
Carbonaceous Materials Porosity Investigation in a Wet State by Low-Field NMR Relaxometry



Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of the Mesoporous Carbon Material
2.2. Thermoporometry Analysis
2.3. Adsorption of Nitrogen
2.4. H-NMR Relaxometry Analysis
2.5. NMR Data Evaluation Procedure
3. Results and Discussion
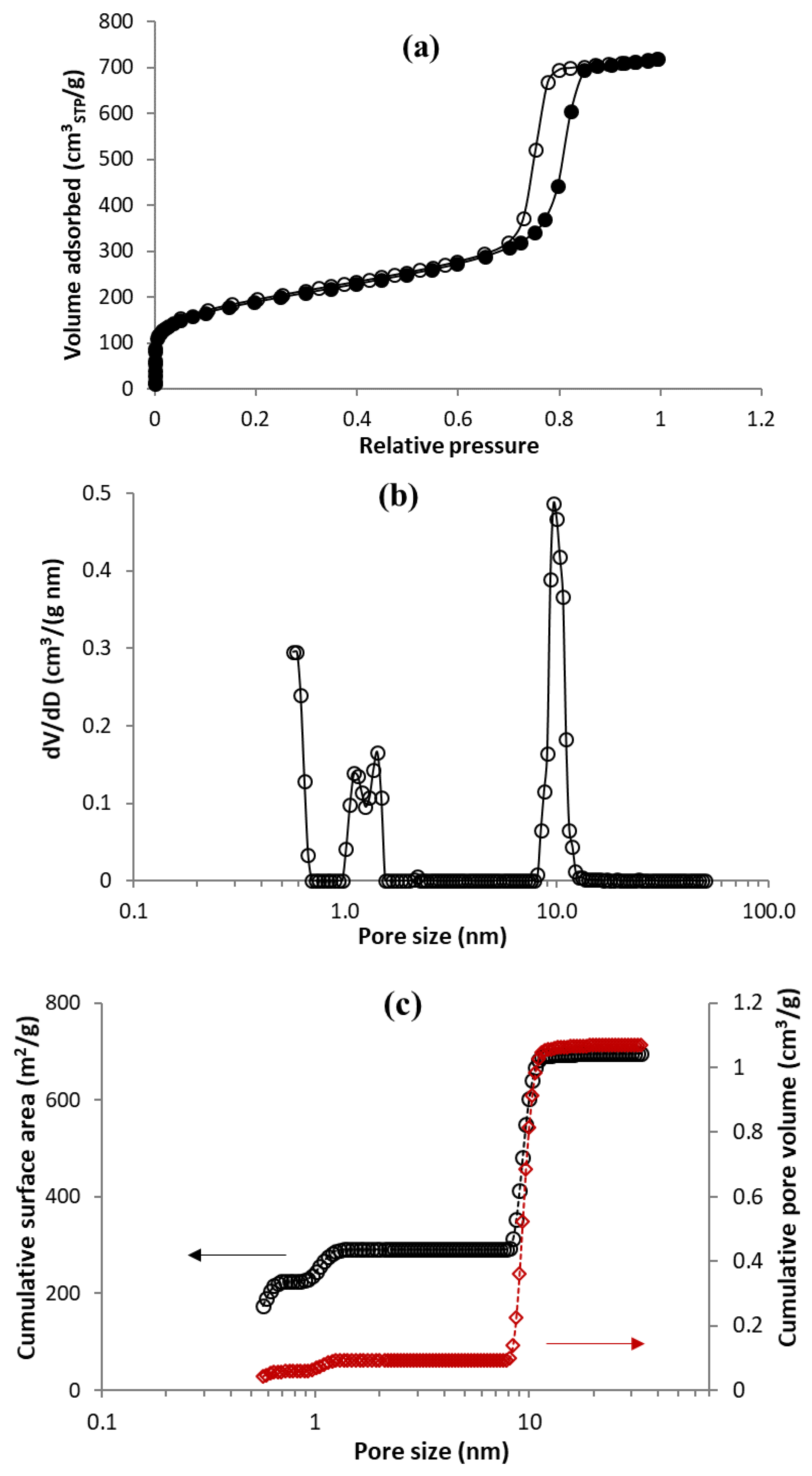
3.1. Characterization of MC Sample by TPM and N2 Physisorption
3.2. 1H-NMR Relaxation Measurements
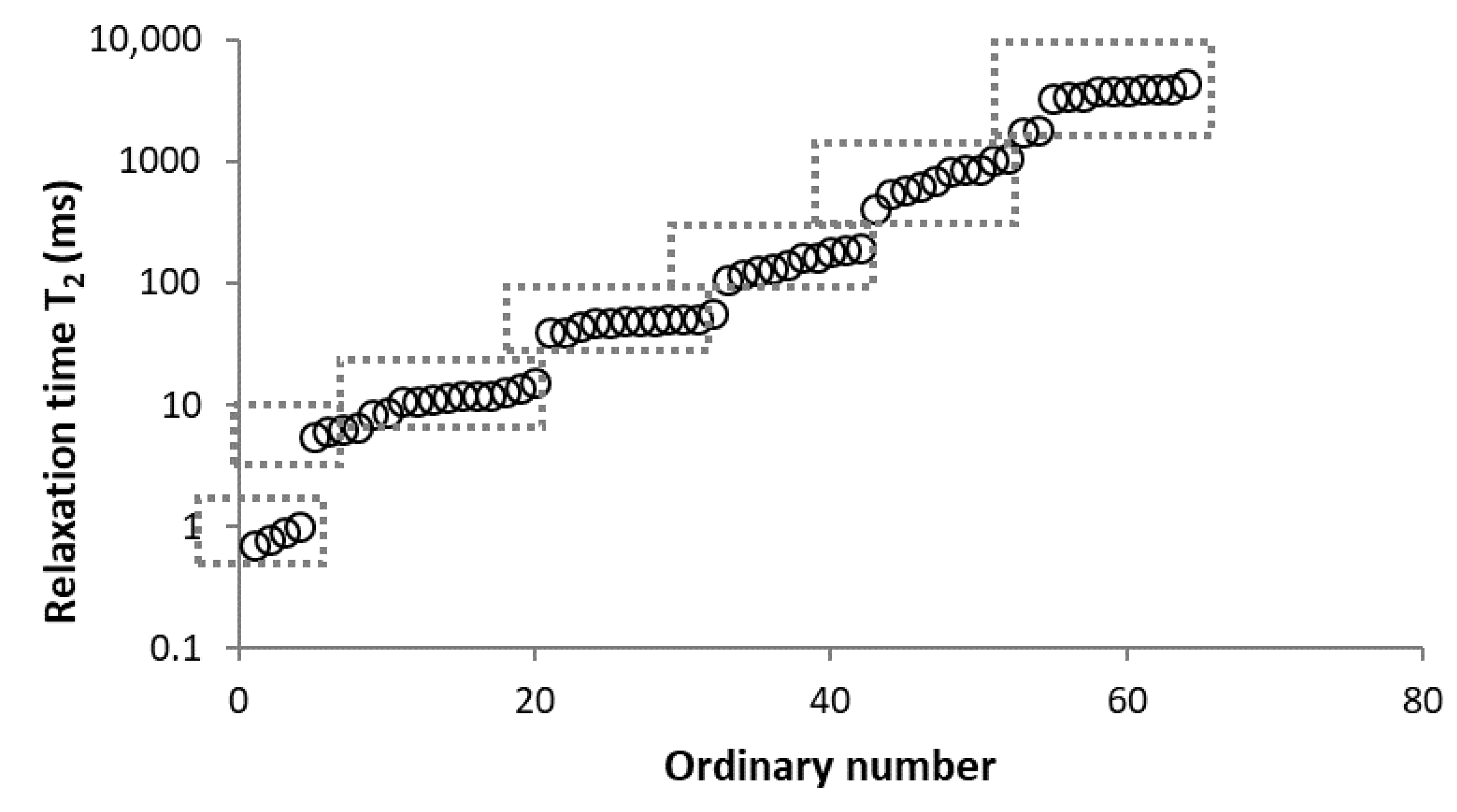
3.2.1. Determination of Number of Components
3.2.2. Analysis of Found Components
3.2.3. Estimation of Surface Relaxivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Stein, A.; Wang, Z.; Fierke, M.A. Functionalization of Porous Carbon Materials with Designed Pore Architecture. Adv. Mater. 2009, 21, 265–293. [Google Scholar] [CrossRef]
- Perrier, L.; Pijaudier-Cabot, G.; Grégoire, D. Extended Poromechanics for Adsorption-Induced Swelling Prediction in Double Porosity Media: Modeling and Experimental Validation on Activated Carbon. Int. J. Solids Struct. 2018, 146, 192–202. [Google Scholar] [CrossRef]
- Bulavová, P.; Parmentier, J.; Slovák, V. Facile Synthesis of Soft-Templated Carbon Monoliths with Hierarchical Porosity for Fast Adsorption from Liquid Media. Microporous Mesoporous Mater. 2018, 272, 155–165. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Springer Science & Business Media: Dordrecht, The Netherlands, 2006. [Google Scholar] [CrossRef]
- Balzer, C.; Braxmeier, S.; Neimark, A.V.; Reichenauer, G. Deformation of Microporous Carbon during Adsorption of Nitrogen, Argon, Carbon Dioxide, and Water Studied by in Situ Dilatometry. Langmuir 2015, 31, 12512–12519. [Google Scholar] [CrossRef]
- Thommes, M.; Cychosz, K.A. Physical Adsorption Characterization of Nanoporous Materials: Progress and Challenges. Adsorption 2014, 20, 233–250. [Google Scholar] [CrossRef]
- Veselá, P.; Riikonen, J.; Nissinen, T.; Lehto, V.-P.; Slovák, V. Optimisation of Thermoporometry Measurements to Evaluate Mesoporous Organic and Carbon Xero-, Cryo- and Aerogels. Thermochim. Acta 2015, 621, 81–89. [Google Scholar] [CrossRef]
- Landry, M.R. Thermoporometry by Differential Scanning Calorimetry: Experimental Considerations and Applications. Thermochim. Acta 2005, 433, 27–50. [Google Scholar] [CrossRef]
- Dessources, A.H.; Hartmann, S.; Baba, M.; Huesing, N.; Nedelec, J.M. Multiscale Characterization of Hierarchically Organized Porous Hybrid Materials. J. Mater. Chem. 2012, 22, 2713–2720. [Google Scholar] [CrossRef][Green Version]
- Cides da Silva, L.C.; Araújo, G.L.B.; Segismundo, N.R.; Moscardini, E.F.; Mercuri, L.P.; Cosentino, I.C.; Fantini, M.C.A.; Matos, J.R. DSC Estimation of Structural and Textural Parameters of SBA-15 Silica Using Water Probe. J. Therm. Anal. Calorim. 2009, 97, 701–704. [Google Scholar] [CrossRef]
- Kloetstra, K.R.; Zandbergen, H.W.; van Koten, M.A.; van Bekkum, H. Thermoporometry as a New Tool in Analyzing Mesoporous MCM-41 Materials. Catal. Lett. 1995, 33, 145–156. [Google Scholar] [CrossRef]
- Ishikiriyama, K.; Todoki, M.; Motomura, K. Pore Size Distribution (PSD) Measurements of Silica Gels by Means of Differential Scanning Calorimetry: I. Optimization for Determination of PSD. J. Colloid Interface Sci. 1995, 171, 92–102. [Google Scholar] [CrossRef]
- Takuji, Y.; Akira, E.; Takao, O.; Masaru, N. Characterization of Mesoporous Silicas with Uniform and Cylindrical Pores by Thermoporometry. In Asian Pacific Confederation of Chemical Engineering Congress Program and Abstracts Asian Pacific Confederation of Chemical Engineers Congress Program and Abstracts; The Society of Chemical Engineers: Tsukuba, Japan, 2004; p. 73. [Google Scholar]
- Zelenková, G.; Zelenka, T.; Slovák, V. Thermoporometry of Porous Carbon: The Effect of the Carbon Surface Chemistry on the Thickness of Non-Freezable Pore Water Layer (Delta Layer). Microporous Mesoporous Mater. 2021, 326, 111358. [Google Scholar] [CrossRef]
- Krutyeva, M.; Grinberg, F.; Furtado, F.; Galvosas, P.; Kärger, J.; Silvestre-Albero, A.; Sepulveda-Escribano, A.; Silvestre-Albero, J.; Rodríguez-Reinoso, F. Characterization of Carbon Materials with the Help of NMR Methods. Microporous Mesoporous Mater. 2009, 120, 91–97. [Google Scholar] [CrossRef]
- Aksnes, D.W.; Førland, K.; Kimtys, L. Pore Size Distribution in Mesoporous Materials as Studied by 1H NMR. Phys. Chem. Chem. Phys. 2001, 3, 3203–3207. [Google Scholar] [CrossRef]
- Camaiti, M.; Bortolotti, V.; Fantazzini, P. Stone Porosity, Wettability Changes and Other Features Detected by MRI and NMR Relaxometry: A More than 15-Year Study: Magnetic Resonance for Fluids in Porous Media and Cultural Heritage. Magn. Reson. Chem. 2015, 53, 34–47. [Google Scholar] [CrossRef]
- Schlienger, S.; Ducrot-Boisgontier, C.; Delmotte, L.; Guth, J.-L.; Parmentier, J. History of the Micelles: A Key Parameter for the Formation Mechanism of Ordered Mesoporous Carbons via a Polymerized Mesophase. J. Phys. Chem. C 2014, 118, 11919–11927. [Google Scholar] [CrossRef]
- Tananuwong, K.; Reid, D. DSC and NMR Relaxation Studies of Starch? Water Interactions during Gelatinization. Carbohydr. Polym. 2004, 58, 345–358. [Google Scholar] [CrossRef]
- Meng, X.; Foston, M.; Leisen, J.; DeMartini, J.; Wyman, C.E.; Ragauskas, A.J. Determination of Porosity of Lignocellulosic Biomass before and after Pretreatment by Using Simons’ Stain and NMR Techniques. Bioresour. Technol. 2013, 144, 467–476. [Google Scholar] [CrossRef]
- Hansen, E.W.; Fonnum, G.; Weng, E. Pore Morphology of Porous Polymer Particles Probed by NMR Relaxometry and NMR Cryoporometry. J. Phys. Chem. B 2005, 109, 24295–24303. [Google Scholar] [CrossRef] [PubMed]
- Stingaciu, L.R.; Pohlmeier, A.; Blümler, P.; Weihermüller, L.; van Dusschoten, D.; Stapf, S.; Vereecken, H. Characterization of Unsaturated Porous Media by High-Field and Low-Field NMR Relaxometry: Porous media investigation by NMR. Water Resour. Res. 2009, 45, 8412. [Google Scholar] [CrossRef]
- Kleinberg, R.L. Pore Size Distributions, Pore Coupling, and Transverse Relaxation Spectra of Porous Rocks. Magn. Reson. Imaging 1994, 12, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, F.; Bowe, S.; Van As, H.; Schaumann, G.E. Evaluation of 1H NMR Relaxometry for the Assessment of Pore-Size Distribution in Soil Samples. Eur. J. Soil Sci. 2009, 60, 1052–1064. [Google Scholar] [CrossRef]
- Bayer, J.V.; Jaeger, F.; Schaumann, G.E. Proton Nuclear Magnetic Resonance (NMR) Relaxometry in Soil Science Applications. Open Magn. Reson. J. 2010, 3, 15–26. [Google Scholar] [CrossRef]
- Hinedi, Z.R.; Kabala, Z.J.; Skaggs, T.H.; Borchardt, D.B.; Lee, R.W.K.; Chang, A.C. Probing Soil and Aquifer Material Porosity with Nuclear Magnetic Resonance. Water Resour. Res. 1993, 29, 3861–3866. [Google Scholar] [CrossRef]
- Votrubová, J.; Šanda, M.; Císlerová, M.; Gao Amin, M.H.; Hall, L.D. The Relationships between MR Parameters and the Content of Water in Packed Samples of Two Soils. Geoderma 2000, 95, 267–282. [Google Scholar] [CrossRef]
- Bardenhagen, I.; Dreher, W.; Fenske, D.; Wittstock, A.; Bäumer, M. Fluid Distribution and Pore Wettability of Monolithic Carbon Xerogels Measured by 1H NMR Relaxation. Carbon 2014, 68, 542–552. [Google Scholar] [CrossRef]
- Norinaga, K.; Hayashi, J.; Kudo, N.; Chiba, T. Evaluation of Effect of Predrying on the Porous Structure of Water-Swollen Coal Based on the Freezing Property of Pore Condensed Water. Energy Fuels 1999, 13, 1058–1066. [Google Scholar] [CrossRef]
- Fairhurst, D.; Cosgrove, T.; Prescott, S.W. Relaxation NMR as a Tool to Study the Dispersion and Formulation Behavior of Nanostructured Carbon Materials: Relaxation NMR for Nanostructured Carbons. Magn. Reson. Chem. 2016, 54, 521–526. [Google Scholar] [CrossRef]
- Krzyżak, A.T.; Habina, I. Low Field 1H NMR Characterization of Mesoporous Silica MCM-41 and SBA-15 Filled with Different Amount of Water. Microporous Mesoporous Mater. 2016, 231, 230–239. [Google Scholar] [CrossRef]
- Krzyżak, A.T.; Mazur, W.; Matyszkiewicz, J.; Kochman, A. Identification of Proton Populations in Cherts as Natural Analogues of Pure Silica Materials by Means of Low Field NMR. J. Phys. Chem. C 2020, 124, 5225–5240. [Google Scholar] [CrossRef]
- Matei Ghimbeu, C.; Le Meins, J.-M.; Zlotea, C.; Vidal, L.; Schrodj, G.; Latroche, M.; Vix-Guterl, C. Controlled Synthesis of NiCo Nanoalloys Embedded in Ordered Porous Carbon by a Novel Soft-Template Strategy. Carbon 2014, 67, 260–272. [Google Scholar] [CrossRef]
- Bakhmutov, V.I. Practical NMR Relaxation for Chemists; Wiley: Chichester, UK; Hoboken, NJ, USA, 2004. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Fleury, M.; Kohler, E.; Norrant, F.; Gautier, S.; M’Hamdi, J.; Barré, L. Characterization and Quantification of Water in Smectites with Low-Field NMR. J. Phys. Chem. C 2013, 117, 4551–4560. [Google Scholar] [CrossRef]
- Belotti, M.; Martinelli, A.; Gianferri, R.; Brosio, E. A Proton NMR Relaxation Study of Water Dynamics in Bovine Serum Albumin Nanoparticles. Phys Chem Chem Phys 2010, 12, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Kleinberg, R.L. Utility of NMR T2 Distributions, Connection with Capillary Pressure, Clay Effect, and Determination of the Surface Relaxivity Parameter Rho 2. Magn. Reson. Imaging 1996, 14, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Liaw, H.-K.; Kulkarni, R.; Chen, S.; Watson, A.T. Characterization of Fluid Distributions in Porous Media by NMR Techniques. AIChE J. 1996, 42, 538–546. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mass of Dry Sample (g) | Mass of H2O (g) | Mass Ratio H2O/C |
|---|---|---|---|
| MC-0-A | 0.1368 | 0 | 0 |
| MC-0-B | 0.1407 | 0 | 0 |
| MC-30-A | 0.1108 | 0.0024 | 0.02 |
| MC-30-B | 0.1142 | 0.0024 | 0.02 |
| MC-100-A | 0.1536 | 0.0391 | 0.25 |
| MC-100-B | 0.1520 | 0.0387 | 0.25 |
| MC-1:2 | 0.1451 | 0.2939 | 2.03 |
| MC-1:4-A | 0.1099 | 0.4341 | 3.95 |
| MC-1:4-B | 0.1173 | 0.4643 | 3.96 |
| MC-1:4-C | 0.1080 | 0.4342 | 4.02 |
| MC-1:6 | 0.0899 | 0.5393 | 6.00 |
| Method | Vultramicro (cm3/g) | Sultramicro (m2/g) | Vsupermicro (cm3/g) | Ssupermicro (m2/g) | Vmeso (cm3/g) | Smeso (m2/g) | Mesopore Diameter (*) (nm) |
|---|---|---|---|---|---|---|---|
| TPM | n/a | n/a | n/a | n/a | 0.92 | n/a | 14 |
| N2 adsorption | 0.06 | 225 | 0.04 | 65 | 0.98 | 405 | 9 |
| Number of Comp. | Relaxation Curve | Residues | Calculated Parameters | |
|---|---|---|---|---|
| A (%) | T2 (ms) | |||
| 1 |  R2 = 0.888 |  | 100 | 2495.5 |
| 2 |  R2 = 0.996 |  | 67.8 32.2 | 49.0 3320.5 |
| 3 |  R2 = 0.999 |  | 43.4 27.9 28.6 | 17.5 113.9 3534.6 |
| 4 |  R2 = 0.999 |  | 32.2 69.5 5.2 27.5 | 11.9 35.1 429.5 3711.6 |
| 5 |  R2 = 0.999 |  | 22.8 30.8 16.7 3.4 26.3 | 8.4 39.9 128.2 1028.2 3861.2 |
| Sample (Measurement) | τ (ms) | Number of Components | T2 (ms) |
|---|---|---|---|
| MC-100 (1) | 0.05 | 2 | 5.5 |
| MC-100 (2) | 0.05 | 2 | 6.2 |
| MC-100 (3) | 0.05 | 2 | 5.5 |
| MC-1:2 (1) | 0.2 | 3 | 20.2 |
| MC-1:2 (2) | 0.05 | 4 | 22.4 |
| MC-1:4-A (1) | 0.2 | 5 | 1353 |
| MC-1:4-A (2) | 0.2 | 5 | 1228 |
| MC-1:4-A (3) | 0.05 | 5 | 1386 |
| MC-1:4-A (4) | 0.05 | 5 | 1413 |
| MC-1:4-B (1) | 0.2 | 5 | 1845 |
| MC-1:4-B (2) | 0.05 | 5 | 1620 |
| MC-1:4-C (1) | 0.2 | 5 | 1299 |
| MC-1:4-C (2) | 0.05 | 5 | 1209 |
| MC-1:6 (1) | 0.2 | 5 | 1086 |
| MC-1:6 (2) | 0.05 | 5 | 1129 |
| Interval T2 (ms) | <2 | 2–7 | 7–20 | 20–80 | 80–350 | 350–1500 | Above 1500 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | A (%) | T2 (ms) | A (%) | T2 (ms) | A (%) | T2 (ms) | A (%) | T2 (ms) | A (%) | T2 (ms) | A (%) | T2 (ms) | A (%) | T2 (ms) |
| MC-100 (1) | 14.1 | 0.9 | 85.9 | 6.2 | ||||||||||
| MC-100 (2) | 7.2 | 0.7 | 92.8 | 6.6 | ||||||||||
| MC-100 (3) | 15.2 | 1.0 | 84.8 | 6.3 | ||||||||||
| MC-1:2 (1) | 18.2 | 0.8 | 55.9 | 13.9 | 24.4 | 50.3 | 1.5 | 1781 | ||||||
| MC-1:2 (2) | 14.9 | 5.6 | 58.0 | 15.4 | 25.3 | 49.8 | 1.8 | 1758 | ||||||
| MC-1:4-A (1) | 18.8 | 11.8 | 25.3 | 50.8 | 15.7 | 195.6 | 7.6 | 823 | 32.6 | 3818 | ||||
| MC-1:4-A (2) | 19.8 | 11.7 | 25.3 | 51.1 | 13.2 | 188.1 | 8.0 | 874 | 33.7 | 3318 | ||||
| MC-1:4-A (3) | 18.4 | 10.9 | 23.9 | 47.4 | 15.0 | 165.1 | 8.5 | 592 | 34.1 | 3806 | ||||
| MC-1:4-A (4) | 19.4 | 11.3 | 25.2 | 48.0 | 14.1 | 162.1 | 7.5 | 697 | 33.8 | 3916 | ||||
| MC-1:4-B (1) | 19.5 | 13.0 | 27.4 | 56.9 | 7.7 | 182.6 | 5.3 | 1051 | 40.1 | 4382 | ||||
| MC-1:4-B (2) | 17.8 | 10.6 | 22.1 | 43.8 | 14.3 | 105.4 | 4.1 | 410 | 41.6 | 3789 | ||||
| MC-1:4-C (1) | 19.1 | 12.1 | 23.4 | 48.9 | 17.6 | 139.8 | 4.3 | 852 | 35.5 | 3449 | ||||
| MC-1:4-C (2) | 19.9 | 12.1 | 23.4 | 49.0 | 17.7 | 132.4 | 5.3 | 551 | 33.7 | 3390 | ||||
| MC-1:6 (1) | 22.8 | 8.4 | 30.8 | 39.9 | 16.7 | 128.2 | 3.4 | 1028 | 26.3 | 3861 | ||||
| MC-1:6 (2) | 21.7 | 8.7 | 30.6 | 39.9 | 17.5 | 117.3 | 3.1 | 633 | 27.1 | 3965 | ||||
| Average | 0.9 | 6.2 | 11.7 | 48.0 | 151.7 | 751 | 3436 | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinnertová, E.; Slovák, V.; Zelenka, T.; Vaulot, C.; Delmotte, L. Carbonaceous Materials Porosity Investigation in a Wet State by Low-Field NMR Relaxometry. Materials 2022, 15, 9021. https://doi.org/10.3390/ma15249021
Kinnertová E, Slovák V, Zelenka T, Vaulot C, Delmotte L. Carbonaceous Materials Porosity Investigation in a Wet State by Low-Field NMR Relaxometry. Materials. 2022; 15(24):9021. https://doi.org/10.3390/ma15249021
Chicago/Turabian StyleKinnertová, Eva, Václav Slovák, Tomáš Zelenka, Cyril Vaulot, and Luc Delmotte. 2022. "Carbonaceous Materials Porosity Investigation in a Wet State by Low-Field NMR Relaxometry" Materials 15, no. 24: 9021. https://doi.org/10.3390/ma15249021
APA StyleKinnertová, E., Slovák, V., Zelenka, T., Vaulot, C., & Delmotte, L. (2022). Carbonaceous Materials Porosity Investigation in a Wet State by Low-Field NMR Relaxometry. Materials, 15(24), 9021. https://doi.org/10.3390/ma15249021