
High-Pressure Phases of SnO and PbO: A Density Functional Theory Combined with an Evolutionary Algorithm Approach



Abstract
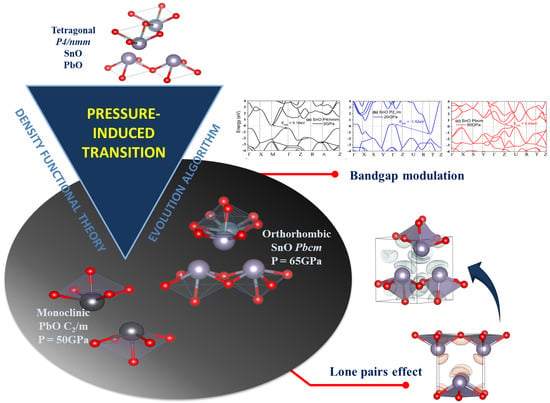
:
1. Introduction
2. Calculation Methods
3. Results
3.1. Genetic Algorithm Prediction of the SnO/PbO Phase at 0 GPa
3.1.1. SnO Metastable Phase at 0 GPa
3.1.2. PbO Metastable Phase at 0 GPa
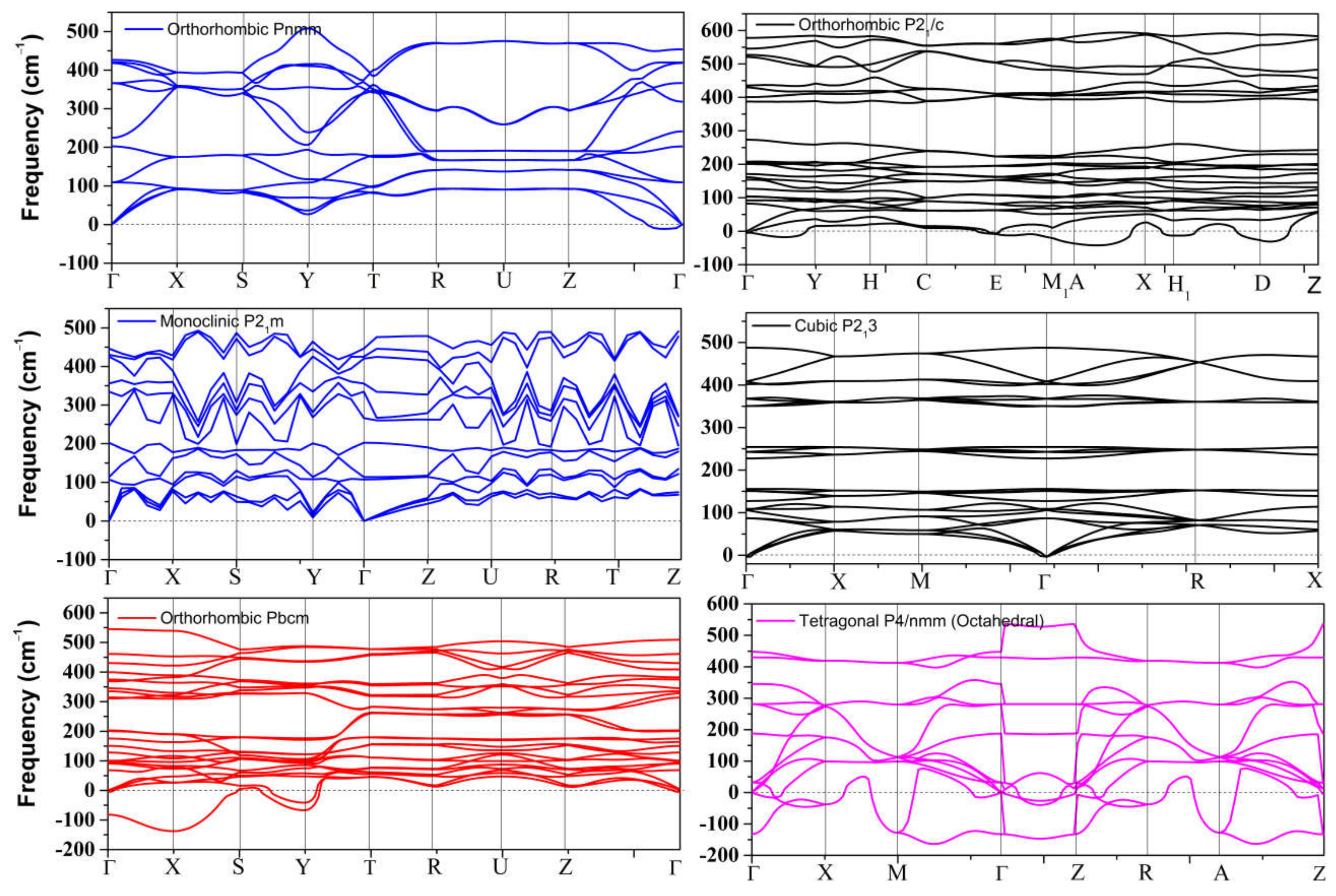
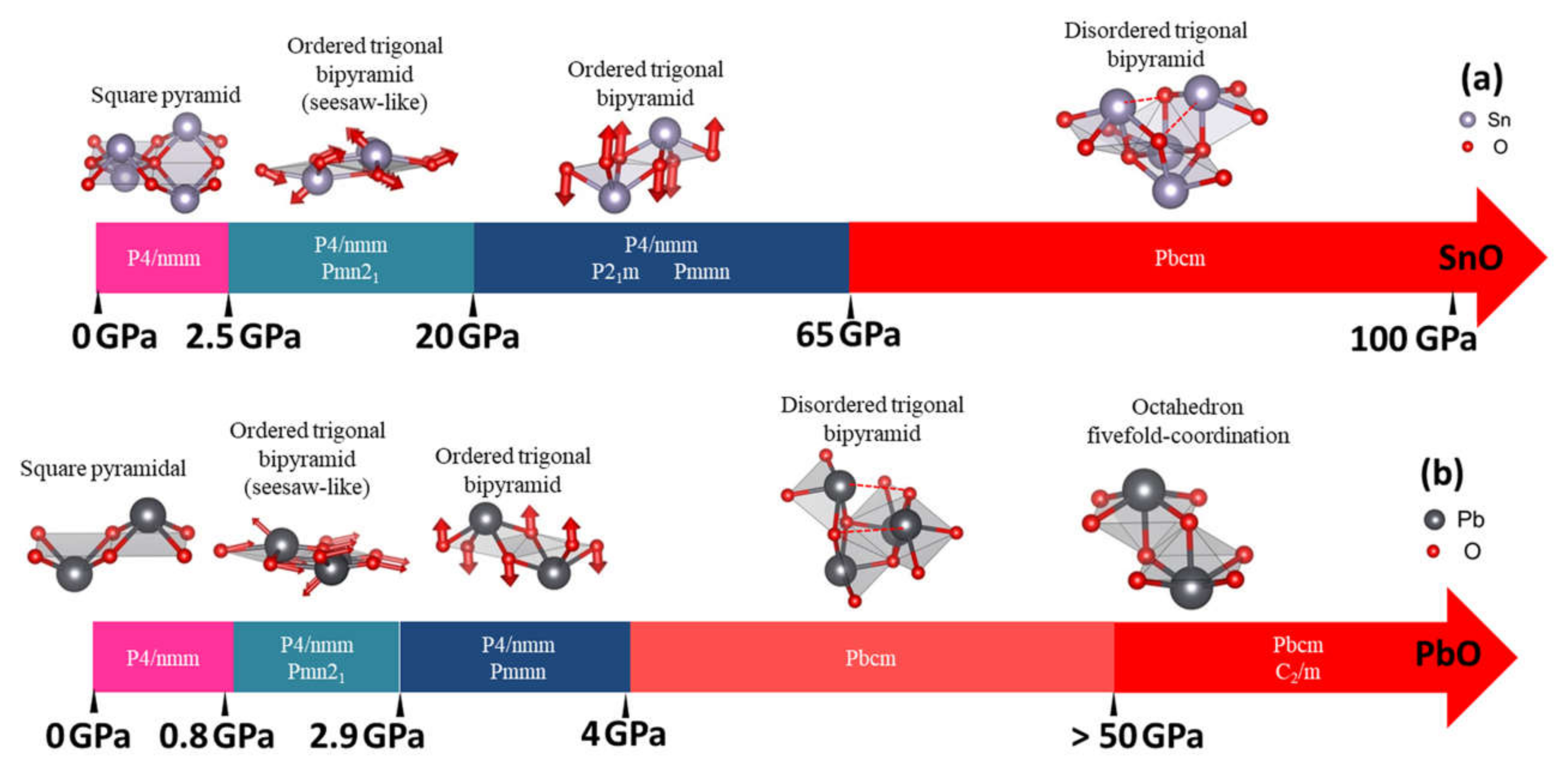
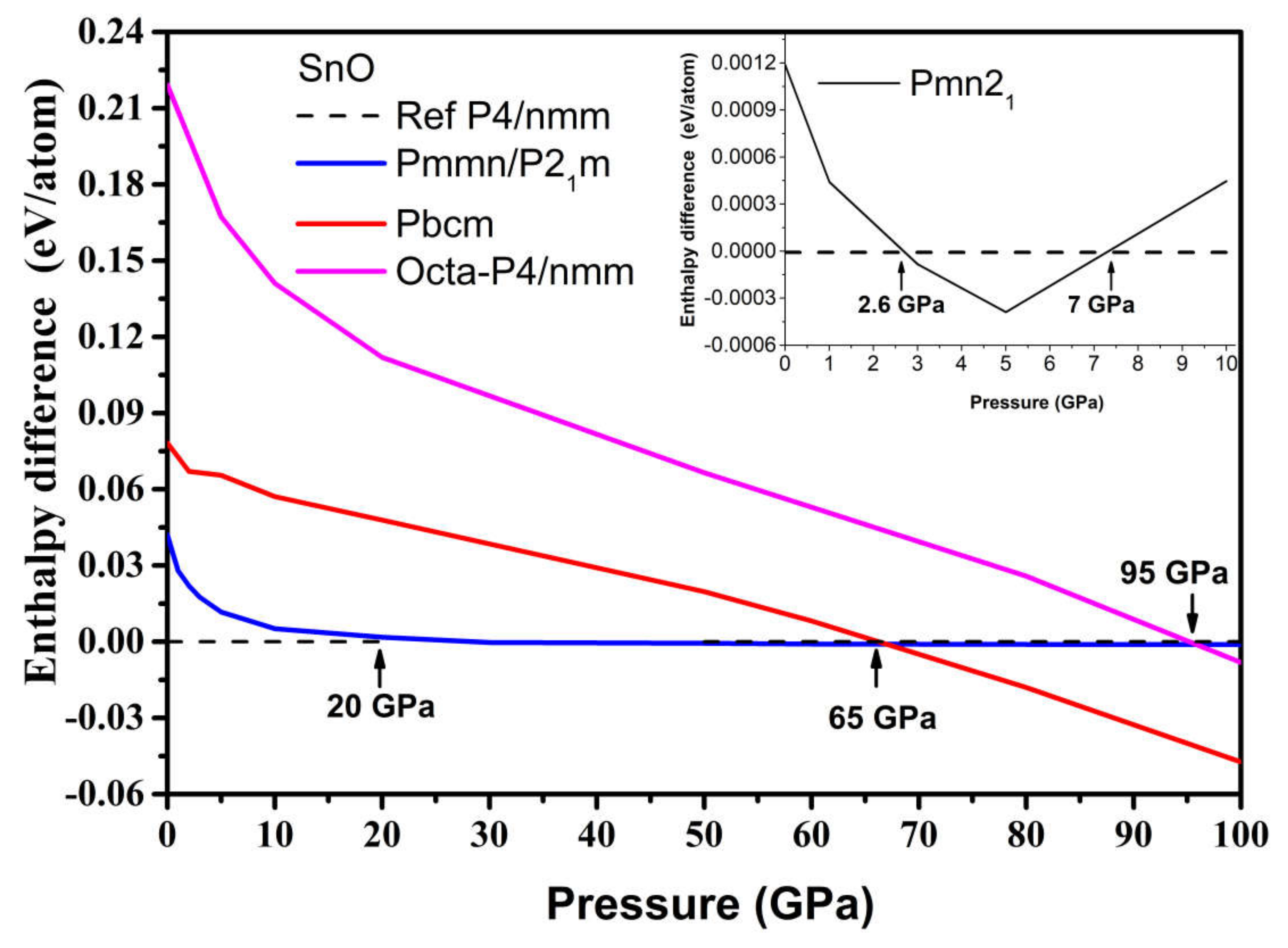
3.2. High Pressure Structures of SnO and PbO
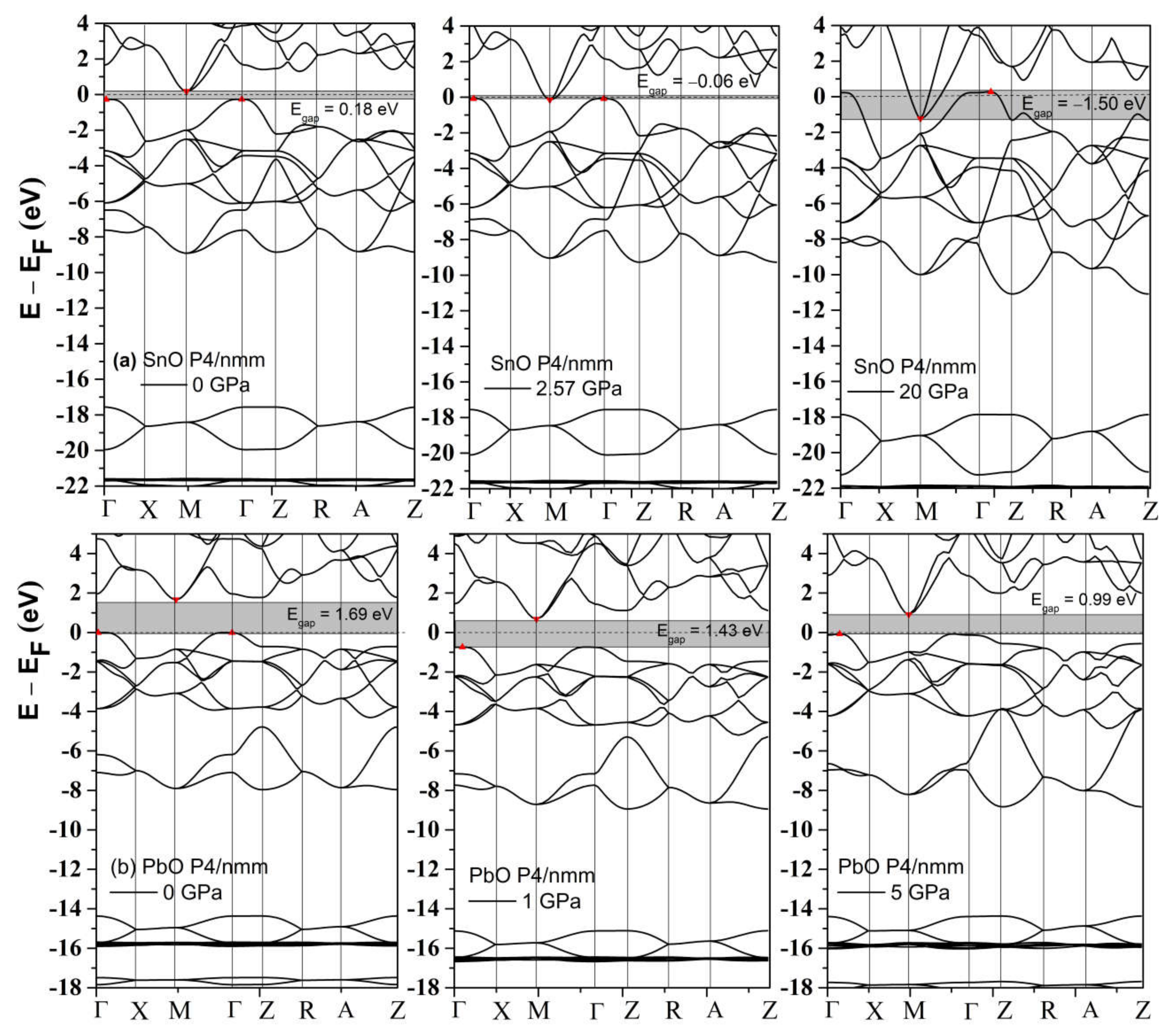
3.2.1. The Structural Variation of Tetragonal SnO and PbO upon Compression
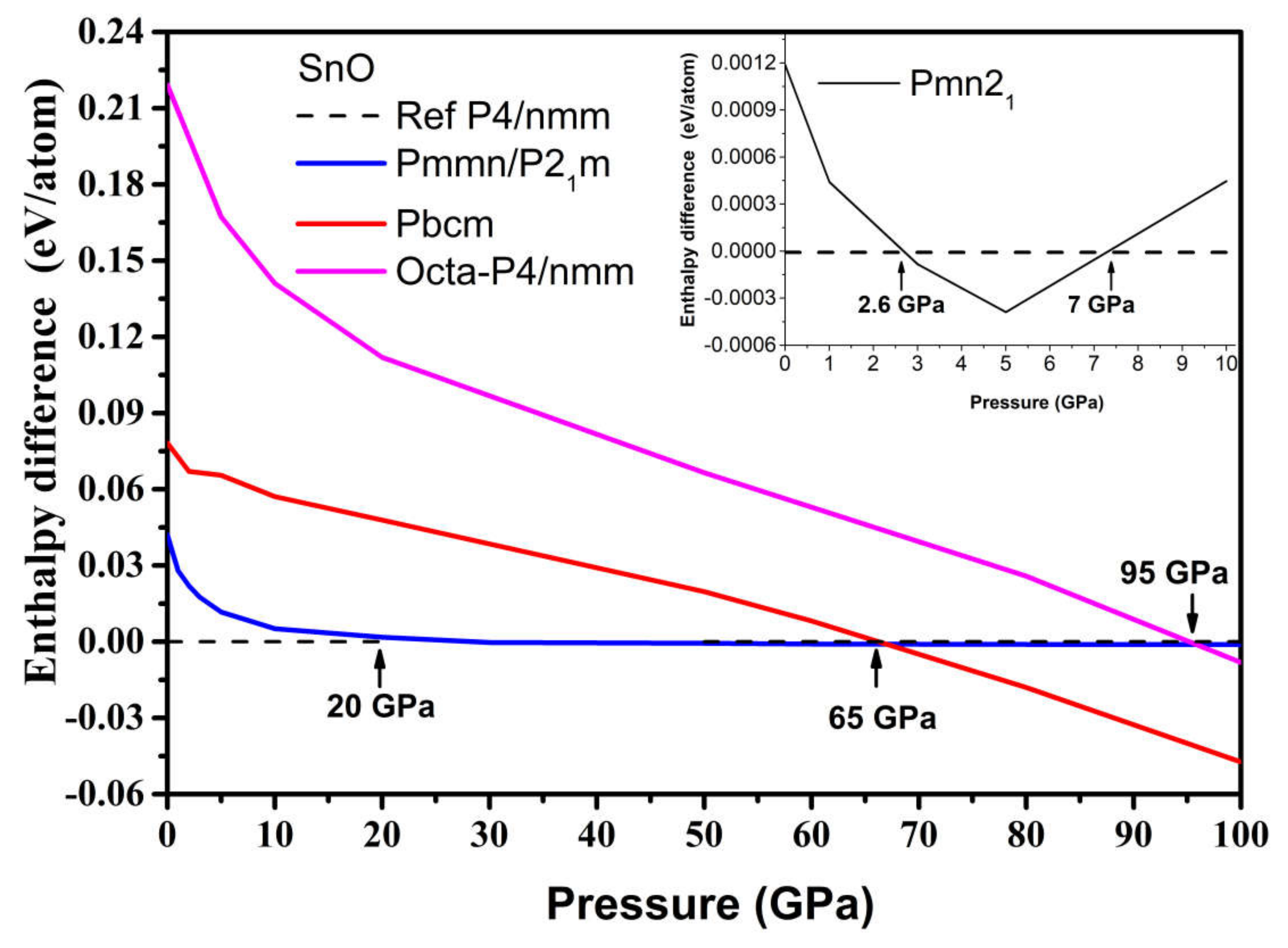
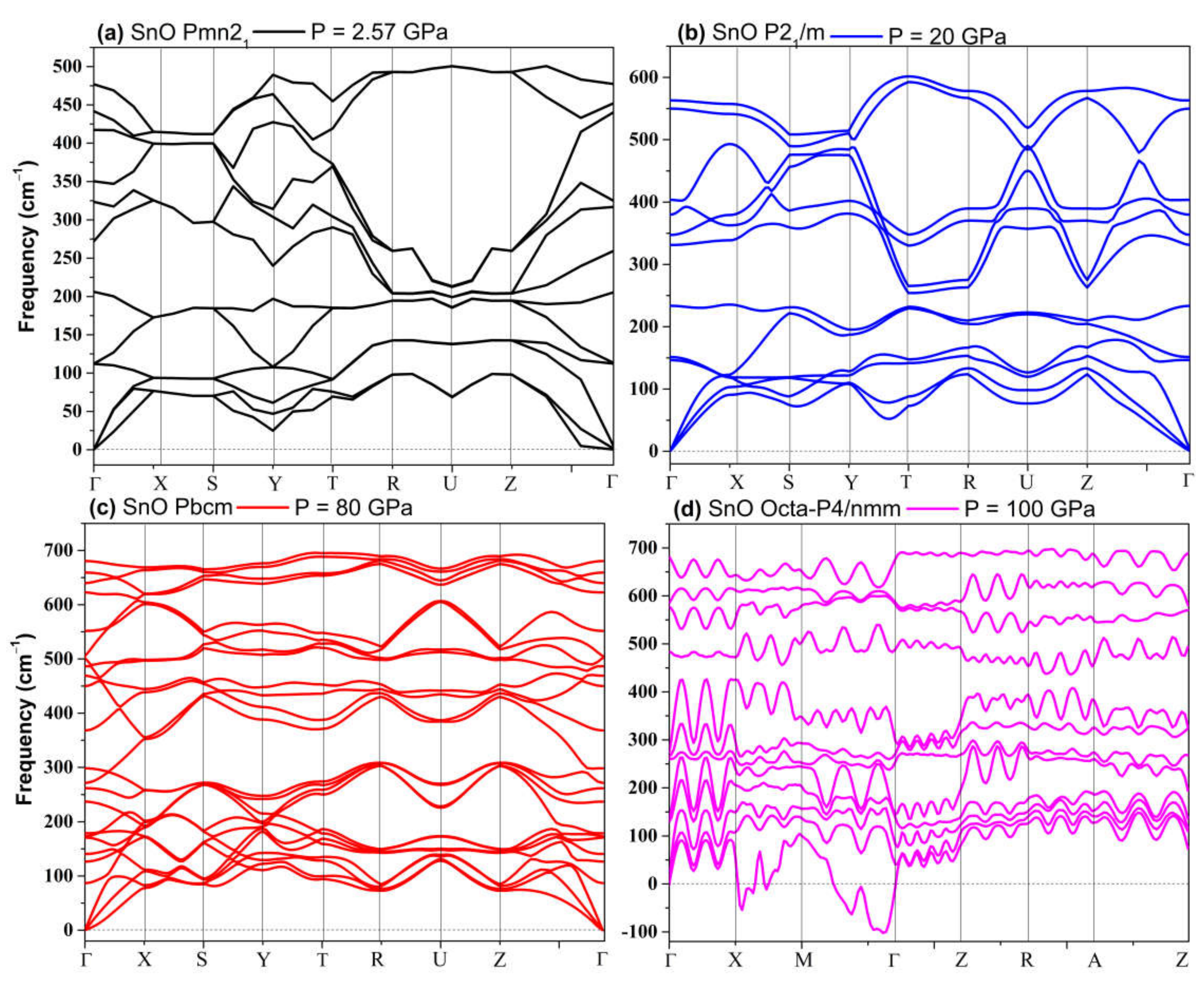
3.2.2. High-Pressure Phases of SnO Predicted by Genetic Algorithm
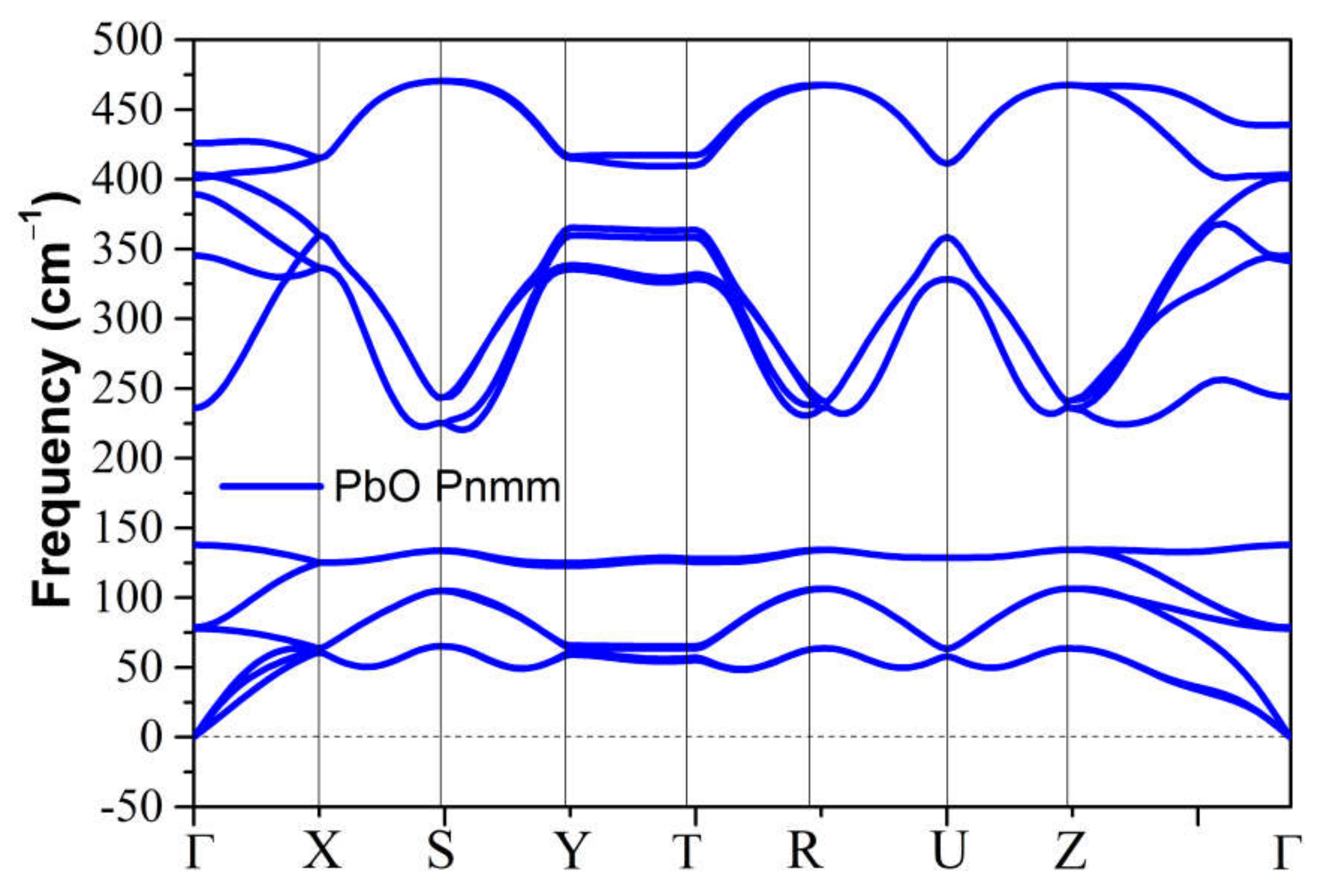
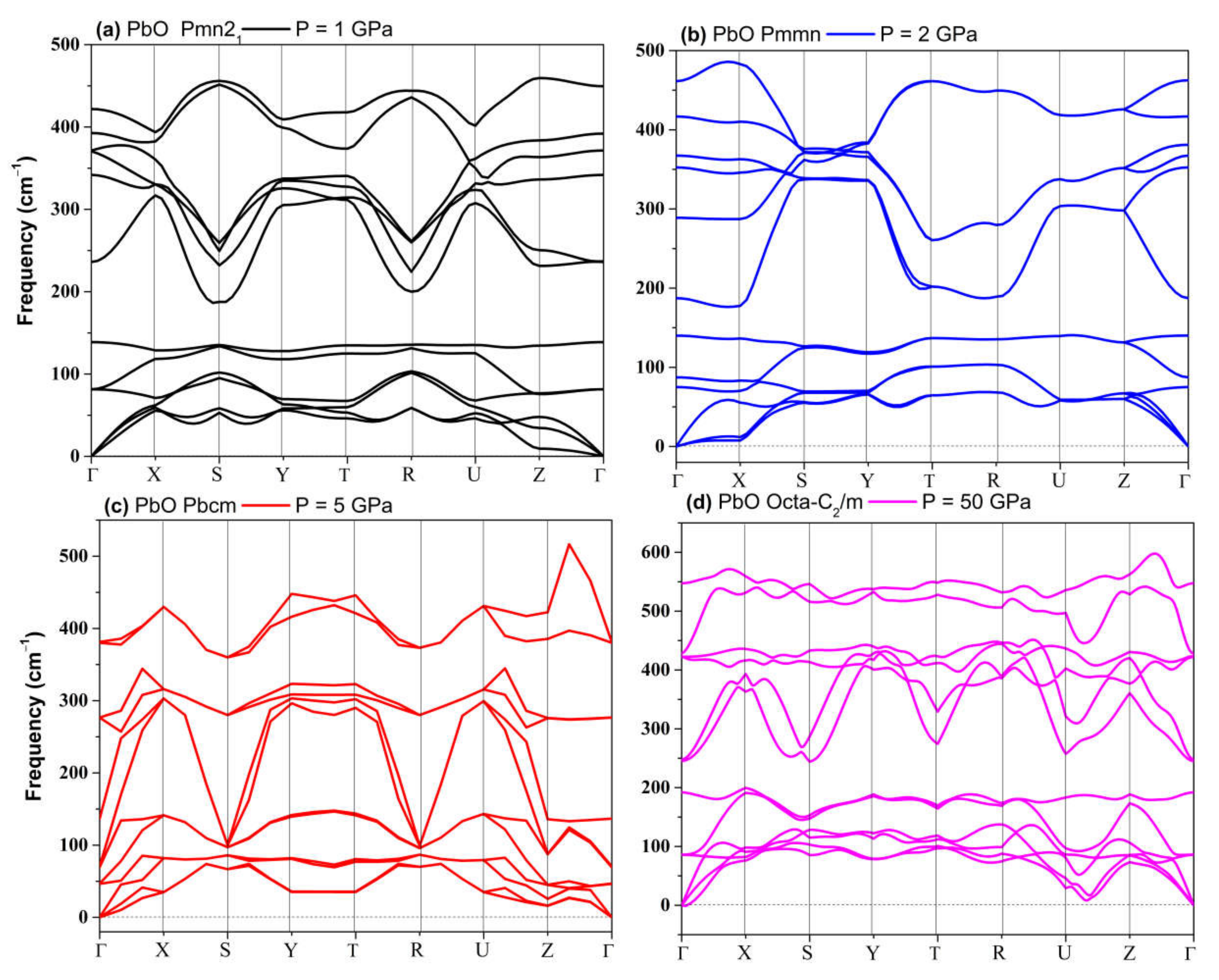
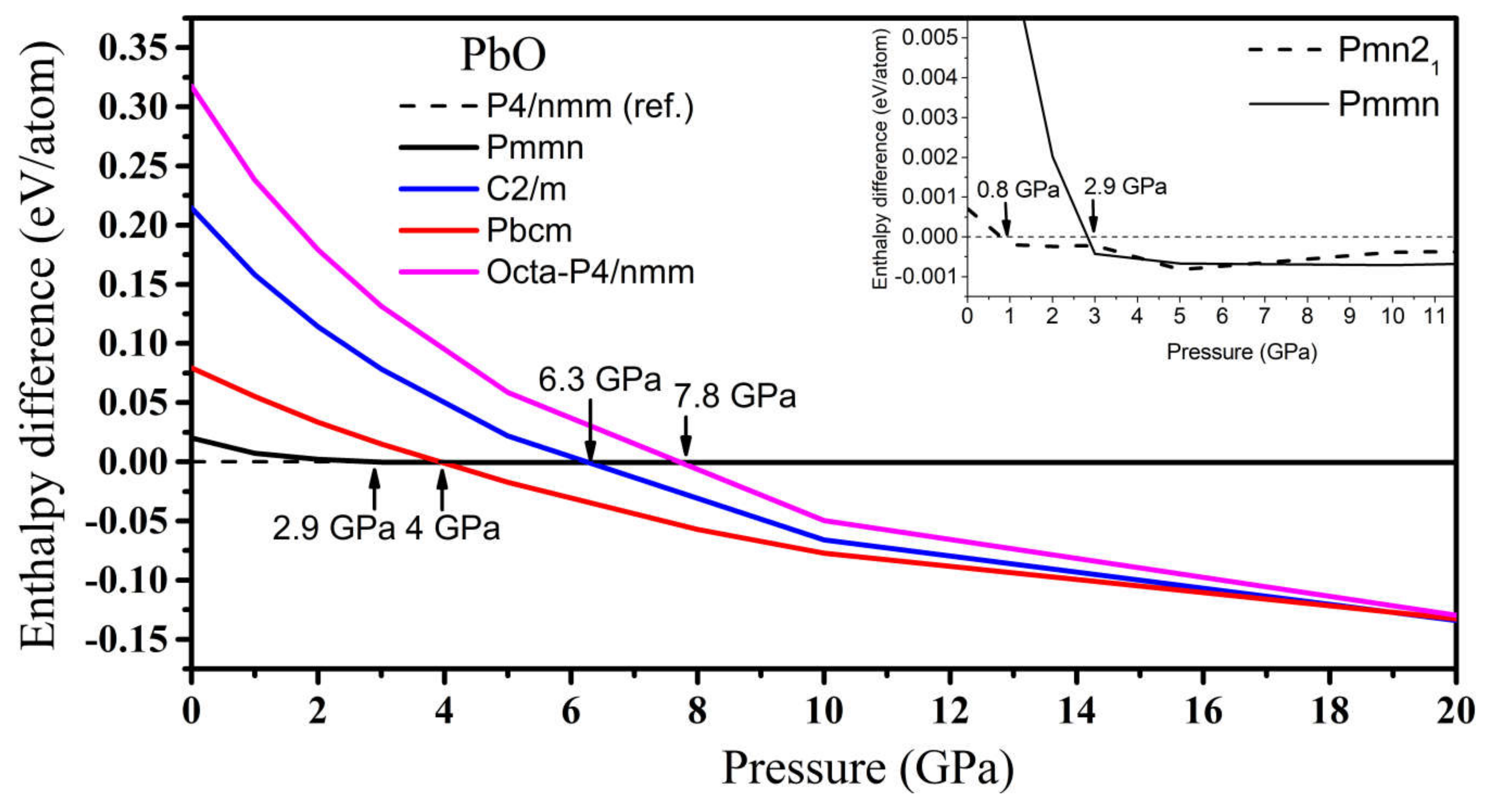
3.2.3. High-Pressure Phases of PbO Predicted by Genetic Algorithm
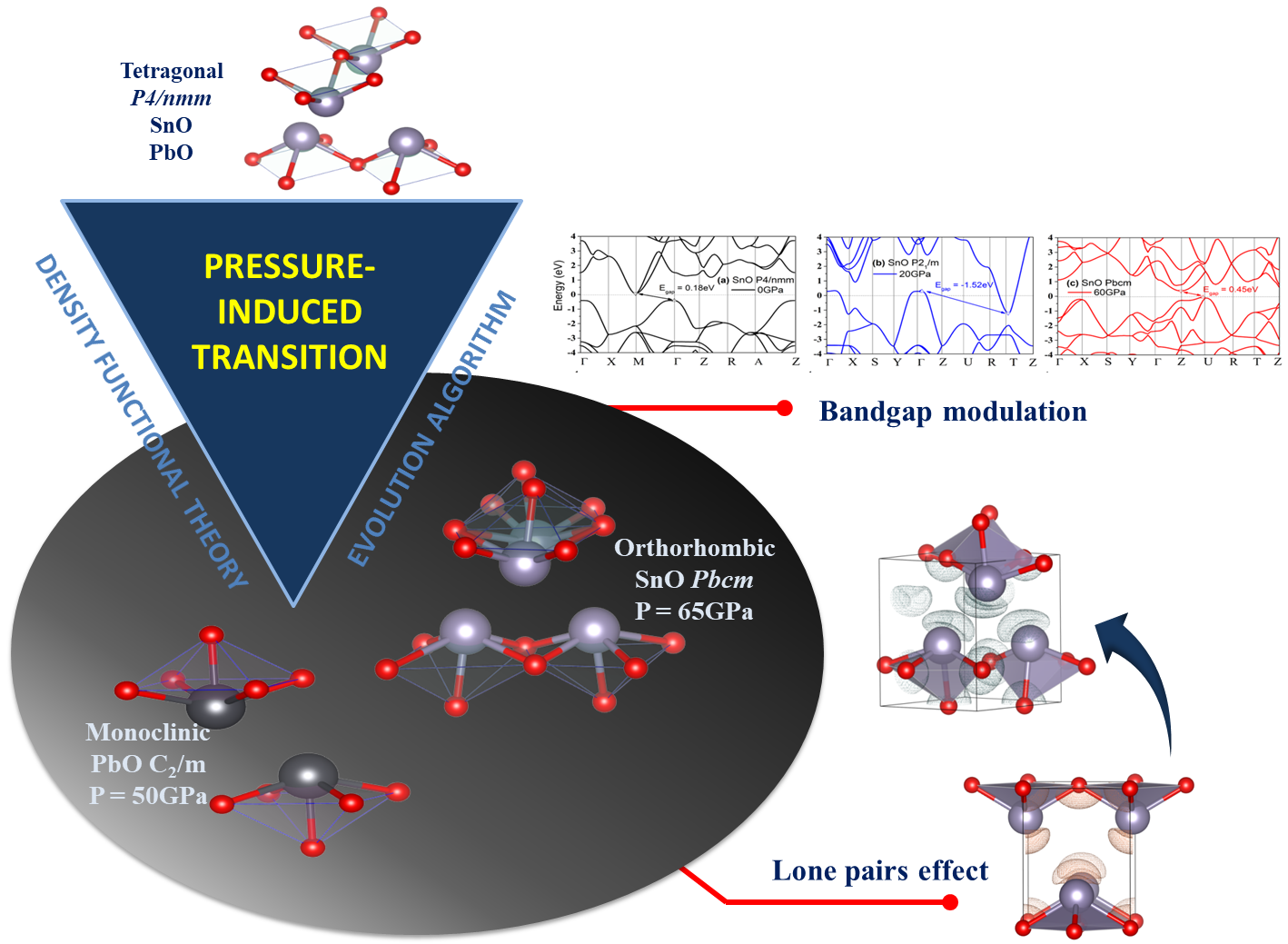
4. Discussion
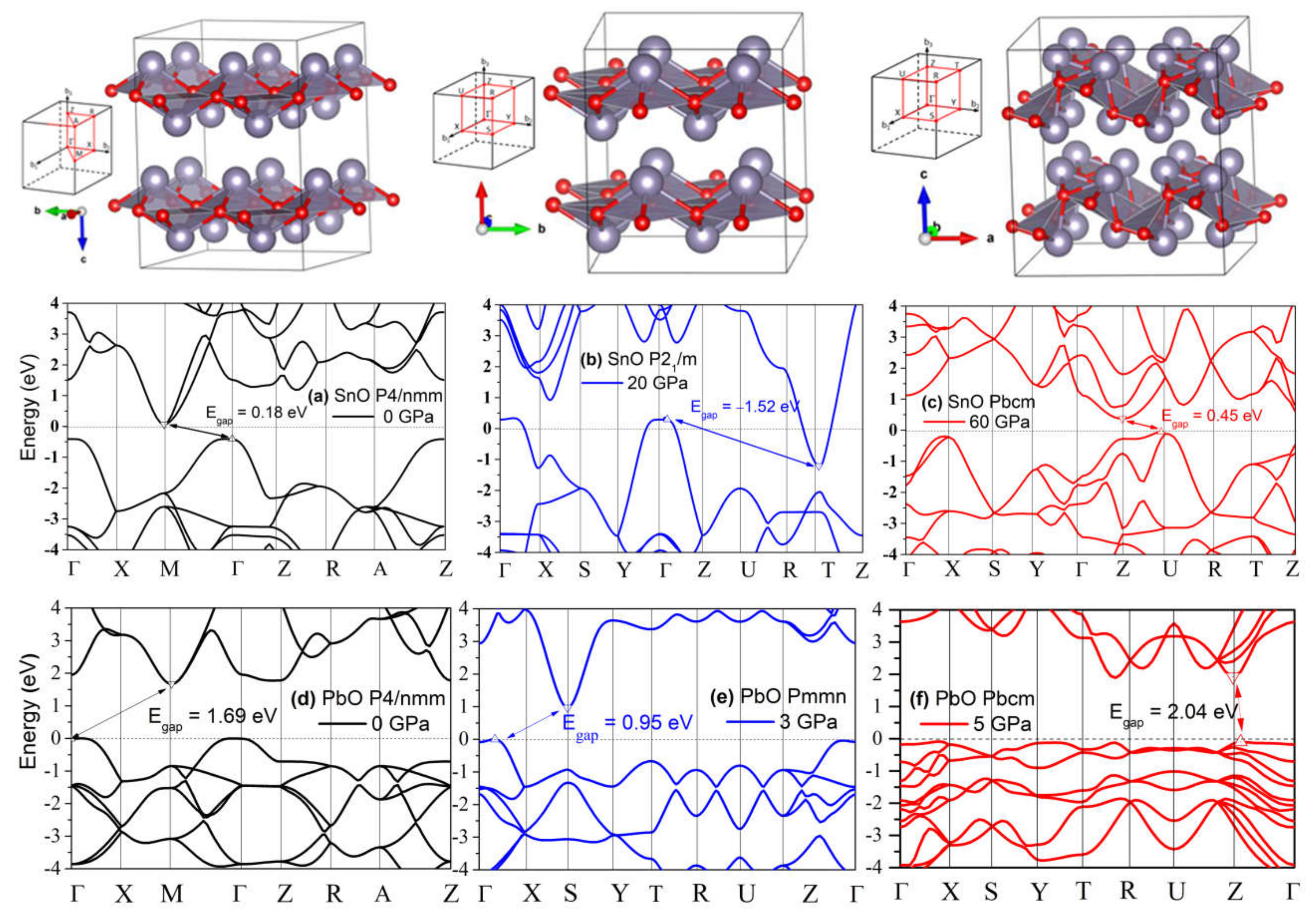
4.1. Structures of SnO and PbO under Pressure—Summarizing Our Results and Their Validity and Comparing Them to Previous Theoretical and Experimental Results
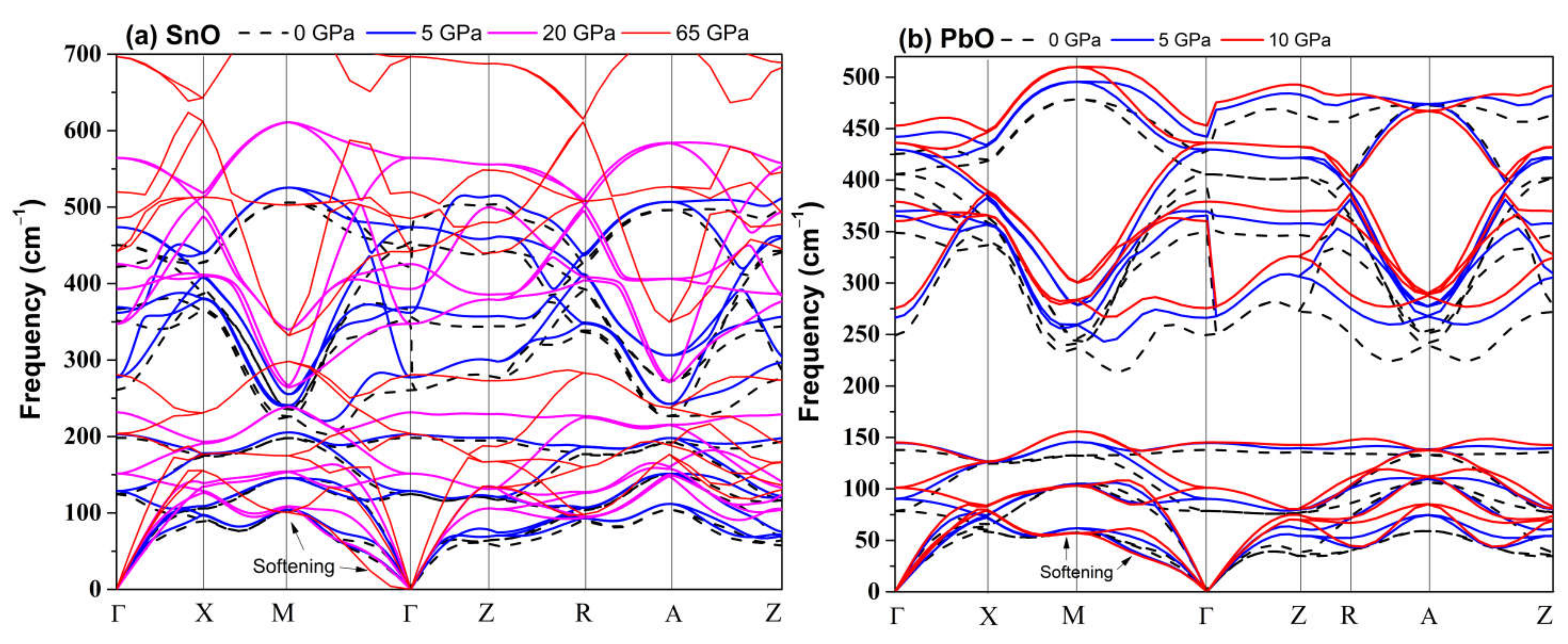
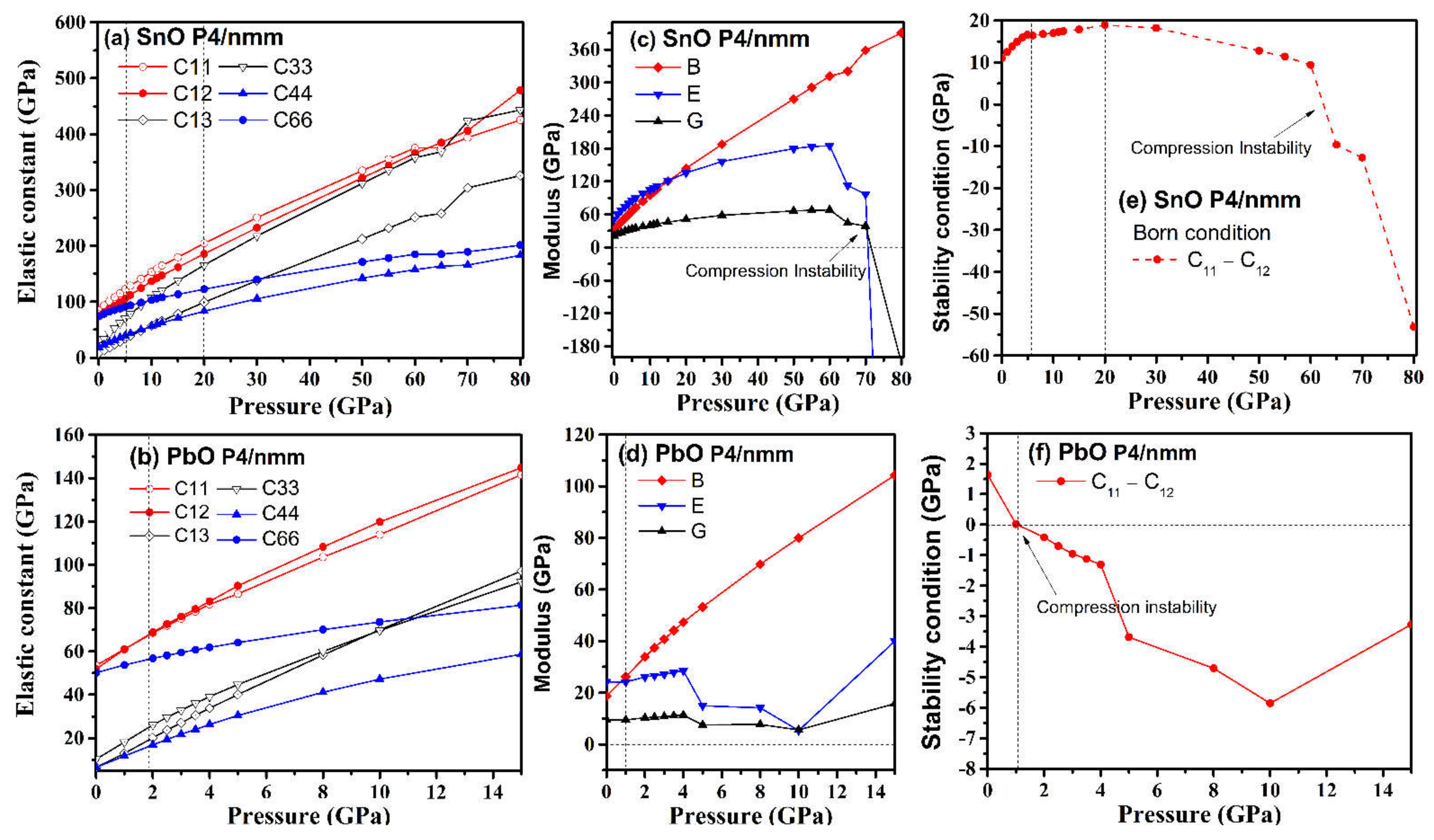
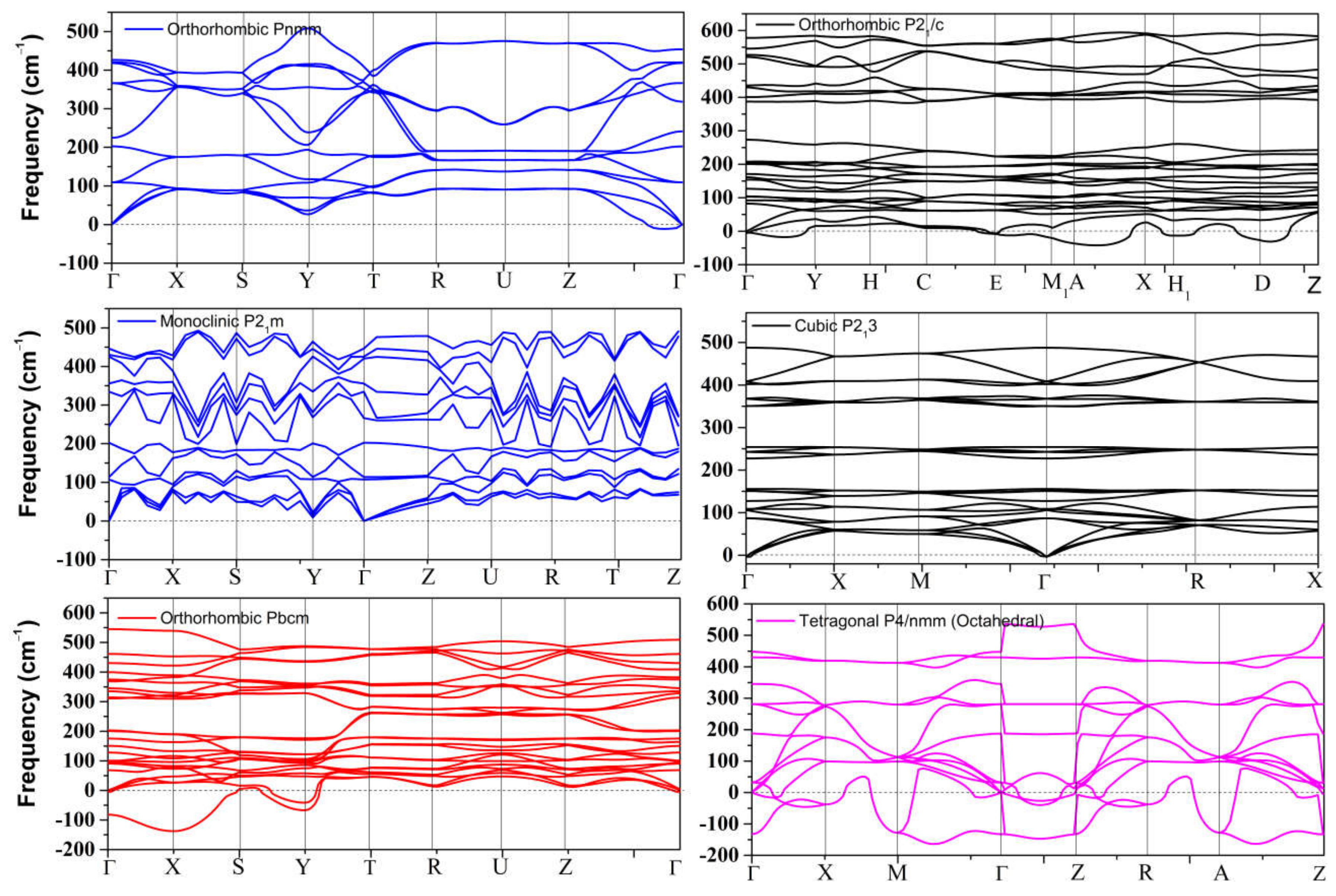
4.2. Elastic Instability of SnO/PbO upon Compression
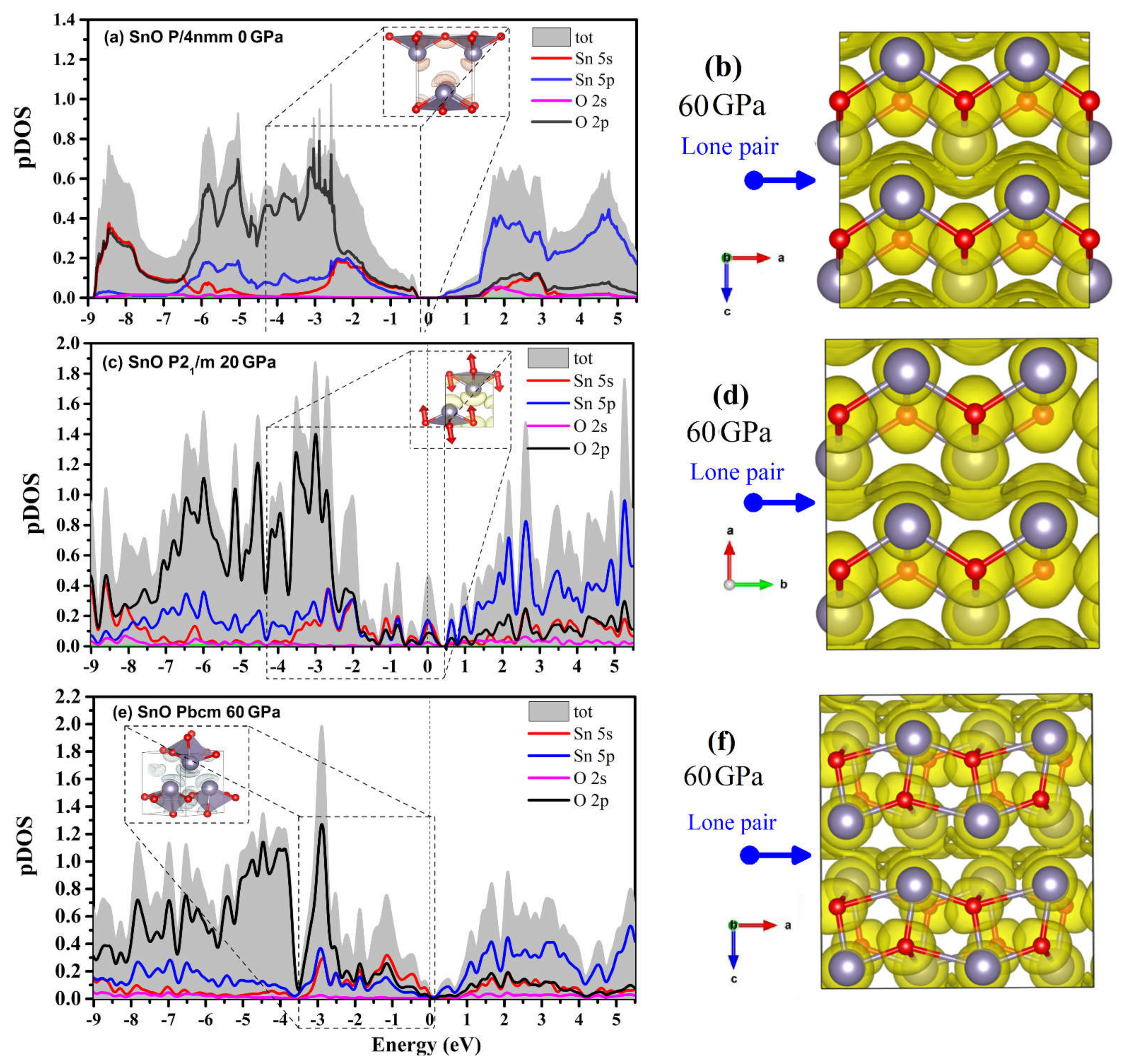
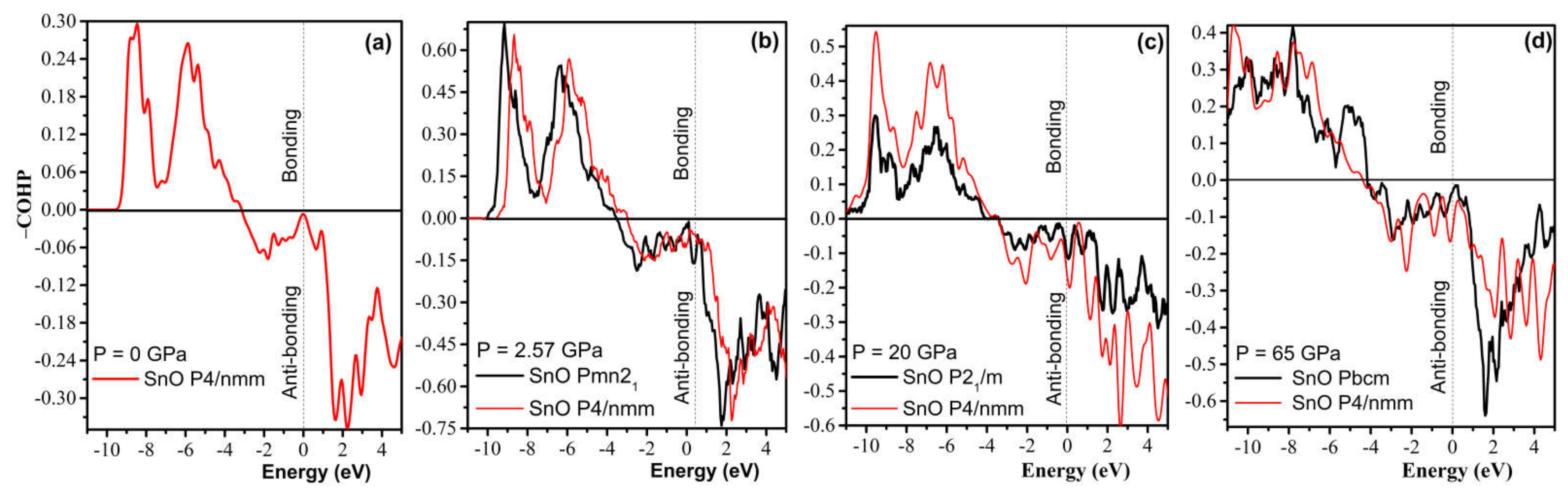
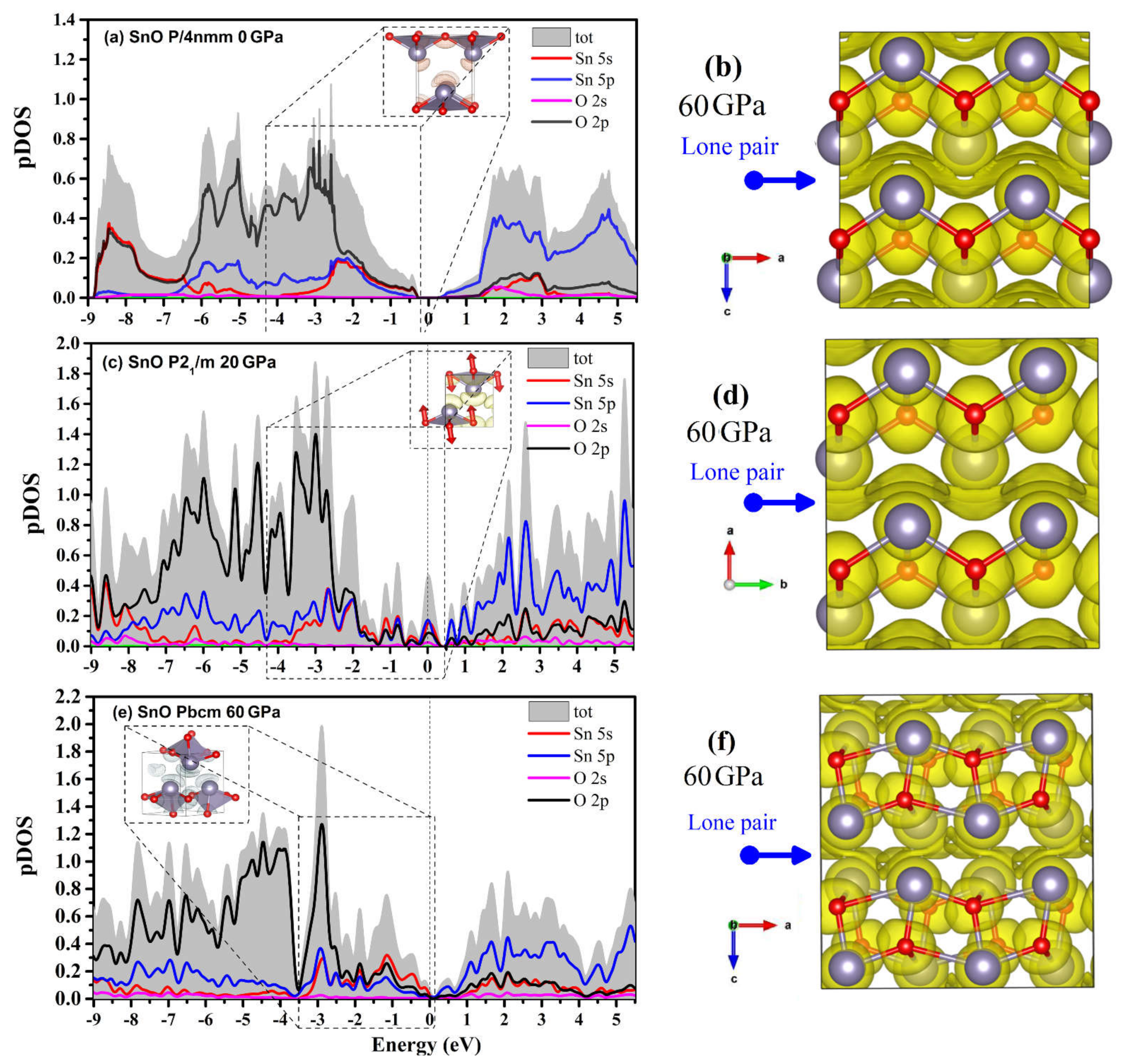
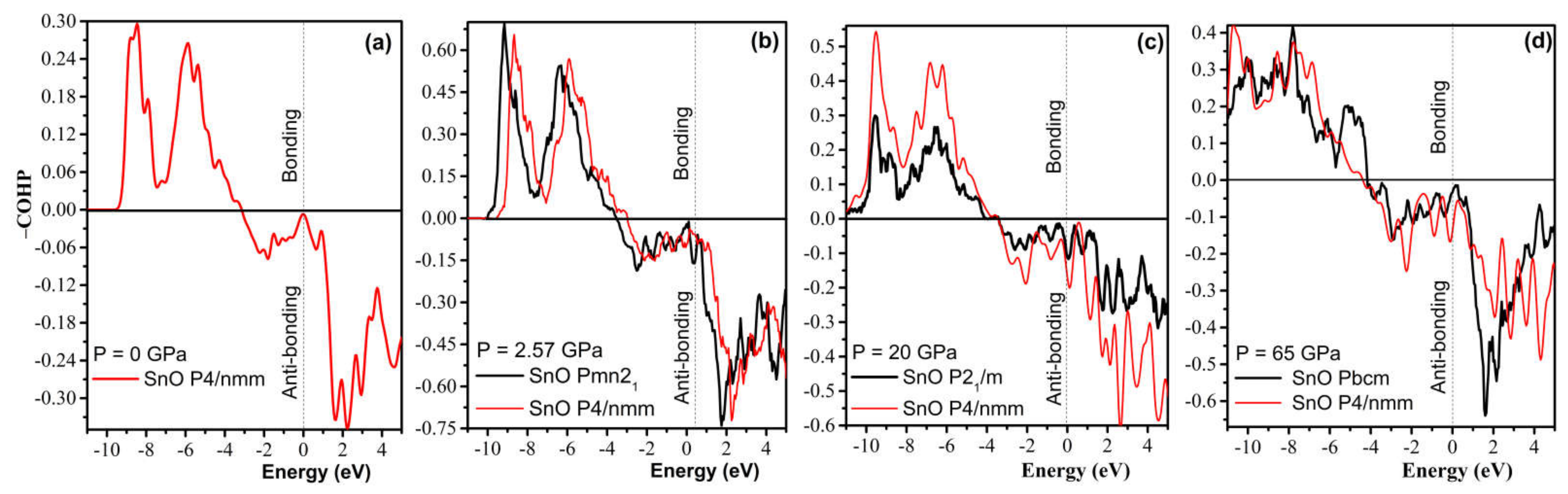
4.3. Electronic Structure and the Role of Lone Pairs upon Compression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, W.; Fu, L.; Zhang, Y.; Zhang, K.; Liang, L.Y.; Liu, Z.M.; Cao, H.T.; Pan, X.Q. Microstructure, optical, and electrical properties of -type SnO thin film. Appl. Phys. Lett. 2010, 96, 042113. [Google Scholar] [CrossRef]
- Saji, K.J.; Tian, K.; Tiwar, A.; Subbaiah, Y.P.V. P-type SnO thin films and SnO/ZnO heterostructures for all-oxide electronic and optoelectronic device applications. Thin Solid Films 2016, 605, 193–220. [Google Scholar] [CrossRef] [Green Version]
- Toyama, T.; Konishi, T.; Okamoto, H.; Morimoto, R.; Nishikawa, Y.; Tsutsumi, Y.; Seo, Y. Optical absorption spectra of P-type Tin monoxide thin films around their indirect fundamental gaps determined using photothermal deflection spectroscopy. Thin Solid Films 2014, 555, 148–152. [Google Scholar] [CrossRef]
- Simon, M.; Ford, R.A.; Franklin, A.R.; Grabowski, S.P.; Menser, B.; Much, G.; Nascetti, A. Analysis of lead oxide (PbO) layers for direct conversion X-ray detection. IEEE Symp. Conf. Rec. Nucl. Sci. 2004, 7, 4268–4272. [Google Scholar] [CrossRef]
- Qamar, A.; LeBlanc, K.; Semeniuk, O.; Reznik, A.; Lin, J.; Pan, Y.; Moewes, A. X-ray spectroscopic study of amorphous and polycrystalline PbO films, α-PbO, and β-PbO for direct conversion imaging. Sci. Rep. 2017, 7, 13159. [Google Scholar] [CrossRef]
- Berashevich, J.; Semeniuk, O.; Rubel, O.; Rowlands, J.A.; Reznik, A. Lead monoxide α-PbO: Electronic properties and point defect formation. J. Phys. Condens. Matter 2013, 25, 075803. [Google Scholar] [CrossRef]
- Ballantyne, A.D.; Hallett, J.P.; Riley, D.J.; Shah, N.; Payne, D.J. Lead acid battery recycling for the twenty-first century. R. Soc. Open Sci. 2018, 5, 171368. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.H.; Wang, J.; Konstantinov, K.; Wexler, D.; Chen, J.; Liu, H.K. Spray Pyrolyzed PbO-Carbon Nanocomposites as Anode for Lithium-Ion Batteries. J. Electrochem. Soc. 2006, 153, A787. [Google Scholar] [CrossRef]
- Christensen, N.E.; Svane, A.; Peltzer, Y.; Blancà, E.L. Electronic and structural properties of SnO under pressure. Phys. Rev. B 2005, 72, 014109. [Google Scholar] [CrossRef]
- Raulot, J.; Baldinozzi, G.; Seshadri, R.; Cortona, P. An ab-initio study of the role of lone pairs in the structure and insulator–metal transition in SnO and PbO. Sol. State Sci. 2002, 4, 467–474. [Google Scholar] [CrossRef]
- Adams, D.M.; Christy, S.A.G.; Haines, J.; Clark, S.M. Second-order phase transition in PbO and SnO at high pressure:Implications for the litharge-massicot phase transformation. Phys. Rev. B 1992, 46, 18. [Google Scholar] [CrossRef]
- Giefers, H.; Porsch, F.; Wortmann, G. Structural study of SnO at high pressure. Phys. B 2006, 373, 76–81. [Google Scholar] [CrossRef]
- Chen, P.J.; Jeng, H.T. Phase diagram of the layered oxide SnO: GW and electronphonon studies. Sci. Rep. 2015, 5, 16359. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.W.; Li, Y.; Cui, T.; Zhang, L.J.; Ma, Y.M.; Zou, G.T. The pressure-induced phase transition in SnO:a first-principles study. J. Phys. Condens. Matter 2007, 19, 425230. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.X.; Loa, I.; Syassen, K.; Hanfland, M.; Mathis, Y.L. Structural properties, infrared reflectivity, and Raman modes of SnO at high pressure. Phys. Status Solidi B 2004, 241, 3168. [Google Scholar] [CrossRef]
- Giefers, H.; Porsch, F. Shear induced phase transition in PbO under high pressure. Phys. B 2007, 400, 53–58. [Google Scholar] [CrossRef]
- Giefers, H.; Porsch, F.; Wortmann, G. High-pressure EXAFS and XRD investigation of unit cell parameters of SnO. Phys. Scr. 2005, 2005, 538. [Google Scholar] [CrossRef]
- Zhang, J.; Han, Y.; Liu, C.; Ren, W.; Li, Y.; Wang, Q.; Su, N.; Li, Y.; Ma, B.; Ma, Y.; et al. Electrical Transport Properties of SnO under High Pressure. J. Phys. Chem. C 2011, 115, 20710–20715. [Google Scholar] [CrossRef]
- McLeod, J.A.; Lukoyanov, A.V.; Kurmaev, E.Z.; Finkelstein, L.D.; Moewes, A. Nature of the electronic states involved in the chemical bonding and superconductivity at high pressure in SnO. JETP Lett. 2011, 94, 142–146. [Google Scholar] [CrossRef] [Green Version]
- Zhuravlev, Y.N.; Korabelnikov, D.V. A first principles study of the mechanical, electronic, and vibrational properties of lead oxide. Phys. Solid State 2017, 59, 2296. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, D.R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 2017, 95, 239906(E). [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlipf, M.; Gygi, F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput. Phys. Commun. 2015, 196, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Avery, P.; Toher, C.; Curtarolo, S.; Zurek, E. XtalOpt Version r12: An open-source evolutionary algorithm for crystal structure prediction. Comput. Phys. Commun. 2019, 237, 274–275. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.; Ertural, C.; George, J.; Deringer, V.L.; Hautier, G.; Dronskowski, R. LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory. J. Comput. Chem. 2020, 41, 1931–1940. [Google Scholar] [CrossRef]
- Segev, E.; Argaman, U.; Abutbul, R.E.; Golan, Y.; Makov, G. A new cubic prototype structure in the IV–VI monochalcogenide system: A DFT study. Cryst. Eng. Comm. 2017, 19, 1751–1761. [Google Scholar] [CrossRef]
- Wang, J.; Umezawa, N.; Hosono, H. Mixed Valence Tin Oxides as Novel van der Waals Materials: Theoretical Predictions and Potential Applications. Adv. Energy Mater. 2016, 6, 1501190. [Google Scholar] [CrossRef]
- Klimeš, J.; Bowler, D.R.; Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef] [Green Version]
- Strehlow, W.H.; Cook, E.L. Compilation of energy band gaps in elemental and binary compound semiconductors and insulators. J. Phys. Chem. Ref. Data 1973, 2, 163. [Google Scholar] [CrossRef] [Green Version]
- Perry, D.L.; Wilkinson, T.J. Synthesis of high-purity α-and β-PbO and possible applications to synthesis and processing of other lead oxide materials. Appl. Phys. A 2007, 89, 77–80. [Google Scholar] [CrossRef]
- Perdew, J.P.; Levy, M. Physical content of the exact Kohn-Sham orbital energies: Band gaps and derivative discontinuities. Phys. Rev. Lett. 1983, 51, 1884. [Google Scholar] [CrossRef]
- Hussermann, U.; Berastegui, P.; Carlson, S.; Haines, J.; Leger, J.M. TlF and PbO under high pressure: Unexpected persistence of the stereochemically active electron pair. Angew. Chem. Int. Ed. 2001, 40, 4426–4429. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamics Theory of Crystal Lattices, 1st ed.; Oxford University Press: London, UK, 1954; pp. 129–154. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phy. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.; Payne, D.J.; Egdell, R.G.; Watson, G.W. Stereochemistry of post-transition metal oxides: Revision of the classical lone pair model. Chem. Soc. Rev. 2011, 40, 4455–4463. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Ef (eV/atom) | Structure | Lattice Parameters (Å) | No. of Atoms Unit Cell | Spacegroup (SG) and Wyckoff Atomic Position |
|---|---|---|---|---|---|
| P4nmm (Tetragonal) | 0 |  | a = b = 3.8699 c = 5.0426 | 4 | SG = 129 Sn 2c 0.25 0.25 0.2296 O 2a 0.75 0.25 0.0 |
| P21m (Monoclinic) | 0.0019 |  | a = 3.9128 b = 3.8225 c = 4.9846 β = 90.339° | 4 | SG = 11 Sn 2e 0.7515 0.25 0.2316 O 2e 0.2499 0.25 0.0045 |
| Pmmn (Orthorhombic) | 0.0043 | 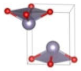 | a = 3.7826 b = 3.9520 c = 4.9417 | 4 | SG = 59 Sn 2b 0.25 0.75 0.2333 O 2a 0.25 0.25 0.0081 |
| Pbcm (Orthorhombic) | 0.0485 | 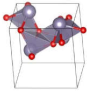 | a = 6.0846 b = 5.6618 c = 4.5270 | 8 | SG = 57 Sn 4d 0.7179 0.4774 0.25 O 4d 0.6161 0.1068 0.25 |
| P21/c (Monoclinic) | 0.0583 | 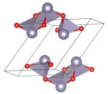 | a = 6.0443 b = 4.2685 c = 6.3114 β = 109.3927° | 8 | SG = 14 Sn 4e 0.7572 −0.0025 0.7682 O 4e 0.8826 0.1828 0.0965 |
| P213 (Cubic) | 0.084 | 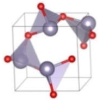 | a = b = c = 5.3448 | 8 | SG = 198 Sn 4a 0.4891 0.4891 0.4891 O 4a 0.8809 0.8809 0.8809 |
| P4/nmm (Tetragonal) | 0.151 |  | a = b = 3.4079 c = 6.2247 | 4 | SG = 198 Sn 2c 0.25 0.25 0.2199 O 2c 0.25 0.25 0.8758 |
| Phase | Ef (eV/Atom) | Structure | Lattice Parameters (Å) | No. of Atoms Unit Cell | Spacegroup (SG) and Wyckoff Atomic Position |
|---|---|---|---|---|---|
| P4nmm (Tetragonal) | 0 | 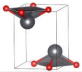 | a = b = 4.0707 c = 5.4175 | 4 | SG = 129 Pb 2c 0.25 0.25 0.2186 O 2a 0.75 0.25 0.0 |
| Pmmn (Orthorhombic) | 0.0009 | 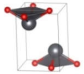 | a = 3.9670 b = 4.1654 c = 5.5171 | 4 | SG = 59 Pb 2a 0.25 0.25 0.7851 O 2b 0.25 0.75 −0.0096 |
| P421m (Tetragonal) | 0.042 | 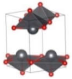 | a = b = 5.134 c = 6.4102 | 8 | SG = 113 Pb 4e 0.7588 0.2588 0.7867 O 4e 0.7145 0.2145 0.1348 |
| C2 (Monoclinic) | 0.043 | 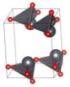 | a = 4.9607 b = 5.2977 c = 6.4035 β = 91.995° | 8 | SG = 5 Pb 4c 0.7414 0.0642 0.7849 O 4c 0.2437 0.1338 0.8664 |
| Pmn21 (Orthorhombic) | 0.044 |  | a = 3.5834 b = 6.2899 c = 3.7034 | 4 | SG = 31 Pb 2a 0.6425 0.6421 0.1415 O 2a 0.7560 0.7437 0.7562 |
| P213 (Cubic) | 0.046 | 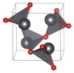 | a = b = c = 5.522 | 8 | SG = 198 Pb 4a 0.2437 0.2437 0.2437 O 4a 0.6420 0.6420 0.6420 |
| P42 (Tetragonal) | 0.0998 | 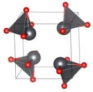 | a = b = 5.5937 c = 5.2962 | 8 | SG = 77 Pb 4d 0.7702 0.7763 0.2180 O 4d 0.8184 0.1902 0.2811 |
| SnO | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | E | ν |
| Pmn21 2.5 GPa | 114.6 | 18.0 | 78.5 | 58.0 | 27.1 | 112.7 | 34.9 | 84.1 | 34.7 | 52.6 | 36.1 | 88.0 | 0.221 |
| Pmmn 20 GPa | 167.2 | 101.5 | 103.6 | 207.4 | 189.6 | 203.5 | 123.6 | 83.8 | 82.7 | 146.5 | 49.1 | 130.5 | 0.330 |
| Pbcm 80 GPa | 241.8 | 143.2 | 113.5 | 585.8 | 252.3 | 396.7 | 85.6 | 85.6 | 132.8 | 224.7 | 94.5 | 248.6 | 0.316 |
| PbO | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | E | ν |
| Pmn21 1 GPa | 20.5 | 13.8 | 15.7 | 73.9 | 45.5 | 75.1 | 53.5 | 17.5 | 16.2 | 27.9 | 19.8 | 47.9 | 0.209 |
| Pmmn 2 GPa | 88.8 | 77.3 | 32.9 | 91.8 | 38.3 | 35.3 | 28.5 | 25.7 | 63.9 | 45.6 | 21.1 | 54.8 | 0.298 |
| Pbcm 5 GPa | 161.1 | 49.1 | 43.0 | 90.8 | 26.2 | 84.5 | 11.4 | 18.9 | 42.3 | 59.8 | 67.4 | 25.6 | 0.311 |
| P4/nmm 50 GPa | 355.3 | 196.9 | 241.6 | - | - | 435.7 | 59.5 | - | 167.9 | 268.2 | 83.6 | 226.9 | 0.358 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, L.T.; Makov, G. High-Pressure Phases of SnO and PbO: A Density Functional Theory Combined with an Evolutionary Algorithm Approach. Materials 2021, 14, 6552. https://doi.org/10.3390/ma14216552
Nguyen LT, Makov G. High-Pressure Phases of SnO and PbO: A Density Functional Theory Combined with an Evolutionary Algorithm Approach. Materials. 2021; 14(21):6552. https://doi.org/10.3390/ma14216552
Chicago/Turabian StyleNguyen, Long Truong, and Guy Makov. 2021. "High-Pressure Phases of SnO and PbO: A Density Functional Theory Combined with an Evolutionary Algorithm Approach" Materials 14, no. 21: 6552. https://doi.org/10.3390/ma14216552
APA StyleNguyen, L. T., & Makov, G. (2021). High-Pressure Phases of SnO and PbO: A Density Functional Theory Combined with an Evolutionary Algorithm Approach. Materials, 14(21), 6552. https://doi.org/10.3390/ma14216552