui L. Reis. 2022. "Surface Functionalization of Ureteral Stents-Based Polyurethane: Engineering Antibacterial Coatings" Materials 15, no. 5: 1676. https://doi.org/10.3390/ma15051676
APA StyleEcevit, K., Silva, E., Rodrigues, L. C., Aroso, I., Barros, A. A., Silva, J. M., & Reis, R. L. (2022). Surface Functionalization of Ureteral Stents-Based Polyurethane: Engineering Antibacterial Coatings. Materials, 15(5), 1676. https://doi.org/10.3390/ma15051676