
First-Principles Study of High-Pressure Phase Stability and Electron Properties of Be-P Compounds

Abstract
1. Introduction
2. Computational Details
3. Results
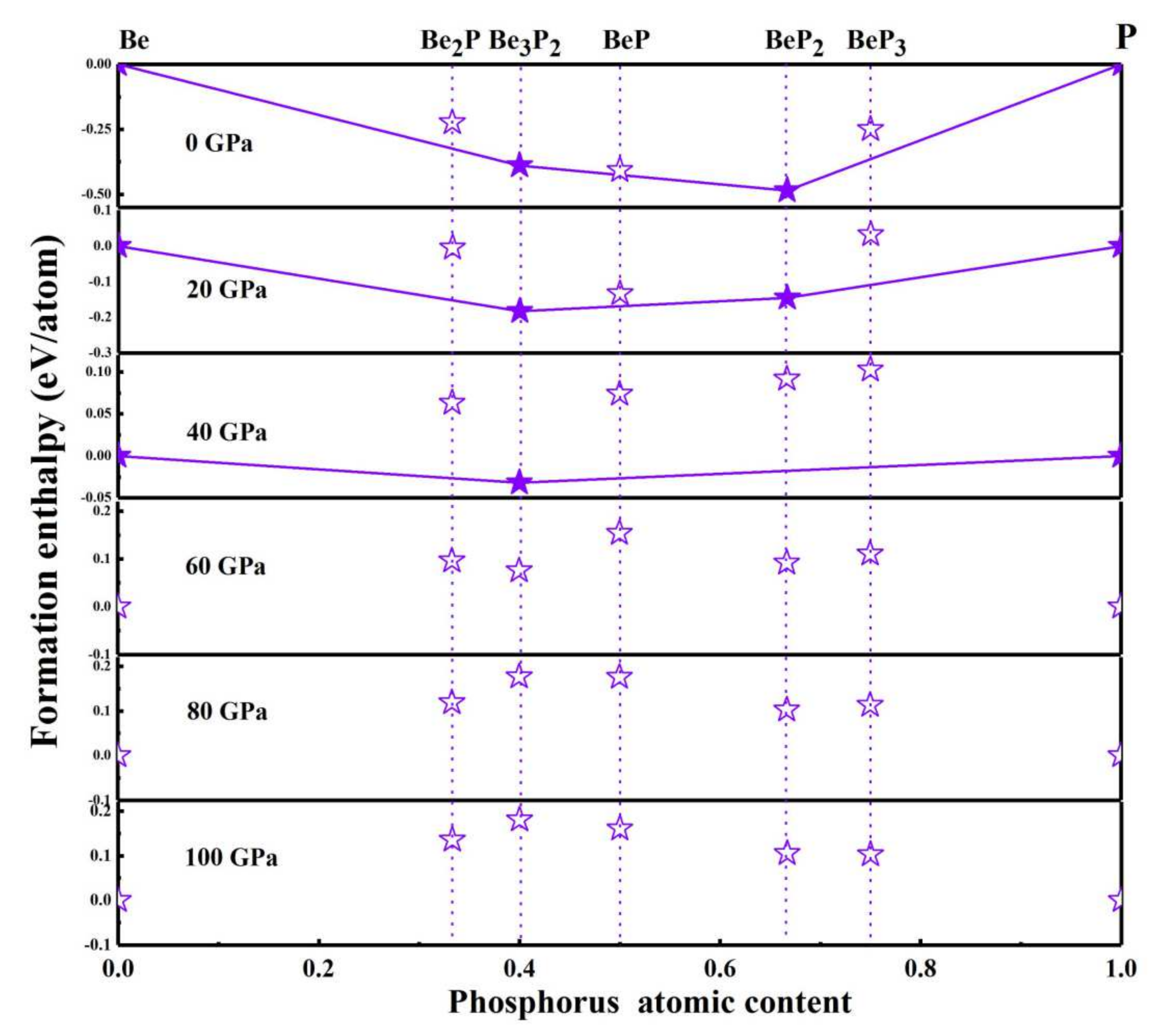
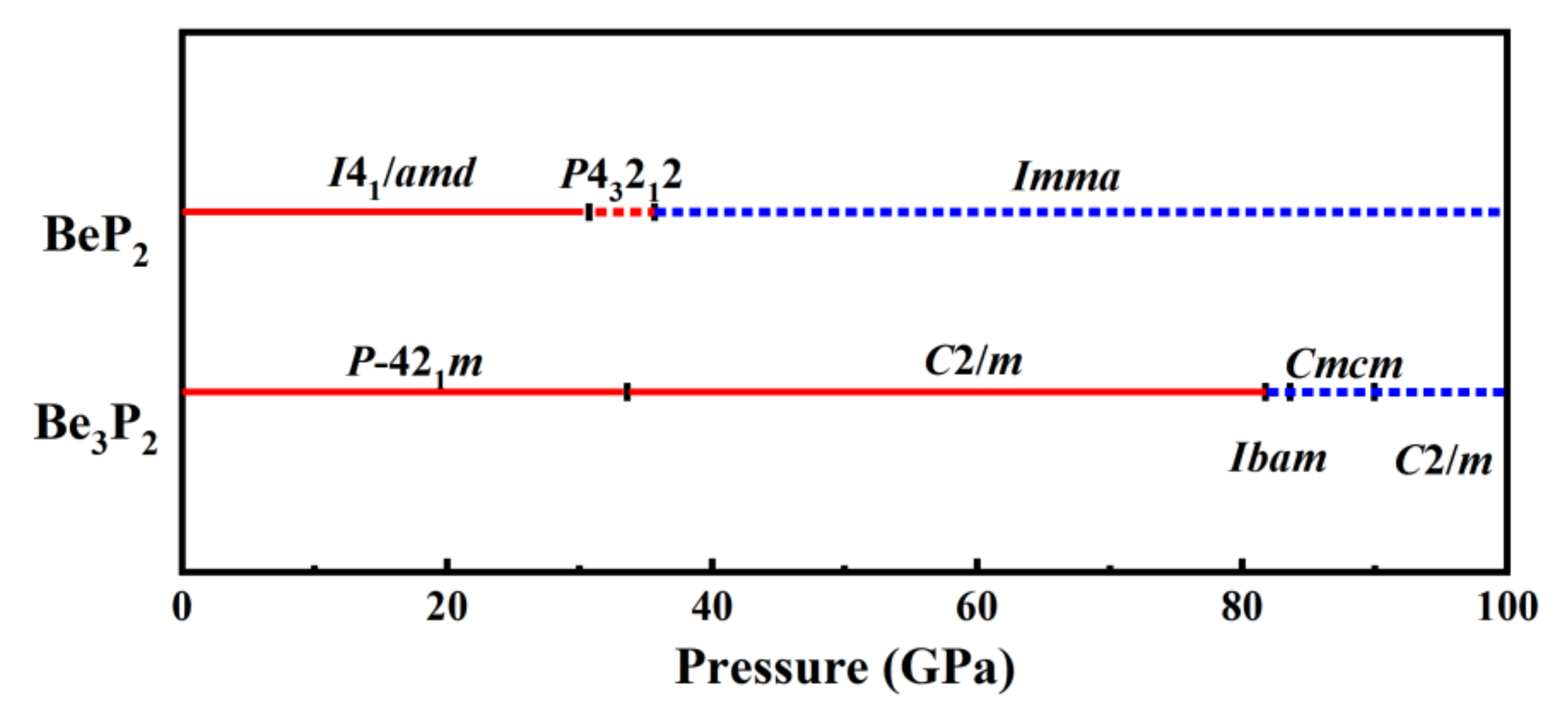
3.1. Crystal Structure
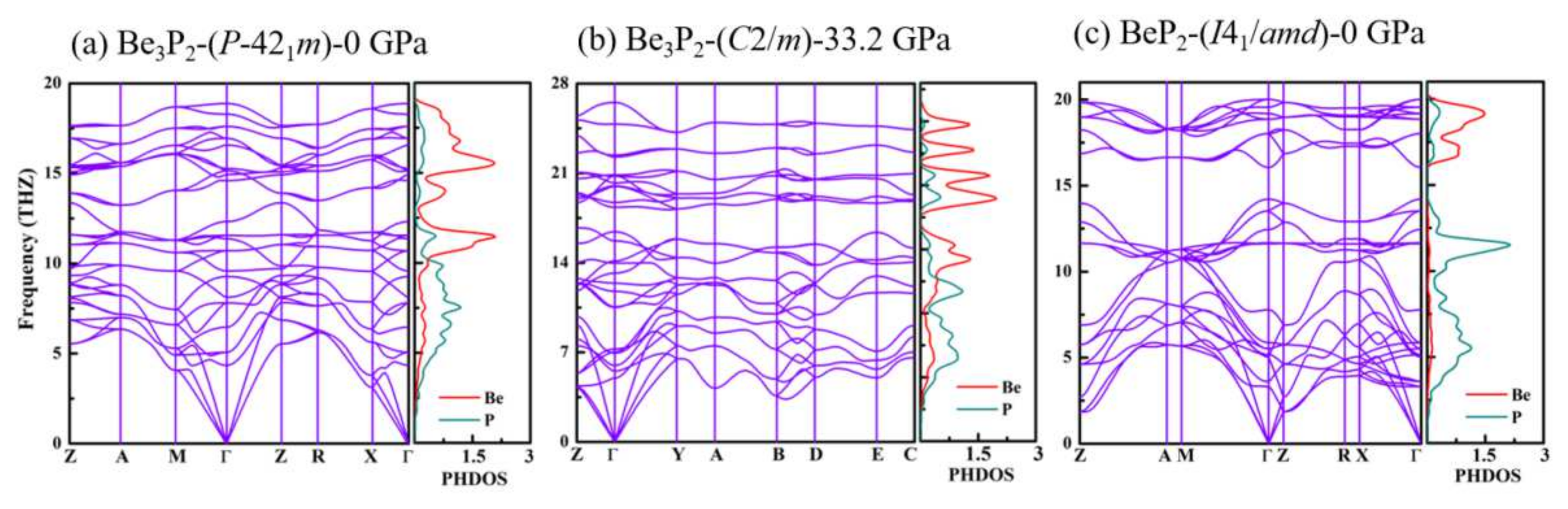
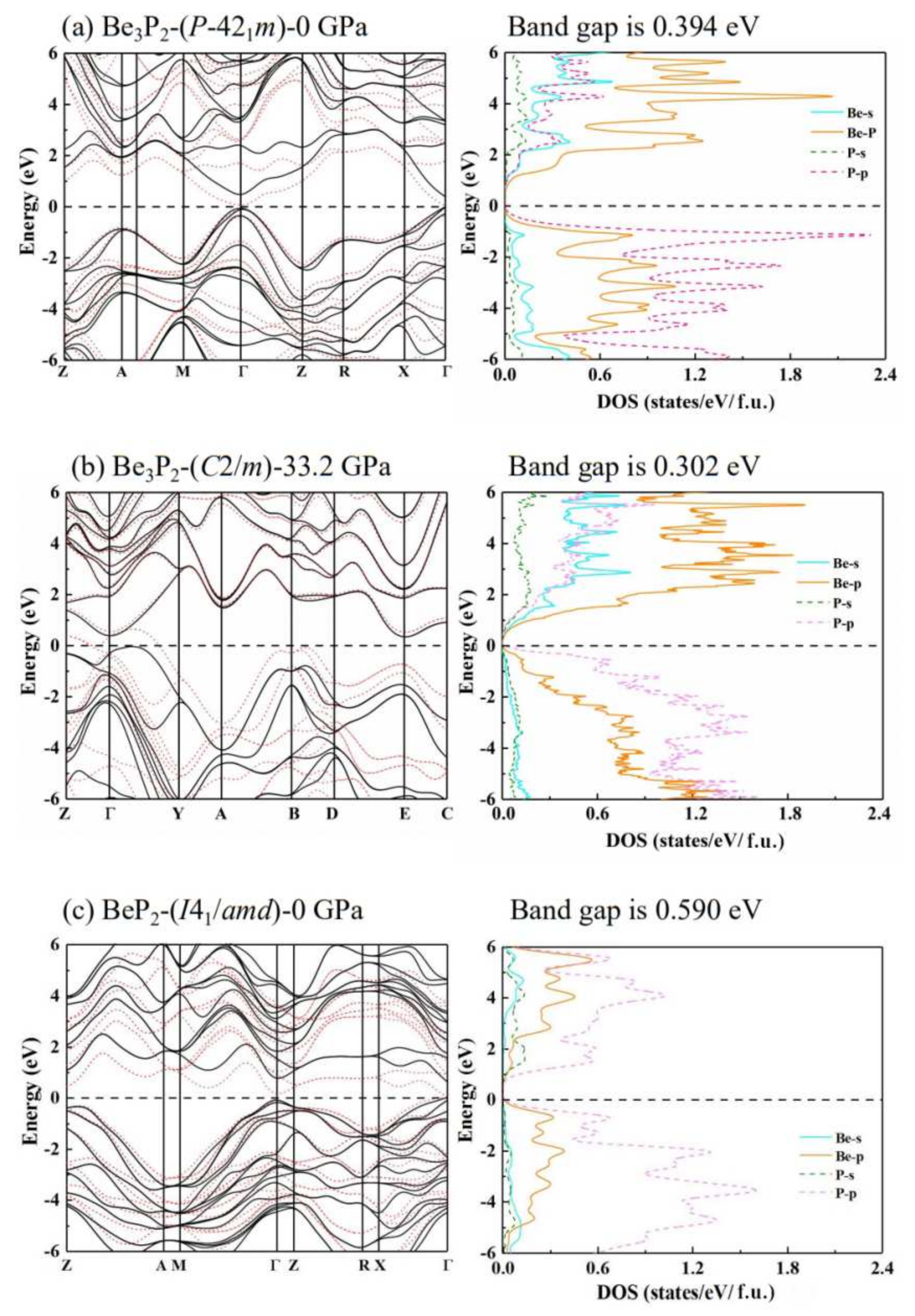
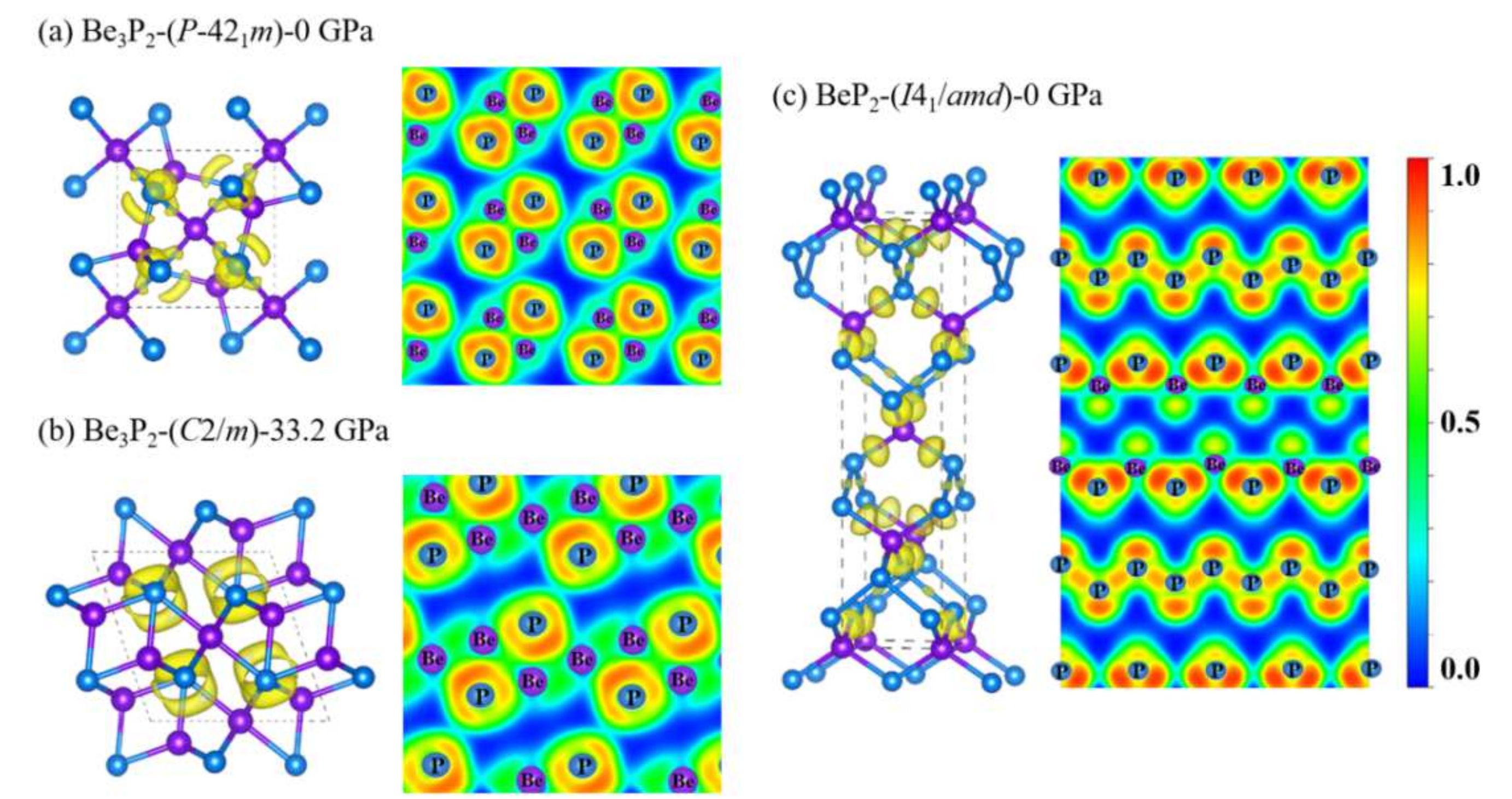
3.2. Electronic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ohno, H.; Shen, N.A.; Matsukura, F.; Oiwa, A.; Endo, A.; Katsumoto, S.; Iye, Y. (Ga, Mn) As: A new diluted magnetic semi-conductor based on GaAs. Appl. Phys. Lett. 1996, 69, 363–365. [Google Scholar] [CrossRef]
- Khramtsov, I.A.; Fedyanin, D.Y. Superinjection of Holes in Homojunction Diodes Based on Wide-Bandgap Semiconductors. Materials 2019, 12, 1972. [Google Scholar] [CrossRef] [PubMed]
- Pereira, N.; Lima, A.C.; Correia, V.M.G.; Peřinka, N.; Lanceros-Mendez, S.; Martins, P. Magnetic Proximity Sensor Based on Magnetoelectric Composites and Printed Coils. Materials 2020, 13, 1729. [Google Scholar] [CrossRef] [PubMed]
- Gamel, M.M.A.; Lee, H.J.; Rashid, W.E.S.W.A.; Ker, P.J.; Yau, L.K.; Hannan, M.A.; Jamaludin, Z. A Review on Thermophotovoltaic Cell and Its Applications in Energy Conversion: Issues and Recommendations. Materials 2021, 14, 4944. [Google Scholar] [CrossRef] [PubMed]
- Dietl, T. A ten-year perspective on dilute magnetic semiconductors and oxides. Nat. Mater. 2010, 9, 965–974. [Google Scholar] [CrossRef]
- L’Haridon, P.; David, J.; Lang, J.; Parthé, E. BeP2: A tetrahedral structure of type order-disorder which obeys a coordination rule for short-range order. J. Solid State Chem. 2020, 19, 287–297. [Google Scholar] [CrossRef]
- Gregory, D. Nitride chemistry of the s-block elements. Co-Ord. Chem. Rev. 2001, 215, 301–345. [Google Scholar] [CrossRef]
- Orhan, E.; Jobic, S.; Brec, R.; Marchand, R.; Saillard, J.Y. Binary nitrides α-M3N2 (M = Be, Mg, Ca): A theoretical study. J. Mater. Chem. 2020, 12, 2475–2479. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Y.; Hu, Z.; Shao, X. Theoretical Study on Thermoelectric Properties and Doping Regulation of Mg3X2 (X = As, Sb, Bi). Metals 2021, 11, 971. [Google Scholar] [CrossRef]
- Mokhtari, A. Density functional study of the group II phosphide semiconductor compounds under hydrostatic pressure. J. Phys. Condens. Matter 2008, 20, 135224. [Google Scholar] [CrossRef]
- Römer, S.R.; Doerfler, T.; Kroll, P.; Schnick, W. Group II element nitrides M3N2 under pressure: A comparative density functional study. Phys. Status Solidi (B) 2020, 246, 1604–1613. [Google Scholar] [CrossRef]
- Mokhtari, A.; Akbarzadeh, H. Ab initio calculations of the electronic and structural properties of beryllium-, magnesium-and calcium-nitrides. J. Phys. Condens. Matter 2003, 337, 122–129. [Google Scholar] [CrossRef]
- Armenta MG, M.; Reyes-Serrato, A.; Borja, M.A. Ab initio determination of the electronic structure of beryllium-, aluminum-, and magnesium-nitrides: A comparative study. Phys. Rev. B 2020, 62, 4890. [Google Scholar] [CrossRef]
- Imai, Y.; Watanabe, A. Electronic structures of Mg3Pn2 (Pn = N, P, As, Sb and Bi) and Ca3N2 calculated by a first-principle pseudopotential method. J. Mater. Sci. 2006, 41, 2435–2441. [Google Scholar] [CrossRef]
- Dobrokhotova, Z.V.; Zaitsev, A.I.; Zemchenko, M.A.; Litvina, A.D.; Yaschenko, S.N. Thermodynamic properties of calcium and barium phosphides. J. Therm. Anal. 1992, 38, 1113–1122. [Google Scholar] [CrossRef]
- Sa, R.; Zha, W.; Liu, D. First-principles insight into the structural, mechanical, electronic and optical properties of Be3X2 (X = N, P, As). J. Phys. Chem. Solids 2020, 145, 109575. [Google Scholar]
- Ullah, M.; Ali, R.; Murtaza, G.; Chen, Y. First principles investigation of Be3X2 (X = N, P, As) and their alloys for solar cell applications. J. Alloy. Compd. 2019, 795, 385–390. [Google Scholar] [CrossRef]
- Joshi, K.B.; Paliwal, U. First-principles study of pressure-induced phase transitions and electronic structure of Be3P2 polymorphs. Philos. Mag. 2012, 92, 1159–1169. [Google Scholar] [CrossRef]
- Joshi, K.B.; Paliwal, U. Pressure dependent electronic properties of α-Be3P2. J. Phys. Conf. Series. IOP Publ. 2012, 377, 012058. [Google Scholar] [CrossRef]
- Stackelberg, Μ.V.; Paulus, R. Untersuchungen über die Kristallstruktur der Nitride und Phosphide zweiwertiger Metalle. Z. Phys. Chem. 1933, 22, 305–322. [Google Scholar] [CrossRef]
- Wyckoff, R.W.G. Crystal Structures, 2nd ed.; Krieger: Malabar, FL, USA, 1986. [Google Scholar]
- Elmaslout, A.; Motte, J.; Courtois, A.; Protas, J.; Gleitzer, C. Crystal- structure of Be3P2. J. Solid State Chem. 1975, 15, 223–228. [Google Scholar] [CrossRef]
- De Carvalho MM, G.; Betinni, J.; Pudenzi, M.A.; Cardoso, L.P.; Cotta, M.A. Evidence of Be3P2 formation during growth of Be-doped phosphorus-based semiconductor compounds. Appl. Phys. Lett. 1999, 74, 3669–3671. [Google Scholar] [CrossRef][Green Version]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Lv, J.; Li, Q.; Zhang, L.; Ma, Y. CALYPSO structure prediction method and its wide application. Comput. Mater. Sci. 2016, 112, 406–415. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, H.; Pickard, C.J.; Zou, G.; Ma, Y. Reactions of xenon with iron and nickel are predicted in the Earth’s inner core. Nat. Chem. 2014, 6, 644–648. [Google Scholar] [CrossRef]
- Li, Y.; Hao, J.; Liu, H.; Li, Y.; Ma, Y. The metallization and superconductivity of dense hydrogen sulfide. J. Chem. Phys. 2014, 140, 174712. [Google Scholar] [CrossRef]
- Zhong, X.; Wang, H.; Zhang, J.; Liu, H.; Zhang, S.; Song, H.; Yang, G.; Zhang, L.; Ma, Y. Tellurium Hydrides at High Pressures: High-Temperature Superconductors. Phys. Rev. Lett. 2016, 116, 057002–057008. [Google Scholar] [CrossRef]
- Zhang, S.; Zhu, L.; Liu, H.; Yang, G. Structure and electronic properties of Fe2SH3 compound under high pressure. Inorg. Chem. 2016, 55, 11434–11439. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Hafnerr, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Thirumalai, D.; Hall, R.W.; Berne, B.J. A path integral Monte Carlo study of liquid neon and the quantum effective pair potential. J. Chem. Phys. 1984, 81, 2523–2527. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Li, X.; Wang, Q. Prediction of a BeP2 monolayer with a compression-induced Dirac semimetal state. Phys. Rev. B 2018, 97, 085418. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Pressure (GPa) | Atom | Number | Charge Value (e) | δ(e) |
|---|---|---|---|---|---|
| Be3P2-P-421m | 0 GPa | Be1 | 4 | 0.466 | −1.534 |
| - | - | Be2 | 2 | 0.457 | −1.543 |
| - | - | P1 | 4 | 7.305 | 2.305 |
| Be3P2-C2/m | 33.2 GPa | Be1 | 4 | 0.452 | −1.548 |
| - | - | Be2 | 2 | 0.408 | −1.592 |
| - | - | P1 | 4 | 7.344 | 2.344 |
| BeP2-I41/amd | 0 GPa | Be1 | 4 | 0.470 | −1.530 |
| - | - | P1 | 8 | 5.765 | 0.765 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Dan, Y.; Zhang, A.; Liu, S.; Yue, J.; Li, J.; Ma, X.; Huang, Y.; Liu, Y.; Cui, T. First-Principles Study of High-Pressure Phase Stability and Electron Properties of Be-P Compounds. Materials 2022, 15, 1255. https://doi.org/10.3390/ma15031255
Liu H, Dan Y, Zhang A, Liu S, Yue J, Li J, Ma X, Huang Y, Liu Y, Cui T. First-Principles Study of High-Pressure Phase Stability and Electron Properties of Be-P Compounds. Materials. 2022; 15(3):1255. https://doi.org/10.3390/ma15031255
Chicago/Turabian StyleLiu, Han, Yaqian Dan, Ao Zhang, Siyuan Liu, Jincheng Yue, Junda Li, Xuejiao Ma, Yanping Huang, Yanhui Liu, and Tian Cui. 2022. "First-Principles Study of High-Pressure Phase Stability and Electron Properties of Be-P Compounds" Materials 15, no. 3: 1255. https://doi.org/10.3390/ma15031255
APA StyleLiu, H., Dan, Y., Zhang, A., Liu, S., Yue, J., Li, J., Ma, X., Huang, Y., Liu, Y., & Cui, T. (2022). First-Principles Study of High-Pressure Phase Stability and Electron Properties of Be-P Compounds. Materials, 15(3), 1255. https://doi.org/10.3390/ma15031255