
Theoretical Investigations into the Different Properties of Al-Based Fluoroperovskite AlMF3 (M = Cr, B) Compounds by the TB-MBJ Potential Method

 , and
, and 
Abstract
:1. Introduction
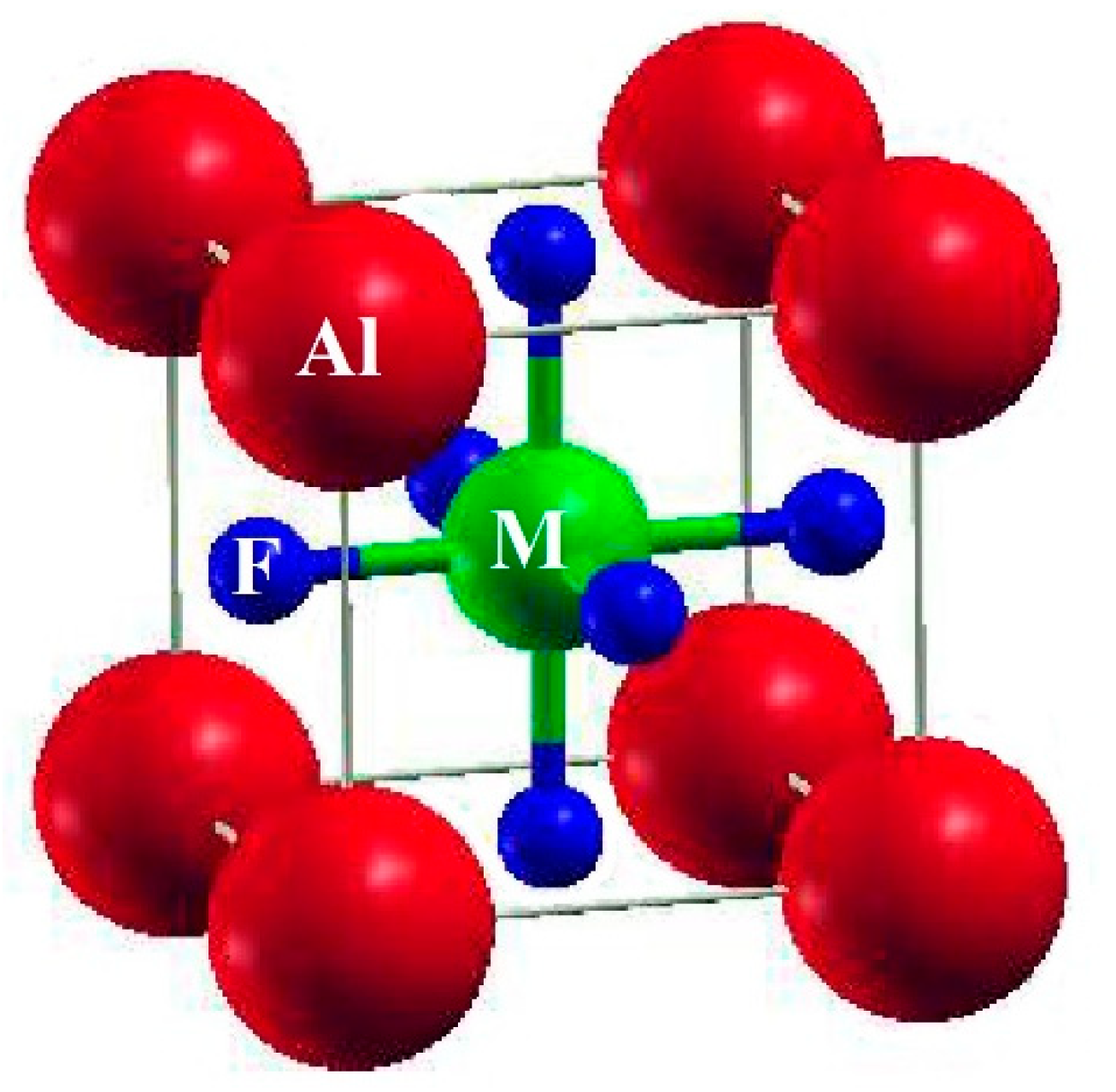
2. Materials and Methods
3. Results
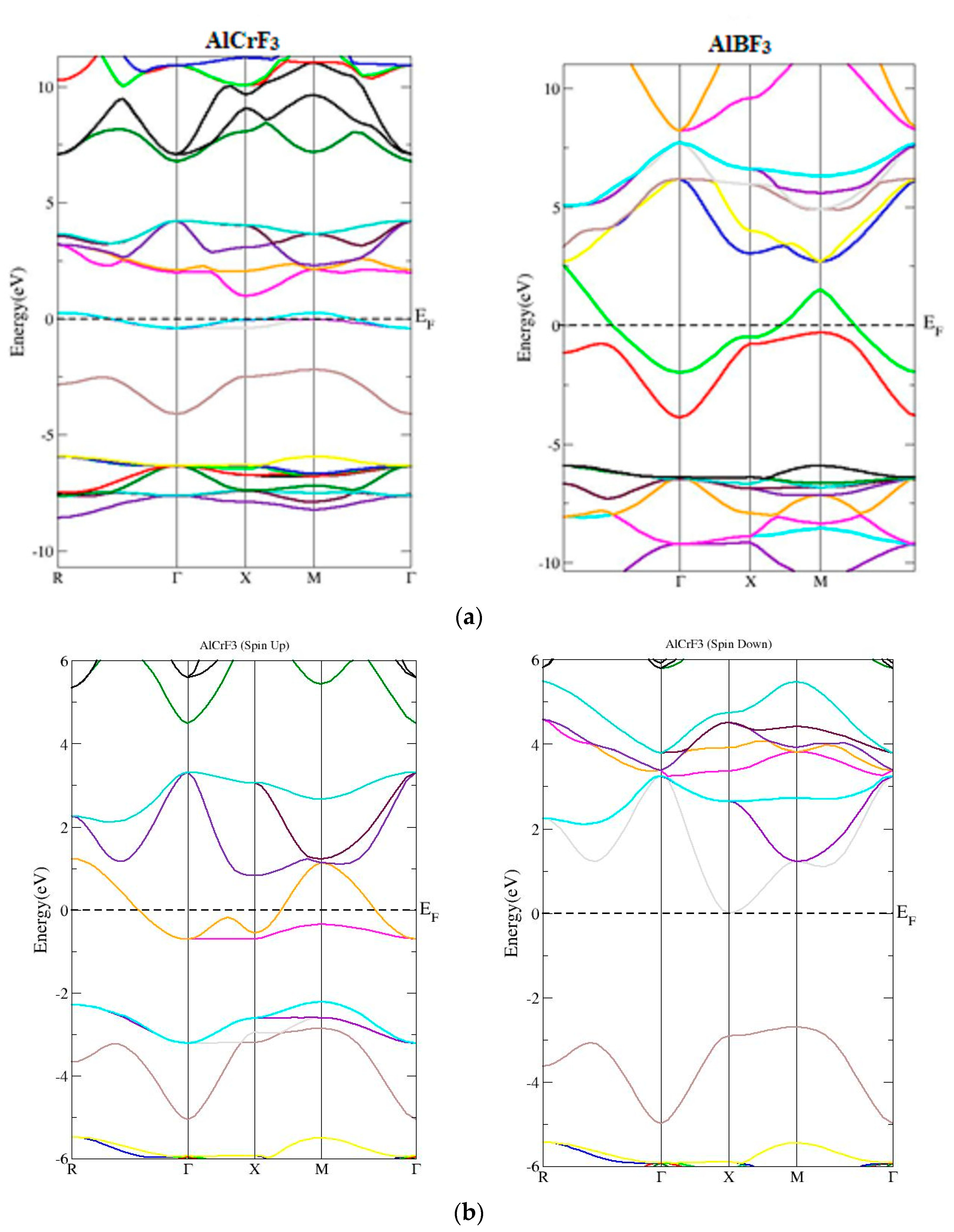
3.1. Electronic Properties
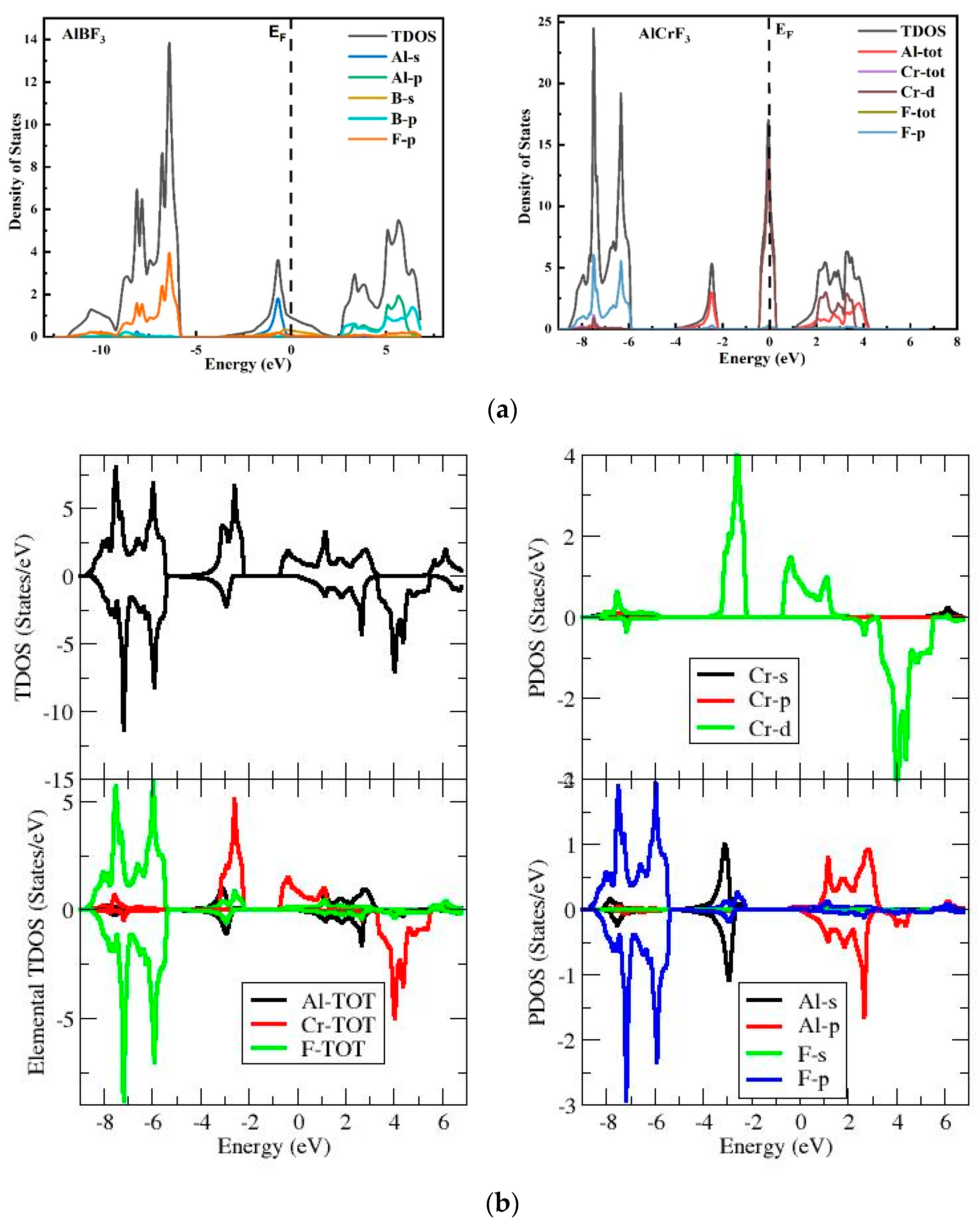
3.2. The DOS (Density of States)
3.3. Elastic Properties
3.4. Optical Properties
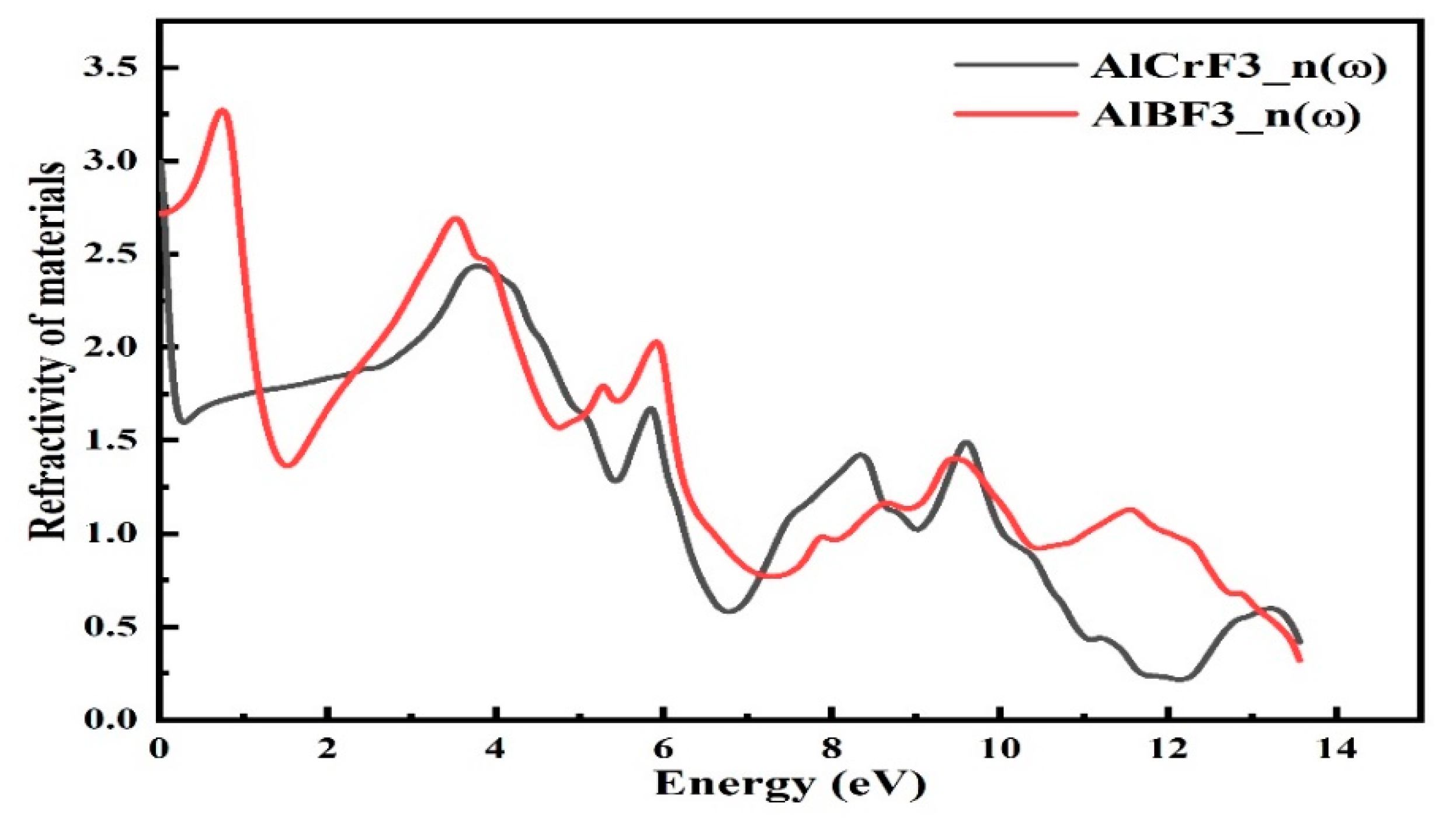
3.4.1. Refractive Index
3.4.2. Absorption Coefficient
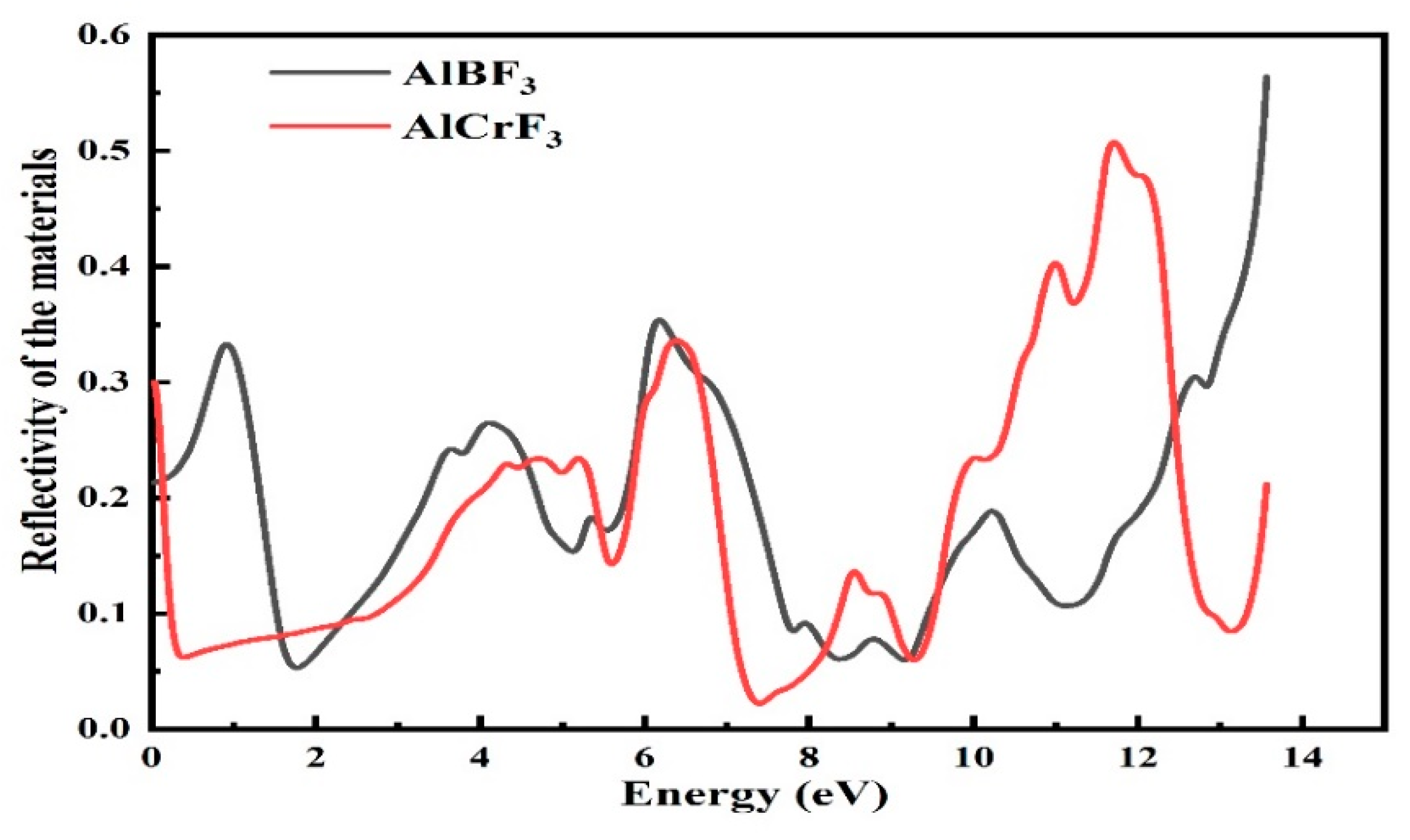
3.4.3. Reflectivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishimatsu, T.; Terakubo, N.; Mizuseki, H.; Kawazoe, Y.; Pawlak, D.A.; Shimamura, K.; Fukuda, T. Band structures of perovskite-like fluorides for vacuum-ultraviolet-transparent lens materials. Jpn. J. Appl. Phys. 2002, 41, L365. [Google Scholar] [CrossRef]
- Husain, M.; Rahman, N.; Reshak, A.H.; Zulfiqar; Habib, A.; Ali, S.; Laref, A.; Al Bakri, A.M.M.; Bila, J. Insight into the physical properties of the inter-metallic titanium-based binary compounds. Eur. Phys. J. Plus 2021, 136, 624. [Google Scholar] [CrossRef]
- Rahman, N.; Husain, M.; Yang, J.; Sajjad, M.; Murtaza, G.; Ul Haq, M.; Habib, A.; Rauf, A.; Karim, A.; Nisar, M.; et al. First principle study of structural, electronic, optical and mechanical properties of cubic fluoro-perovskites: (CdXF3, X = Y, Bi). Eur. Phys. J. Plus 2021, 136, 347. [Google Scholar] [CrossRef]
- Rahman, N.; Yang, J.; Sohail, M.; Khan, R.; Iqbal, A.; Maouche, C.; Khan, A.A.; Husain, M.; Khattak, S.A.; Khan, S.N.; et al. Insight into metallic oxide semiconductor (SnO2, ZnO, CuO, α-Fe2O3, WO3)-carbon nitride (g-C3N4) heterojunction for gas sensing application. Sens. Actuators A Phys. 2021, 332, 113128. [Google Scholar] [CrossRef]
- Murtaza, G.; Sadique, G.; Aliabad, H.R.; Khalid, M.; Naeem, S.; Afaq, A.; Amin, B.; Ahmad, I. First principle study of cubic perovskites: AgTF3 (T = Mg, Zn). Phys. B Condens. Matter 2011, 406, 4584–4589. [Google Scholar] [CrossRef]
- Körbel, S.; Marques, M.A.L.; Botti, S. Stability and electronic properties of new inorganic perovskites from high-throughput ab initio calculations. J. Mater. Chem. C 2016, 4, 3157–3167. [Google Scholar] [CrossRef]
- Dubrovin, R.M.; Alyabyeva, L.N.; Siverin, N.V.; Gorshunov, B.P.; Novikova, N.N.; Boldyrev, K.N.; Pisarev, R.V. Incipient multiferroicity in P n m a fluoroperovskite NaMnF3. Phys. Rev. B 2020, 101, 180403. [Google Scholar] [CrossRef]
- Kumar, Y.A.; Kumar, K.D.; Kim, H.J. A novel electrode for supercapacitors: Efficient PVP-assisted synthesis of Ni3S2 nanostructures grown on Ni foam for energy storage. Dalton Trans. 2020, 49, 4050–4059. [Google Scholar] [CrossRef]
- Pallavolu, M.R.; Kumar, Y.A.; Mani, G.; Alshgari, R.A.; Ouladsmane, M.; Joo, S.W. Facile fabrication of novel heterostructured tin disulfide (SnS2)/tin sulfide (SnS)/N-CNO composite with improved energy storage capacity for high-performance supercapacitors. J. Electroanal. Chem. 2021, 899, 115695. [Google Scholar] [CrossRef]
- Pallavolu, M.R.; Kumar, Y.A.; Mani, G.; Nallapureddy, R.R.; Parvathala, A.; Albaqami, M.D.; Karami, A.M.; Joo, S.W. A novel hybridized needle-like Co3O4/N-CNO composite for superior energy storage asymmetric supercapacitors. J. Alloys Compd. 2022, 908, 164447. [Google Scholar] [CrossRef]
- Mubarak, A.A. Ab initio Study of Ag-Based Fluoroperovskite AgMF3 (M = Co and Ni) Compounds. J. Electron. Mater. 2018, 47, 887–898. [Google Scholar] [CrossRef]
- Bouich, A.; Marí-Guaita, J.; Sahraoui, B.; Palacios, P.; Marí, B. Tetrabutylammonium (TBA)-Doped Methylammonium Lead Iodide: High Quality and Stable Perovskite Thin Films. Front. Energy Res. 2012, 10, 840817. [Google Scholar] [CrossRef]
- Arar, R.; Ouahrani, T.; Varshney, D.; Khenata, R.; Murtaza, G.; Rached, D.; Bouhemadou, A.; Al-Douri, Y.; Bin Omran, S.; Reshak, A. Structural, mechanical and electronic properties of sodium based fluoroperovskites NaXF3 (X = Mg, Zn) from first-principle calculations. Mater. Sci. Semicond. Process. 2015, 33, 127–135. [Google Scholar] [CrossRef]
- Abdullah, A.; Husain, M.; Rahman, N.; Khan, R.; Iqbal, Z.; Zulfiqar, S.; Sohail, M.; Umer, M.; Murtaza, G.; Khan, S.N.; et al. Computational investigation of structural, magnetic, elastic, and electronic properties of Half-Heusler ScVX (X = Si, Ge, Sn, and Pb) compounds. Eur. Phys. J. Plus 2021, 136, 1176. [Google Scholar] [CrossRef]
- Chouit, N.; Korba, S.A.; Slimani, M.; Meradji, H.; Ghemid, S.; Khenata, R. First-principles study of the structural, electronic and thermal properties of CaLiF3. Phys. Scr. 2013, 88, 035702. [Google Scholar] [CrossRef]
- Seddik, T.; Khenata, R.; Merabiha, O.; Bouhemadou, A.; Bin-Omran, S.; Rached, D. Elastic, electronic and thermodynamic properties of fluoro-perovskite KZnF 3 via first-principles calculations. Appl. Phys. A 2012, 106, 645–653. [Google Scholar] [CrossRef]
- Harmel, M.; Khachai, H.; Haddou, A.; Khenata, R.; Murtaza, G.; Abbar, B.; Omran, S.B.; Khalfa, M. Ab initio study of the mechanical, thermal and optoelectronic properties of the cubic CsBaF3. Acta Phys. Pol. 2015, 128, 34–42. [Google Scholar] [CrossRef]
- Koteras, K.; Gawraczyński, J.; Derzsi, M.; Mazej, Z.; Grochala, W. Lattice Dynamics of KAgF3 Perovskite, Unique 1D Antiferromagnet. Chemistry 2021, 3, 94–103. [Google Scholar] [CrossRef]
- Dubrovin, R.M.; Garcia-Castro, A.C.; Siverin, N.V.; Novikova, N.N.; Boldyrev, K.N.; Romero, A.H.; Pisarev, R.V. Incipient geometric lattice instability of cubic fluoroperovskites. Phys. Rev. B 2021, 104, 144304. [Google Scholar] [CrossRef]
- Dubrovin, R.M.; Siverin, N.V.; Syrnikov, P.P.; Novikova, N.N.; Boldyrev, K.N.; Pisarev, R.V. Lattice dynamics and microscopic mechanisms of the spontaneous magnetodielectric effect in the antiferromagnetic fluoroperovskites KCoF3 and RbCoF3. Phys. Rev. B 2019, 100, 24429. [Google Scholar] [CrossRef]
- Vaitheeswaran, G.; Kanchana, V.; Zhang, X.; Ma, Y.; Svane, A.; Christensen, N.E. Calculated high-pressure structural properties, lattice dynamics and quasi particle band structures of perovskite fluorides KZnF3, CsCaF3 and BaLiF3. J. Phys. Condens. Matter 2016, 28, 315403. [Google Scholar] [CrossRef] [PubMed]
- Vaitheeswaran, G.; Kanchana, V.; Kumar, R.S.; Cornelius, A.L.; Nicol, M.F.; Svane, A.; Christensen, N.E.; Eriksson, O. High-pressure structural study of fluoro-perovskite CsCdF3 up to 60 GPa: A combined experimental and theoretical study. Phys. Rev. B 2010, 81, 075105. [Google Scholar] [CrossRef]
- Vaitheeswaran, G.; Kanchana, V.; Kumar, R.S.; Cornelius, A.L.; Nicol, M.F.; Svane, A.; Delin, A.; Johansson, B. High-pressure structural, elastic, and electronic properties of the scintillator host material KMgF3. Phys. Rev. B 2007, 76, 014107. [Google Scholar] [CrossRef]
- Korba, S.A.; Meradji, H.; Ghemid, S.; Bouhafs, B. First principles calculations of structural, electronic and optical properties of BaLiF3. Comput. Mater. Sci. 2009, 44, 1265–1271. [Google Scholar] [CrossRef]
- Furetta, C.; Santopietro, F.; Sanipoli, C.; Kitis, G. Thermoluminescent (TL) properties of the perovskite KMgF3 activated by Ce and Er impurities. Appl. Radiat. Isot. 2001, 55, 533–542. [Google Scholar] [CrossRef]
- Khan, I.; Shehzad, N.; Ahmad, I.; Ali, Z.; Jalali-Asadabadi, S. First-principle studies of the optoelectronic properties of ASnF3 (A = Na, K, Rb and Cs). Int. J. Mod. Phys. B 2017, 31, 1750148. [Google Scholar] [CrossRef]
- Hamioud, F.; AlGhamdi, G.S.; Al-Omari, S.; Mubarak, A.A. Ab initio investigation of the structural, electronic, magnetic and optical properties of the perovskite TlMnX3 (X = F, Cl) compounds. Int. J. Mod. Phys. B 2016, 30, 1650031. [Google Scholar] [CrossRef]
- Cheriet, A.; Lagoun, B.; Halit, M.; Zaabat, M.; Abdelhakim, C.; Hamza, L. First-principles study of structural, electronic, optical and elastic properties of cadmium based Fluoro-Perovskite MCdF3 (M = Rb, Tl). Solid State Phenom. 2019, 297, 173–186. [Google Scholar] [CrossRef]
- Kim, H.; Rooh, G.; Park, H.; Kim, S. Luminescence and scintillation properties of the new Ce-doped Tl2LiGdCl6 single crystals. J. Lumin. 2015, 164, 86–89. [Google Scholar] [CrossRef]
- Khan, A.; Rooh, G.; Kim, H.; Kim, S. Ce3+-activated Tl2GdCl5: Novel halide scintillator for X-ray and γ-ray detection. J. Alloys Compd. 2018, 741, 878–882. [Google Scholar] [CrossRef]
- Blaha, P.; Schwarz, K.; Madsen, G.K.; Kvasnicka, D.; Luitz, J. Wien2k: An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties; Vienna University of Technology: Vienna, Austria, 2001; Volume 60. [Google Scholar]
- Roy, A.; Mukherjee, S.; Sarkar, S.; Auluck, S.; Prasad, R.; Gupta, R.; Garg, A. Effects of site disorder, off-stoichiometry and epitaxial strain on the optical properties of magnetoelectric gallium ferrite. J. Phys. Condens. Matter 2012, 24, 435501. [Google Scholar] [CrossRef] [PubMed]
- Han, M.J.; Ozaki, T.; Yu, J. Rapid Communication Magnetic ordering and exchange interactions in multiferroic GaFeO3. Phys. Rev. B 2007, 75, 060404. [Google Scholar] [CrossRef]
- Ambrosch-Draxl, C.; Sofo, J.O. Linear optical properties of solids within the full-potential linearized augmented planewave method. Comput. Phys. Commun. 2006, 175, 1–14. [Google Scholar] [CrossRef]
- Reshak, A.H.; Jamal, M. DFT calculation for elastic constants of orthorhombic structure within WIEN2K code: A new package (ortho-elastic). J. Alloys Compd. 2012, 543, 147–151. [Google Scholar] [CrossRef]
- Jamal, M.; Bilal, M.; Ahmad, I.; Jalali-Asadabadi, S. IRelast package. J. Alloys Compd. 2018, 735, 569–579. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef]
- Berger, J.; Hauret, G.; Rousseau, M. Brillouin scattering investigation of the structural phase transition of TlCdF3 and RbCaF3. Solid State Commun. 1978, 25, 569–571. [Google Scholar] [CrossRef]
- Grimvall, G. Thermophysical Properties of Materials; Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- SAzam, S.; Khan, S.A. A first principles study of electronic and optical properties of the polar quaternary chalcogenides β-A2Hg3Ge2S8 (A= K and Rb). Mater. Sci. Semicond. Process. 2015, 34, 250–259. [Google Scholar]
- Sohail, M.; Husain, M.; Rahman, N.; Althubeiti, K.; Algethami, M.; Khan, A.A.; Iqbal, A.; Ullah, A.; Khan, A.; Khan, R. First-Principal Investigations of Electronic, Structural, Elastic, and Optical Properties of the Fluoroperovskite TlLF3 (L = Ca, Cd) Compounds using TB-mBJ Potential method. RSC Adv. 2022, 30, 7002–7008. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
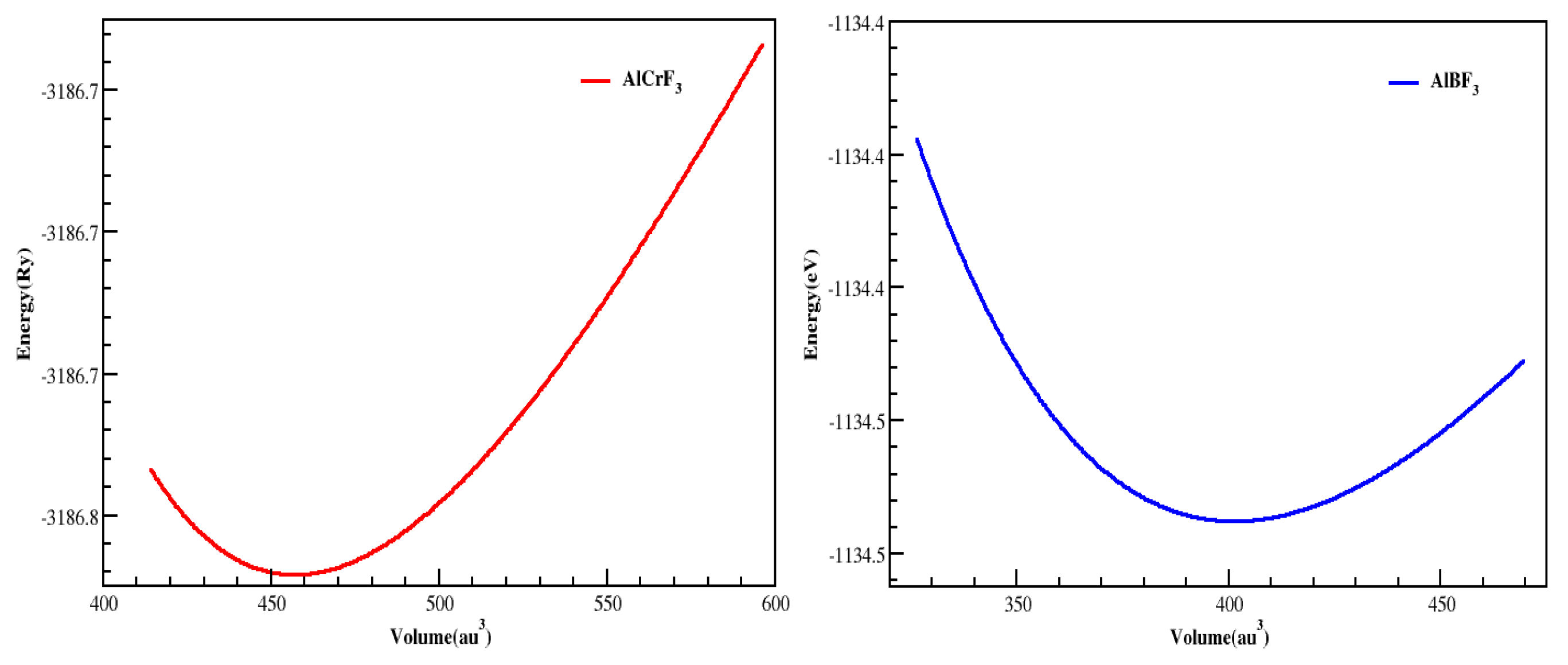
| Compounds | ao (Lattice Constant in Å) | B (Bulk Modulus in GPa) | B’ (Derivative of Bulk Modulus) | V0 (Ground State Volume in a.u3) | E0 (Ground State Energy in Ry) |
|---|---|---|---|---|---|
| AlCrF3 | 7.96 | 89.57 | 4.80 | 457.18 | −3186.77 |
| AlBF3 | 7.70 | 81.15 | 4.66 | 401.27 | −1134.47 |
| Compounds | AlCrF3 | AlBF3 |
|---|---|---|
| C11 | 79.11 | 77.00 |
| C12 | 44.85 | 94.22 |
| C44 | 11.86 | 31.92 |
| B | 89.57 | 81.15 |
| G | 13.74 | 10.22 |
| E | 40.80 | 45.74 |
| A | 0.69 | 3.71 |
| v | 0.46 | 0.46 |
| B/G | 12.51 | 16.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, H.; Sohail, M.; Khan, R.; Raman, N.; Ullah, A.; Khan, A.; Alataway, A.; Dewidar, A.Z.; Elansary, H.O.; Yessoufou, K. Theoretical Investigations into the Different Properties of Al-Based Fluoroperovskite AlMF3 (M = Cr, B) Compounds by the TB-MBJ Potential Method. Materials 2022, 15, 5942. https://doi.org/10.3390/ma15175942
Khan H, Sohail M, Khan R, Raman N, Ullah A, Khan A, Alataway A, Dewidar AZ, Elansary HO, Yessoufou K. Theoretical Investigations into the Different Properties of Al-Based Fluoroperovskite AlMF3 (M = Cr, B) Compounds by the TB-MBJ Potential Method. Materials. 2022; 15(17):5942. https://doi.org/10.3390/ma15175942
Chicago/Turabian StyleKhan, Hukam, Mohammad Sohail, Rajwali Khan, Nasir Raman, Asad Ullah, Aurangzeb Khan, Abed Alataway, Ahmed Z. Dewidar, Hosam O. Elansary, and Kowiyou Yessoufou. 2022. "Theoretical Investigations into the Different Properties of Al-Based Fluoroperovskite AlMF3 (M = Cr, B) Compounds by the TB-MBJ Potential Method" Materials 15, no. 17: 5942. https://doi.org/10.3390/ma15175942
APA StyleKhan, H., Sohail, M., Khan, R., Raman, N., Ullah, A., Khan, A., Alataway, A., Dewidar, A. Z., Elansary, H. O., & Yessoufou, K. (2022). Theoretical Investigations into the Different Properties of Al-Based Fluoroperovskite AlMF3 (M = Cr, B) Compounds by the TB-MBJ Potential Method. Materials, 15(17), 5942. https://doi.org/10.3390/ma15175942