
Supercritical CO2-Induced Evolution of Alkali-Activated Slag Cements

 , , ,
, , , 
 and
and
Abstract
:1. Introduction
2. Experimental Procedure
2.1. Materials and Sample Preparation
2.2. Test Methods
3. Results
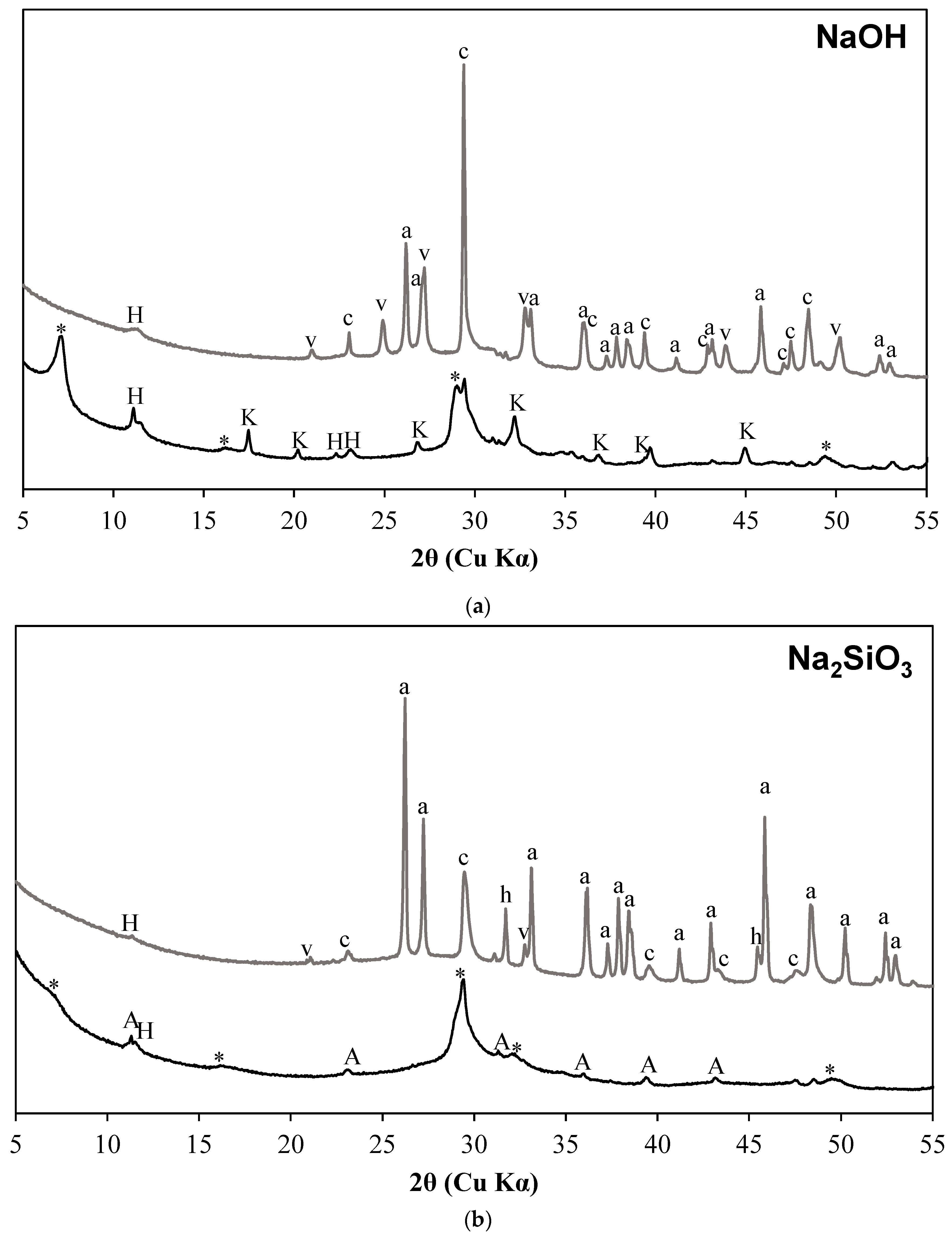
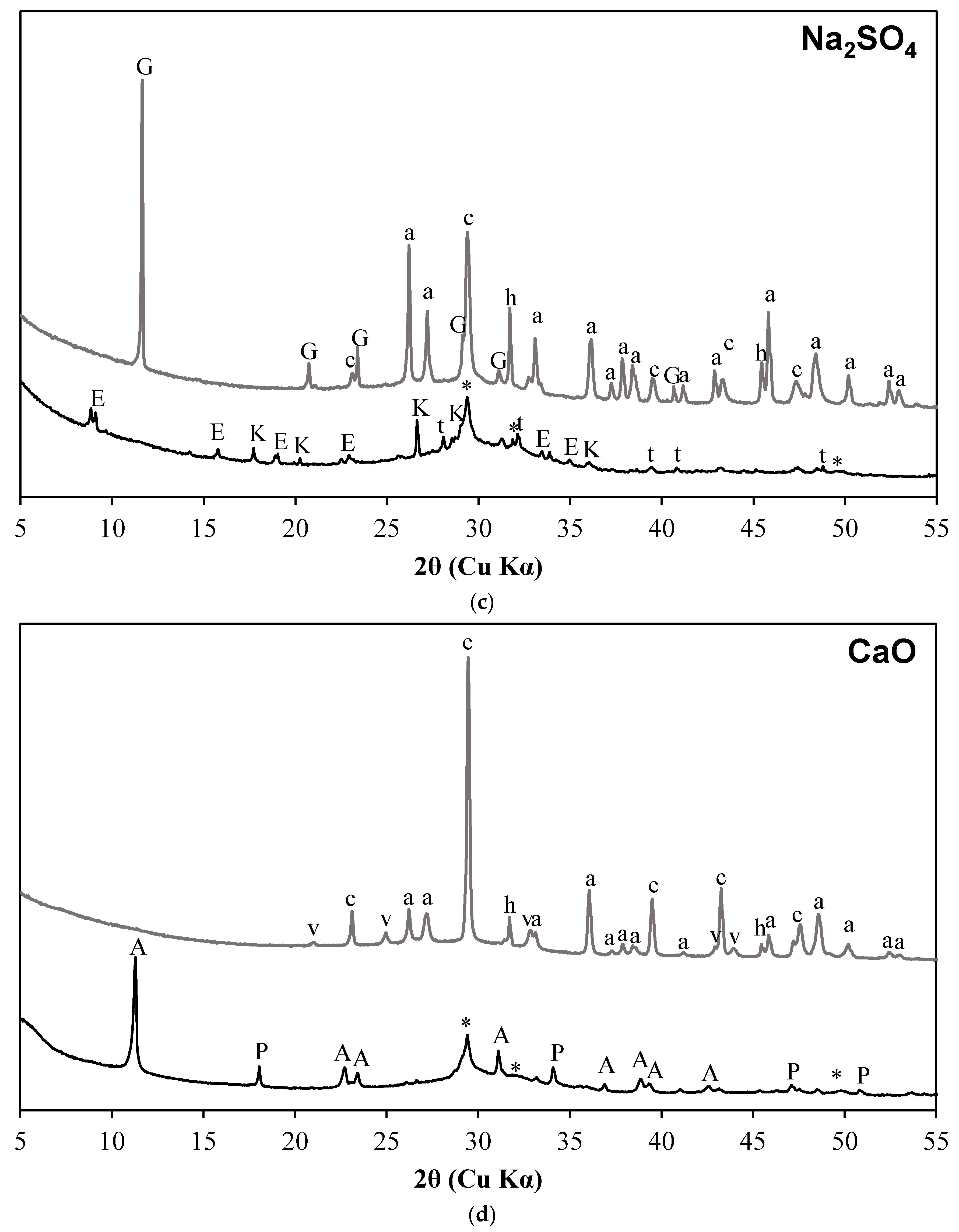
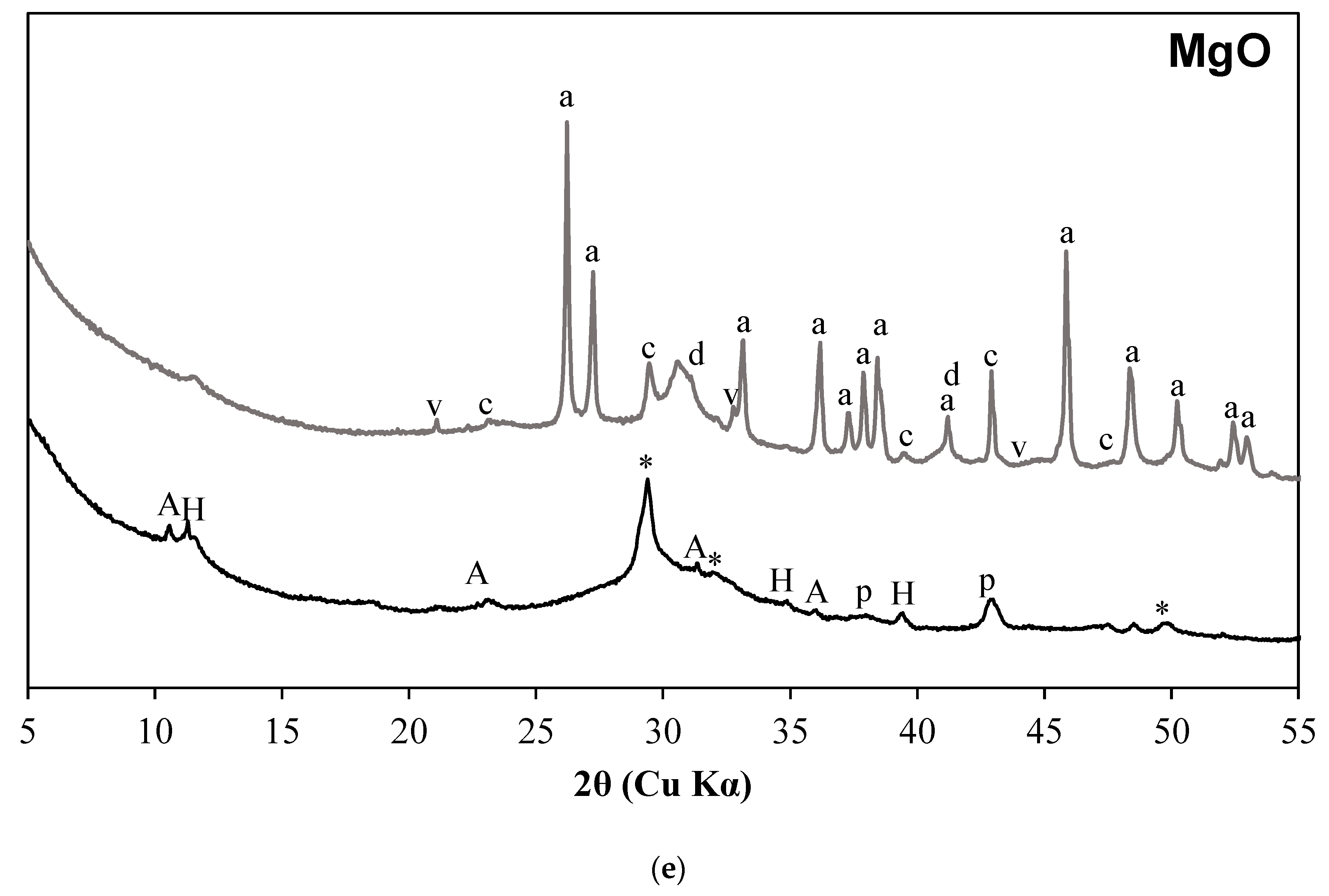
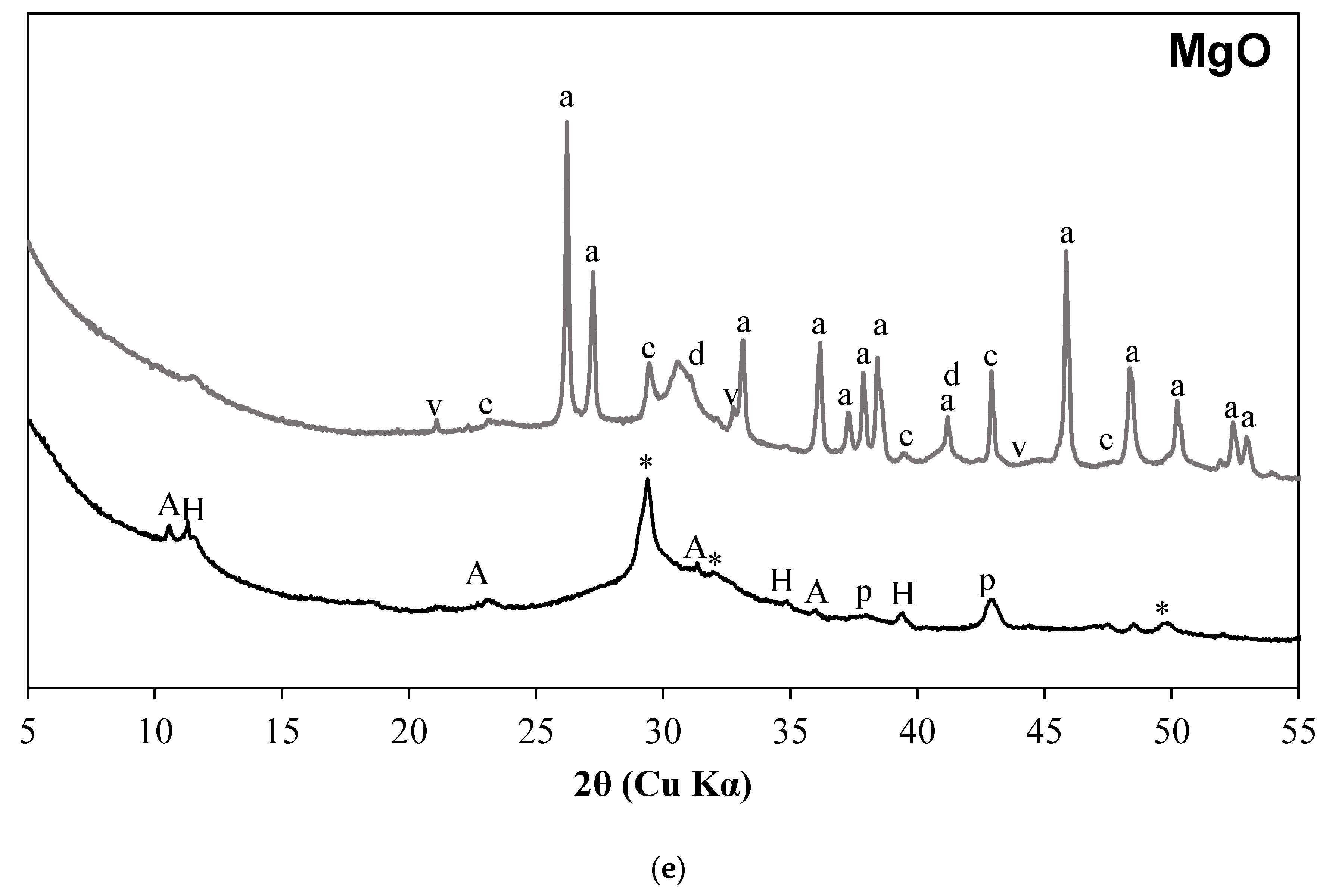
3.1. X-ray Diffraction Results
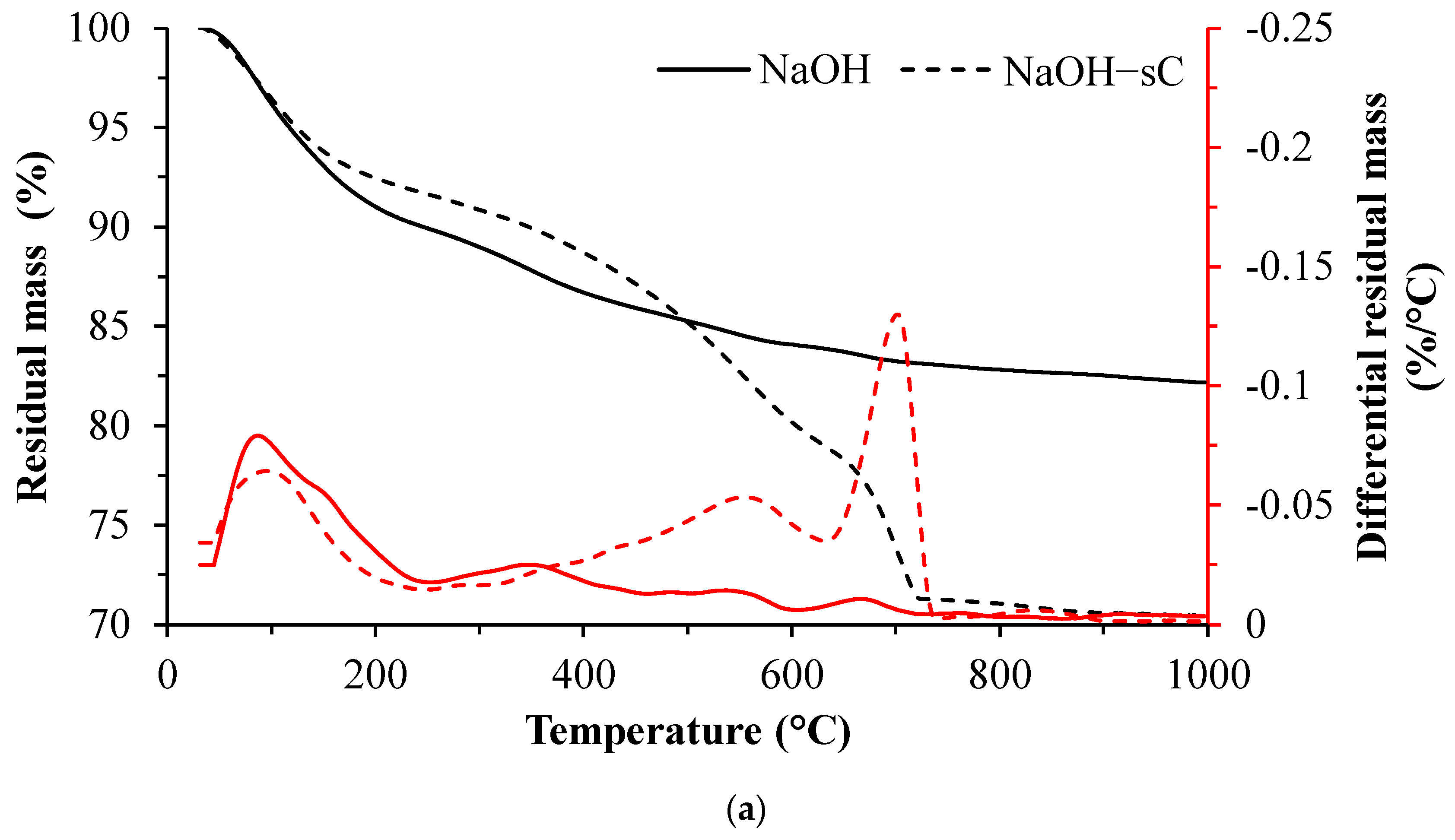
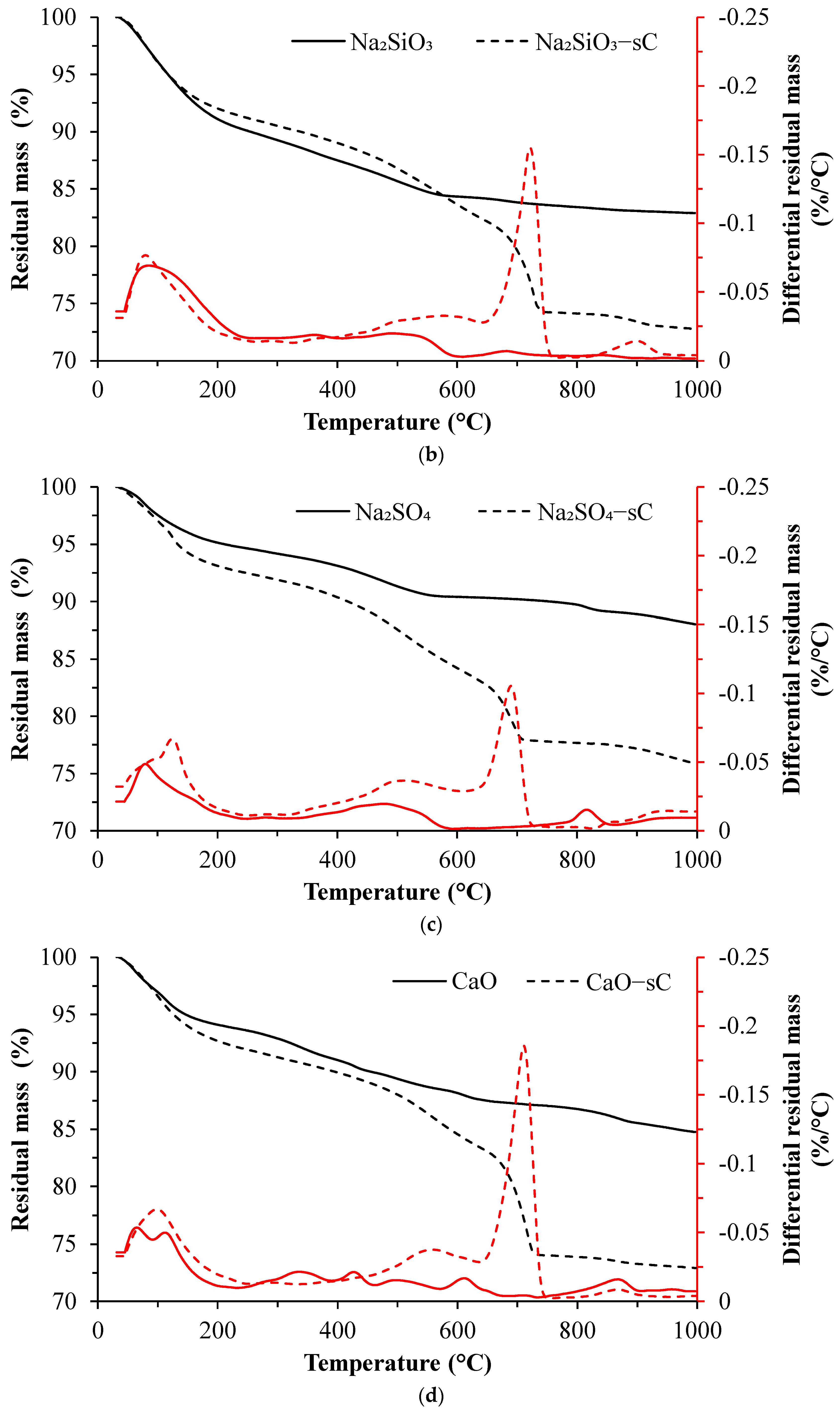
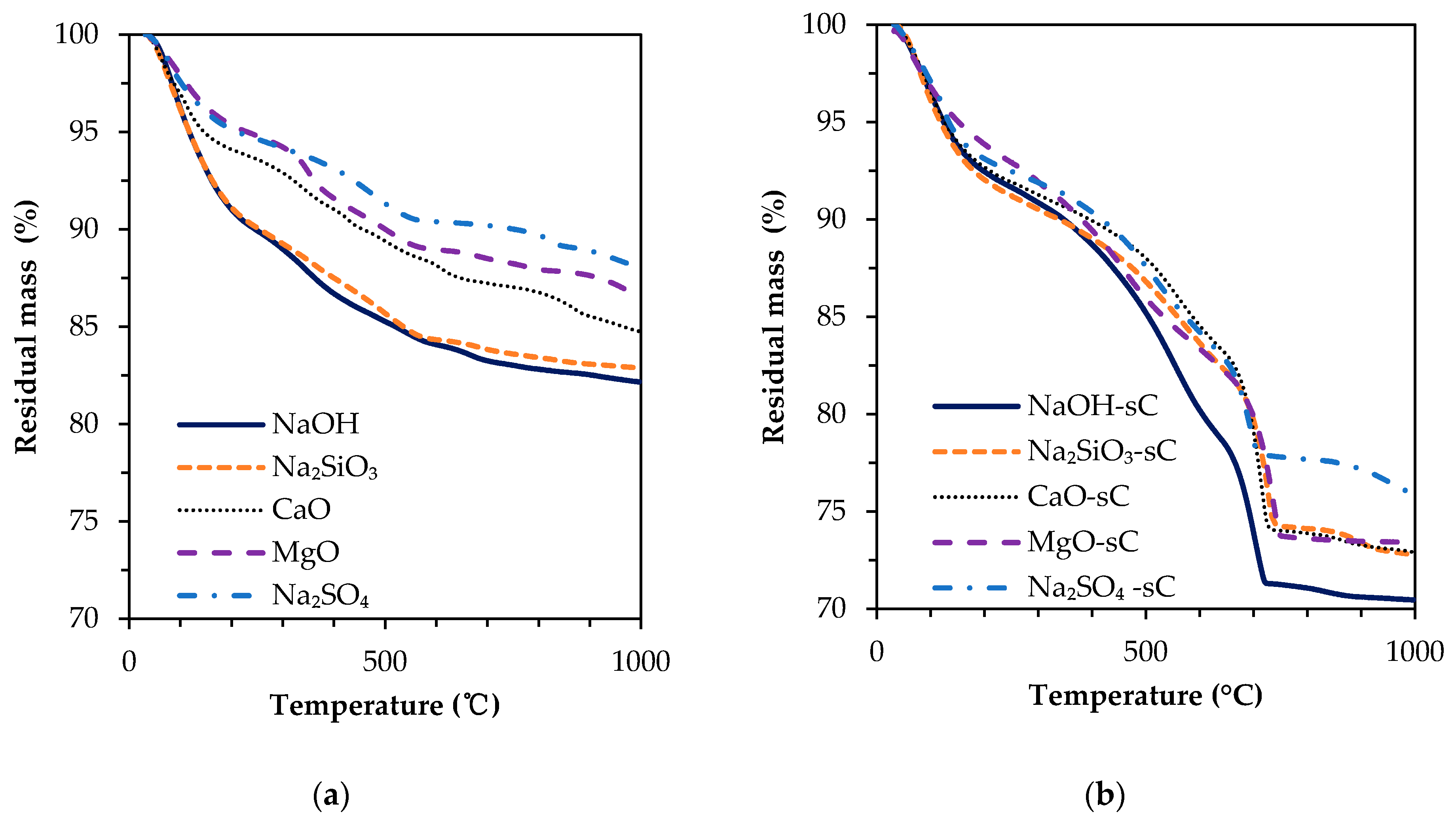
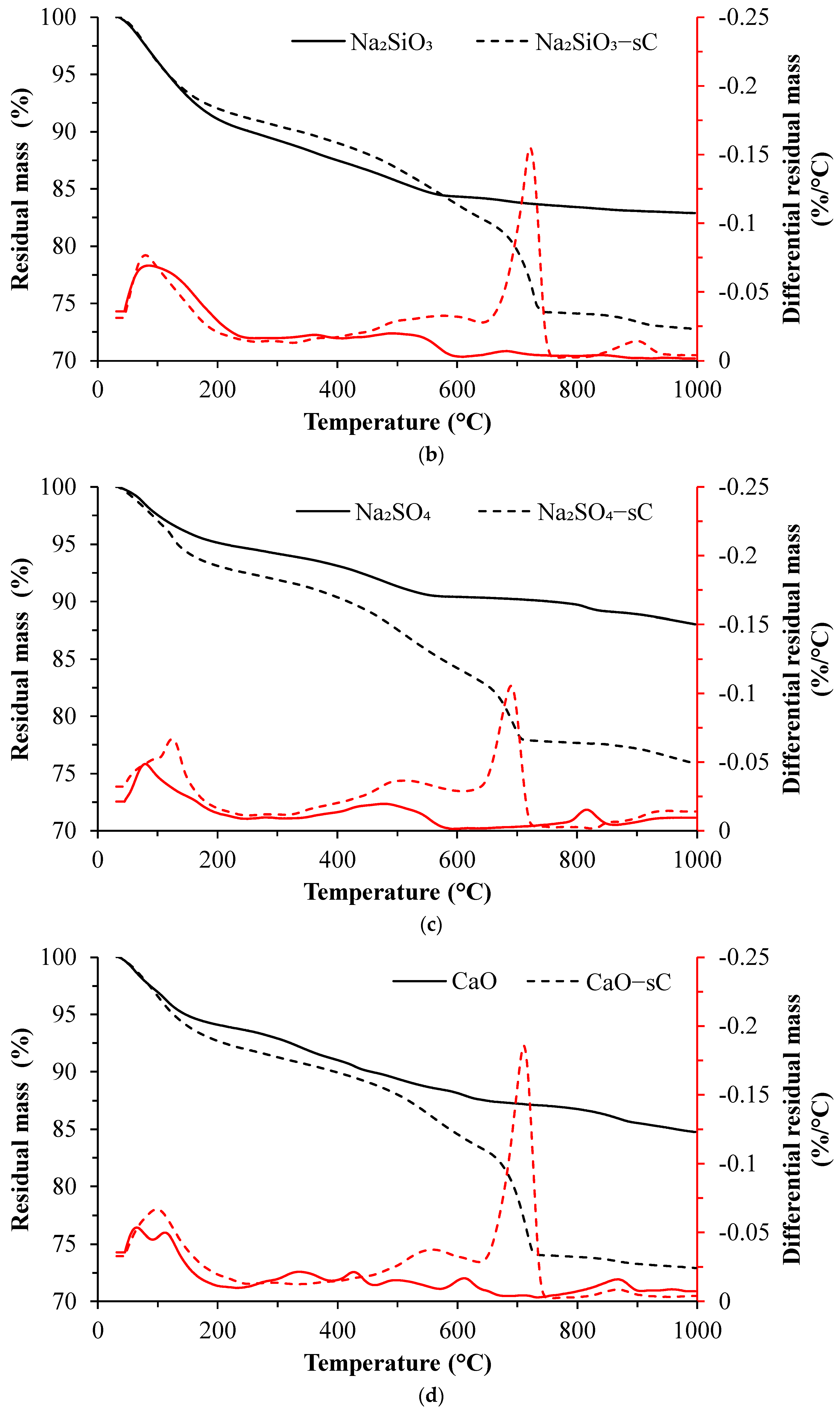
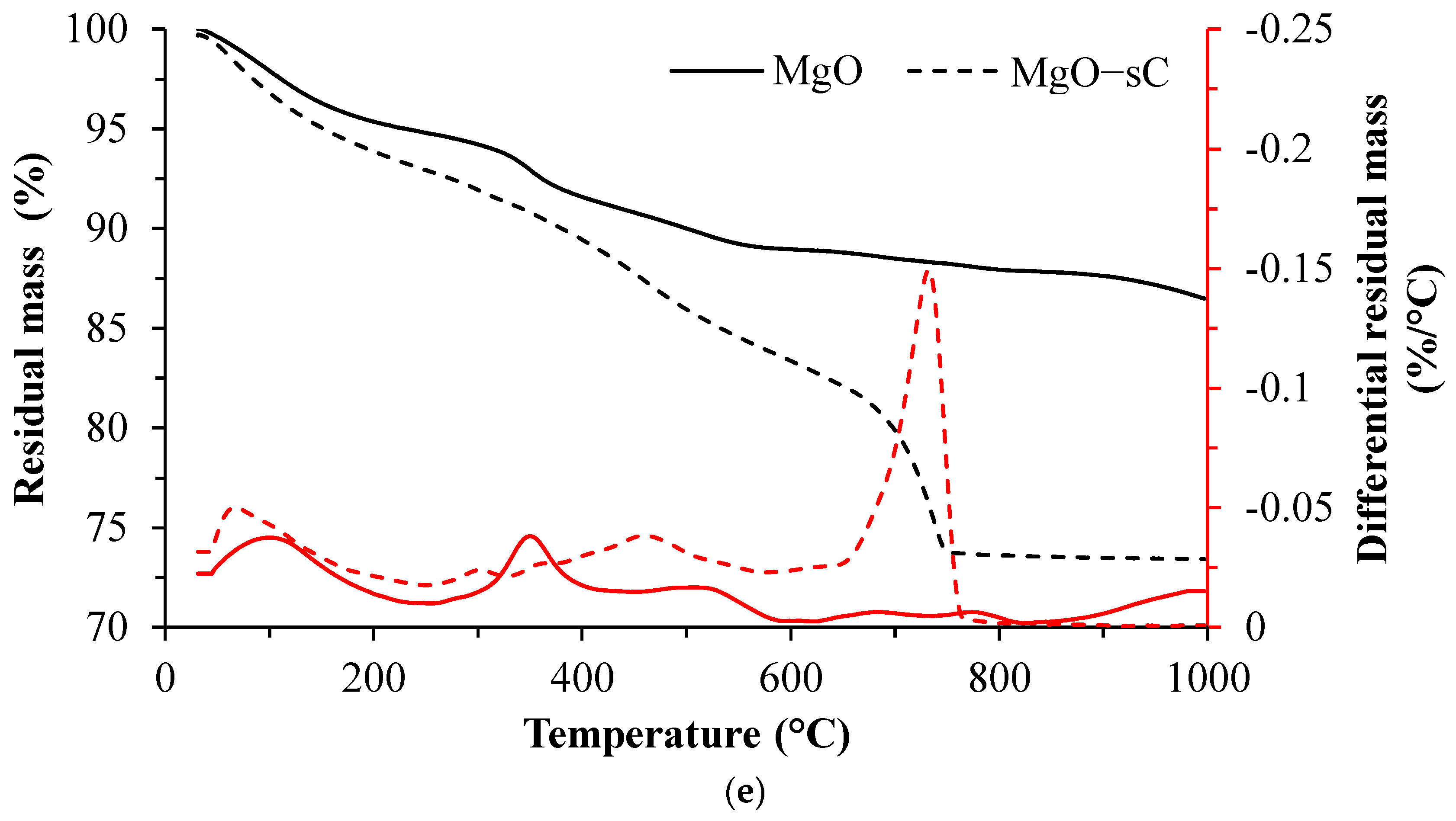
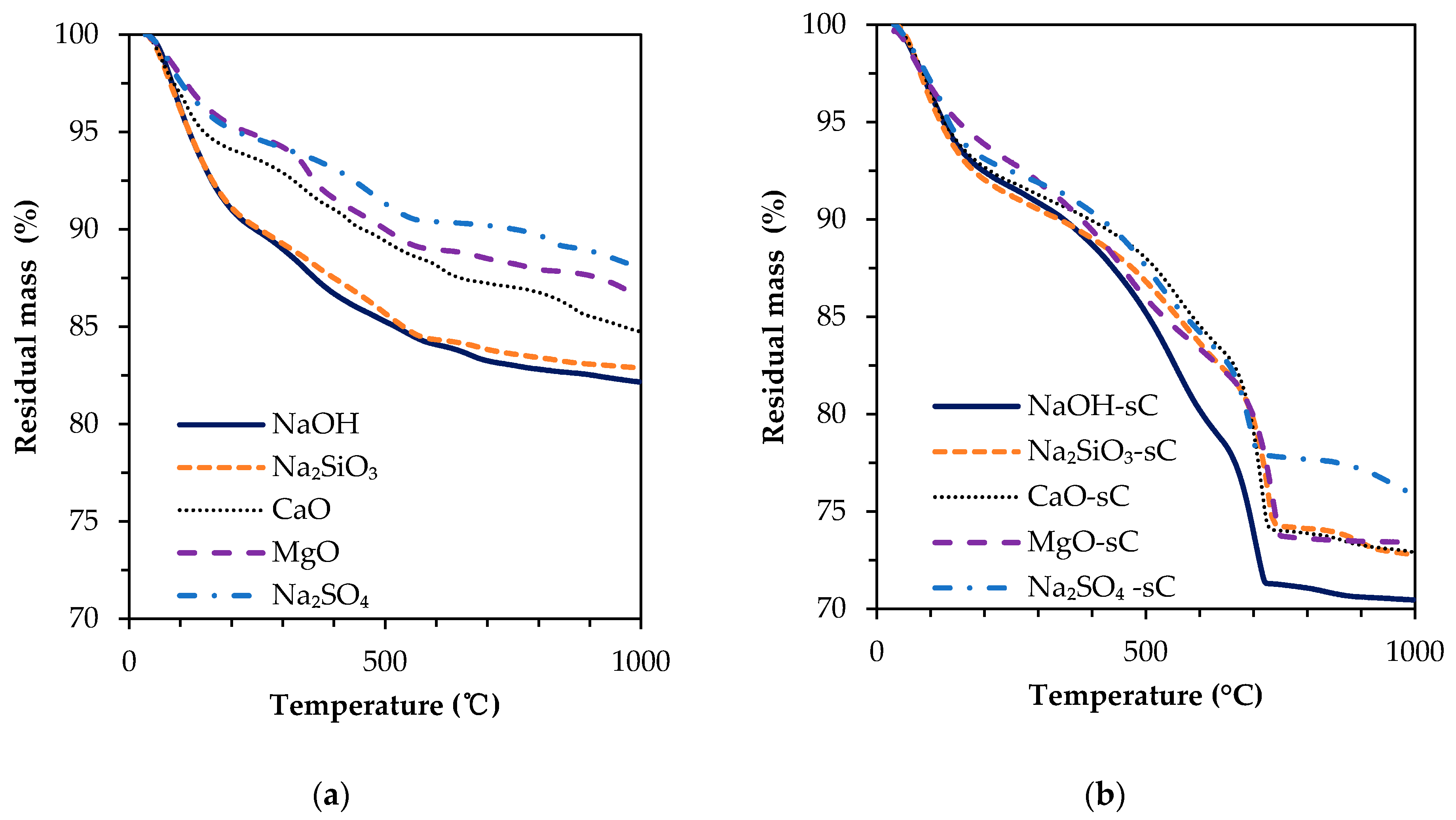
3.2. TGA Results
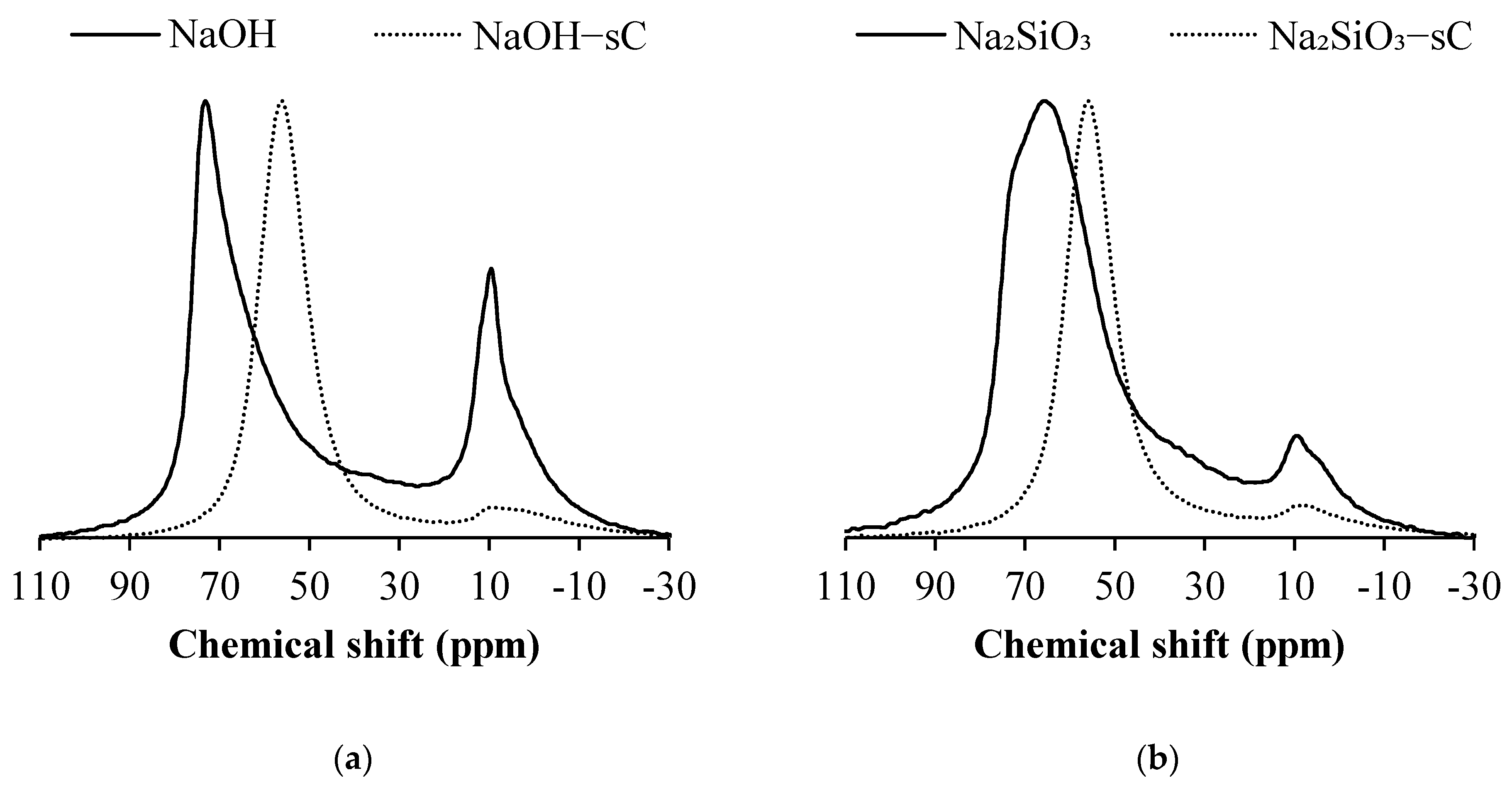
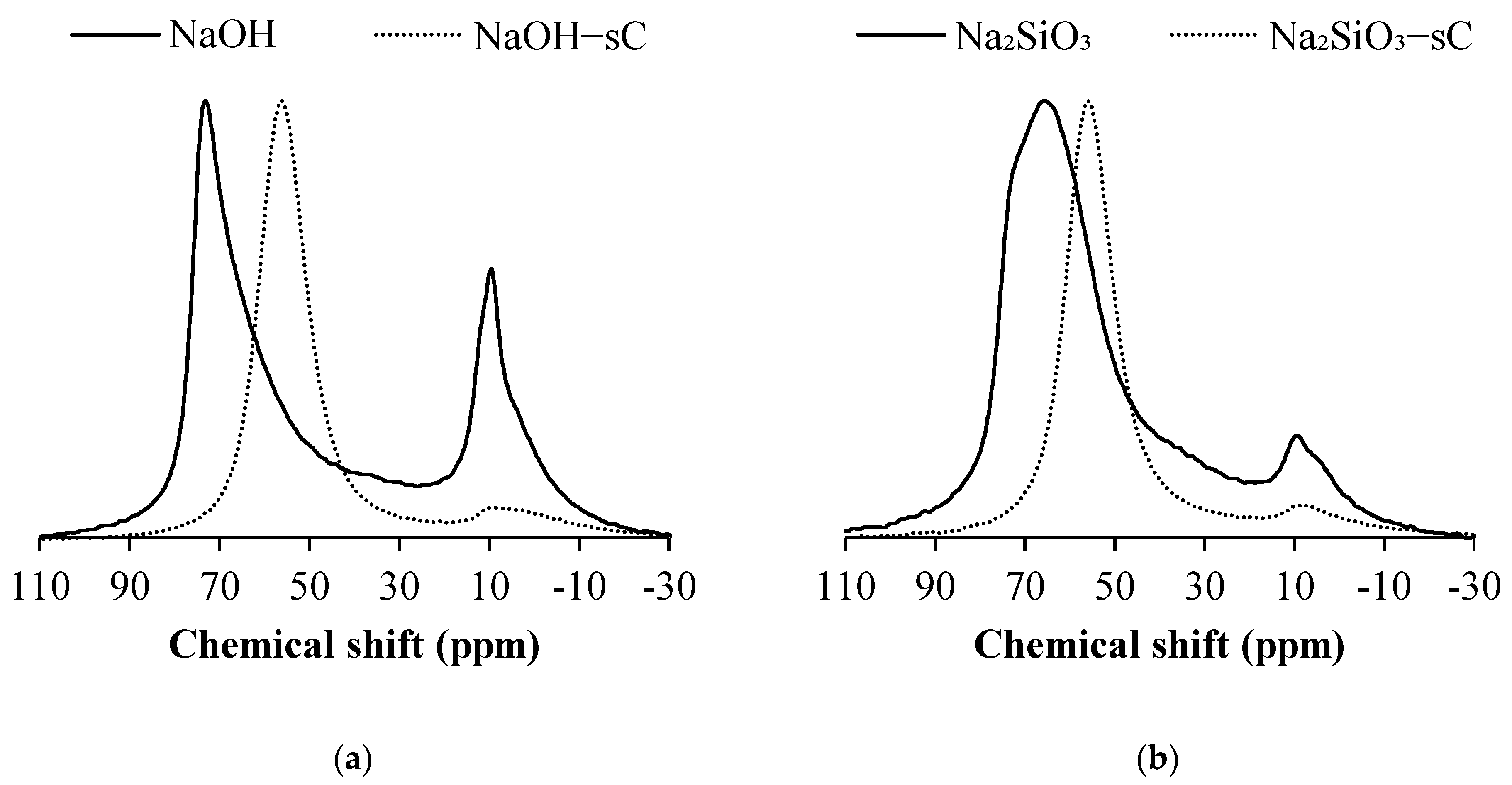
3.3. 29Si MAS NMR
3.4. 27Al MAS NMR
3.5. Discussion
4. Conclusions
- (1)
- C-S-H with varying degrees of Al incorporation commonly detected in alkali-activated slag systems is observed as the prominent reaction product regardless of the type of activators used in the study.
- (2)
- C-S-H is not observed in the carbonated samples suggesting its complete carbonation. The CaCO3 polymorphs such as calcite, aragonite, and vaterite are observed as carbonation products upon exposure to sCO2. The relative proportions of different CaCO3 polymorphs differed with the change in activator type.
- (3)
- The unreacted slag content decreased drastically with carbonation. About 20–30 g of CO2/100 g of reacted slag was consumed for the carbonation of alkali-activated slag samples in sCO2 conditions.
- (4)
- The extent of carbonation per reacted slag based on the activator is as follows CaO > MgO > NaOH > Na2SiO3 > Na2SO4. The higher C-S-H contents in NaOH-activated slag and the additional carbonation phases resulting from the carbonation of activator in CaO- and MgO-activated slag systems resulted in higher CO2 contents.
- (5)
- A decrease in Q1, Q2 sites and a corresponding increase in the quantity of Q3 and Q4 units were observed in AAS samples when subjected to sCO2, indicating the formation of a highly cross-linked aluminosilicates upon carbonation for all types of activators considered in the study. The formation of highly cross-linked aluminosilicates was also reported previously.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bernal, S.A.; Provis, J.L.; Rose, V.; De Gutierrez, R.M. Evolution of binder structure in sodium silicate-activated slag-metakaolin blends. Cem. Concr. Compos. 2011, 33, 46–54. [Google Scholar] [CrossRef]
- Bernal, S.A.; de Gutiérrez, R.M.; Pedraza, A.L.; Provis, J.L.; Rodriguez, E.D.; Delvasto, S. Effect of binder content on the performance of alkali-activated slag concretes. Cem. Concr. Res. 2011, 41, 1–8. [Google Scholar] [CrossRef]
- de Azevedo, A.R.; Cruz, A.S.; Marvila, M.T.; de Oliveira, L.B.; Monteiro, S.N.; Vieira, C.M.F.; Fediuk, R.; Timokhin, R.; Vatin, N.; Daironas, M. Natural Fibers as an Alternative to Synthetic Fibers in Reinforcement of Geopolymer Matrices: A Comparative Review. Polymers 2021, 13, 2493. [Google Scholar] [CrossRef] [PubMed]
- Puertas, F.; Gutierrez, R.; Fernández-Jiménez, A.; Delvasto, S.; Maldonado, J. Alkaline cement mortars. Chemical resistance to sulfate and seawater attack. Mater. Constr. 2002, 52, 55–71. [Google Scholar] [CrossRef]
- Bakharev, T.; Sanjayan, J.G.; Cheng, Y.-B. Resistance of alkali-activated slag concrete to acid attack. Cem. Concr. Res. 2003, 33, 1607–1611. [Google Scholar] [CrossRef]
- Runci, A.; Serdar, M. Effect of curing time on the chloride diffusion of alkali-activated slag. Case Stud. Constr. Mater. 2022, 16, e00927. [Google Scholar] [CrossRef]
- Abdel-Gawwad, H.A.; Sikora, P.; Mohammed, A.H.; Arif, M.A.; Al-Kroom, H.; Elrahman, M.A. Resistance of alkali-activated slag mixed with wastewater towards biogenic sulfuric acid attack. Case Stud. Constr. Mater. 2022, 17, e01164. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, C.; Zhang, Z.; Ou, Z. Durability of alkali-activated materials in aggressive environments: A review on recent studies. Constr. Build. Mater. 2017, 152, 598–613. [Google Scholar] [CrossRef]
- Provis, J.L. Alkali-activated materials. Cem. Concr. Res. 2018, 114, 40–48. [Google Scholar] [CrossRef]
- Xu, H.; Provis, J.L.; van Deventer, J.S.J.; Krivenko, P.V. Characterization of Aged Slag Concretes. ACI Mater. J. 2008, 105, 131–139. [Google Scholar] [CrossRef]
- Bernal, S.A.; San Nicolas, R.; Provis, J.L.; De Gutiérrez, R.M.; van Deventer, J.S. Natural carbonation of aged alkali-activated slag concretes. Mater. Struct. 2014, 47, 693–707. [Google Scholar] [CrossRef]
- Shi, C.; Roy, D.; Krivenko, P. Alkali-Activated Cements and Concretes; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Song, K.-I.; Song, J.-K.; Lee, B.Y.; Yang, K.-H. Carbonation characteristics of alkali-activated blast-furnace slag mortar. Adv. Mater. Sci. Eng. 2014, 2014, 326458. [Google Scholar] [CrossRef]
- Bilim, C.; Atiş, C.D. Alkali activation of mortars containing different replacement levels of ground granulated blast furnace slag. Constr. Build. Mater. 2012, 28, 708–712. [Google Scholar] [CrossRef]
- Shi, Z.; Shi, C.; Wan, S.; Li, N.; Zhang, Z. Effect of alkali dosage and silicate modulus on carbonation of alkali-activated slag mortars. Cem. Concr. Res. 2018, 113, 55–64. [Google Scholar] [CrossRef]
- Byfors, K.; Klingstedt, G.; Lehtonen, V.; Pyy, H.; Romben, L. Durability of Concrete Made with Alkali-Activated Slag. Int. Concr. Abstr. Portal 1989, 114, 1429–1466. [Google Scholar] [CrossRef]
- Al-Otaibi, S. Durability of concrete incorporating GGBS activated by water-glass. Constr. Build. Mater. 2008, 22, 2059–2067. [Google Scholar] [CrossRef]
- Rashad, A.M. A synopsis of carbonation of alkali-activated materials. Green Mater. 2019, 7, 118–136. [Google Scholar] [CrossRef]
- Zhang, X.; Long, K.; Liu, W.; Li, L.; Long, W.-J. Carbonation and Chloride Ions’ Penetration of Alkali-Activated Materials: A Review. Molecules 2020, 25, 5074. [Google Scholar] [CrossRef]
- Puertas, F.; Palacios, M.; Vázquez, T. Carbonation process of alkali-activated slag mortars. J. Mater. Sci. 2006, 41, 3071–3082. [Google Scholar] [CrossRef]
- Law, D.W.; Adam, A.A.; Molyneaux, T.K.; Patnaikuni, I. Durability assessment of alkali activated slag (AAS) concrete. Mater. Struct. 2012, 45, 1425–1437. [Google Scholar] [CrossRef]
- Bernal, S.A.; Provis, J.L.; Brice, D.G.; Kilcullen, A.; Duxson, P.; van Deventer, J.S. Accelerated carbonation testing of alkali-activated binders significantly underestimates service life: The role of pore solution chemistry. Cem. Concr. Res. 2012, 42, 1317–1326. [Google Scholar] [CrossRef]
- Nedeljković, M.; Ghiassi, B.; van der Laan, S.; Li, Z.; Ye, G. Effect of curing conditions on the pore solution and carbonation resistance of alkali-activated fly ash and slag pastes. Cem. Concr. Res. 2019, 116, 146–158. [Google Scholar] [CrossRef]
- Ye, H.; Cai, R.; Tian, Z. Natural carbonation-induced phase and molecular evolution of alkali-activated slag: Effect of activator composition and curing temperature. Constr. Build. Mater. 2020, 248, 118726. [Google Scholar] [CrossRef]
- Bernal, S.A.; de Gutierrez, R.M.; Provis, J.L.; Rose, V. Effect of silicate modulus and metakaolin incorporation on the carbonation of alkali silicate-activated slags. Cem. Concr. Res. 2010, 40, 898–907. [Google Scholar] [CrossRef]
- Lee, N.K.; Koh, K.T.; Kim, M.O.; An, G.H.; Ryu, G.S. Physicochemical changes caused by reactive MgO in alkali-activated fly ash/slag blends under accelerated carbonation. Ceram. Int. 2017, 43, 12490–12496. [Google Scholar] [CrossRef]
- Bernal, S.A.; San Nicolas, R.; Myers, R.J.; de Gutiérrez, R.M.; Puertas, F.; van Deventer, J.S.; Provis, J.L. MgO content of slag controls phase evolution and structural changes induced by accelerated carbonation in alkali-activated binders. Cem. Concr. Res. 2014, 57, 33–43. [Google Scholar] [CrossRef]
- Park, S.M.; Jang, J.G.; Lee, H.K. Unlocking the role of MgO in the carbonation of alkali-activated slag cement. Inorg. Chem. Front. 2018, 5, 1661–1670. [Google Scholar] [CrossRef]
- McCaslin, E.R.; White, C.E. A parametric study of accelerated carbonation in alkali-activated slag. Cem. Concr. Res. 2021, 145, 106454. [Google Scholar] [CrossRef]
- Palacios, M.; Puertas, F. Effect of Carbonation on Alkali-Activated Slag Paste. J. Am. Ceram. Soc. 2006, 89, 3211–3221. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Phung, Q.T.; Yu, Z.; Frederickx, L.; Jacques, D.; Sakellariou, D.; Dauzeres, A.; Elsen, J.; Pontikes, Y. Alteration in molecular structure of alkali activated slag with various water to binder ratios under accelerated carbonation. Sci. Rep. 2022, 12, 5524. [Google Scholar] [CrossRef]
- Zhao, J.; Shumuye, E.D.; Wang, Z.; Bezabih, G.A. Performance of GGBS Cement Concrete under Natural Carbonation and Accelerated Carbonation Exposure. J. Eng. 2021, 2021, 6659768. [Google Scholar] [CrossRef]
- Barlet-Gouedard, V.; Rimmele, G.; Goffe, B.; Porcherie, O. Mitigation strategies for the risk of CO2 migration through wellbores. In Proceedings of the IADC/SPE Drilling Conference, Miami, FL, USA, 21–23 February 2006. [Google Scholar]
- Thomas, D.C.; Benson, S.M. Carbon Dioxide Capture for Storage in Deep Geologic Formations-Results from the CO2 Capture Project: Vol 2-Geologic Storage of Carbon Dioxide with Monitoring and Verification; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Park, S.; Moon, H.; Kim, J.-H.; Lee, M.; Chung, C.-W. Reaction of hydrated cement paste with supercritical carbon dioxide. Constr. Build. Mater. 2021, 281, 122615. [Google Scholar] [CrossRef]
- García-González, C.A.; el Grouh, N.; Hidalgo, A.; Fraile, J.; López-Periago, A.M.; Andrade, C.; Domingo, C. New insights on the use of supercritical carbon dioxide for the accelerated carbonation of cement pastes. J. Supercrit. Fluids 2008, 43, 500–509. [Google Scholar] [CrossRef]
- Vance, K.; Falzone, G.; Pignatelli, I.; Bauchy, M.; Balonis, M.; Sant, G. Direct Carbonation of Ca(OH)2 Using Liquid and Supercritical CO2: Implications for Carbon-Neutral Cementation. Ind. Eng. Chem. Res. 2015, 54, 8908–8918. [Google Scholar] [CrossRef]
- Short, N.R.; Purnell, P.; Page, C.L. Preliminary investigations into the supercritical carbonation of cement pastes. J. Mater. Sci. 2001, 36, 35–41. [Google Scholar] [CrossRef]
- Park, S.; Park, H.M.; Yoon, H.; Seo, J.; Yang, C.-M.; Provis, J.L.; Yang, B. Hydration kinetics and products of MgO-activated blast furnace slag. Constr. Build. Mater. 2020, 249, 118700. [Google Scholar] [CrossRef]
- Park, S.M.; Jang, J. Carbonation-induced weathering effect on cesium retention of cement paste. J. Nucl. Mater. 2018, 505, 159–164. [Google Scholar] [CrossRef]
- Seo, J.; Park, S.M.; Lee, H.-K. Evolution of the binder gel in carbonation-cured Portland cement in an acidic medium. Cem. Concr. Res. 2018, 109, 81–89. [Google Scholar] [CrossRef]
- Yoon, H.N.; Seo, J.; Kim, S.; Lee, H.K.; Park, S. Characterization of blast furnace slag-blended Portland cement for immobilization of Co. Cem. Concr. Res. 2020, 134, 106089. [Google Scholar] [CrossRef]
- Myers, R.; Bernal, S.A.; Gehman, J.D.; Van Deventer, J.S.J.; Provis, J.L. The Role of Al in Cross-Linking of Alkali-Activated Slag Cements. J. Am. Ceram. Soc. 2015, 98, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.J.; L’Hôpital, E.; Provis, J.L.; Lothenbach, B. Effect of temperature and aluminium on calcium (alumino)silicate hydrate chemistry under equilibrium conditions. Cem. Concr. Res. 2015, 68, 83–93. [Google Scholar] [CrossRef]
- Bernal, S.A.; Provis, J.L.; Walkley, B.; San Nicolas, R.; Gehman, J.D.; Brice, D.G.; Kilcullen, A.R.; Duxson, P.; Van Deventer, J.S. Gel nanostructure in alkali-activated binders based on slag and fly ash, and effects of accelerated carbonation. Cem. Concr. Res. 2013, 53, 127–144. [Google Scholar] [CrossRef]
- Reddy, K.C.; Subramaniam, K.V.L. Quantitative phase analysis of slag hydrating in an alkaline environment. J. Appl. Crystallogr. 2020, 53, 424–434. [Google Scholar] [CrossRef]
- Reddy, K.C.; Subramaniam, K.V.L. Blast Furnace Slag Hydration in an Alkaline Medium: Influence of Sodium Content and Sodium Hydroxide Molarity. J. Mater. Civ. Eng. 2020, 32, 04020371. [Google Scholar] [CrossRef]
- Zuo, Y.; Nedeljković, M.; Ye, G. Pore solution composition of alkali-activated slag/fly ash pastes. Cem. Concr. Res. 2019, 115, 230–250. [Google Scholar] [CrossRef]
- Ye, H.; Radlińska, A. Quantitative Analysis of Phase Assemblage and Chemical Shrinkage of Alkali-Activated Slag. J. Adv. Concr. Technol. 2016, 14, 245–260. [Google Scholar] [CrossRef]
- Renaudin, G.; Russias, J.; Leroux, F.; Frizon, F.; Cau-dit-Coumes, C. Structural characterization of C–S–H and C–A–S–H samples—part I: Long-range order investigated by Rietveld analyses. J. Solid State Chem. 2009, 182, 3312–3319. [Google Scholar] [CrossRef]
- Haha, M.B.; LE Saout, G.; Winnefeld, F.; Lothenbach, B. Influence of activator type on hydration kinetics, hydrate assemblage and microstructural development of alkali activated blast-furnace slags. Cem. Concr. Res. 2011, 41, 301–310. [Google Scholar] [CrossRef]
- Garcia-Lodeiro, I.; Palomo, A.; Fernández-Jiménez, A.; Macphee, D. Compatibility studies between NASH and CASH gels. Study in the ternary diagram Na2O–CaO–Al2O3–SiO2–H2O. Cem. Concr. Res. 2011, 41, 923–931. [Google Scholar] [CrossRef]
- Ben Haha, M.; Lothenbach, B.; Le Saout, G.; Winnefeld, F. Influence of slag chemistry on the hydration of alkali-activated blast-furnace slag—Part II: Effect of Al2O3. Cem. Concr. Res. 2012, 42, 74–83. [Google Scholar] [CrossRef]
- Myers, R.J.; Bernal, S.A.; Provis, J.L. Phase diagrams for alkali-activated slag binders. Cem. Concr. Res. 2017, 95, 30–38. [Google Scholar] [CrossRef]
- Mobasher, N.; Bernal, S.A.; Hussain, O.H.; Apperley, D.C.; Kinoshita, H.; Provis, J.L. Characterisation of Ba(OH)2–Na2SO4–blast furnace slag cement-like composites for the immobilisation of sulfate bearing nuclear wastes. Cem. Concr. Res. 2014, 66, 64–74. [Google Scholar] [CrossRef]
- Hargis, C.W.; Lothenbach, B.; Müller, C.J.; Winnefeld, F. Carbonation of calcium sulfoaluminate mortars. Cem. Concr. Compos. 2017, 80, 123–134. [Google Scholar] [CrossRef]
- Seo, J.; Kim, S.; Park, S.; Yoon, H.; Lee, H. Carbonation of calcium sulfoaluminate cement blended with blast furnace slag. Cem. Concr. Compos. 2021, 118, 103918. [Google Scholar] [CrossRef]
- Ke, X.; Bernal, S.A.; Provis, J.L. Uptake of chloride and carbonate by Mg-Al and Ca-Al layered double hydroxides in simulated pore solutions of alkali-activated slag cement. Cem. Concr. Res. 2017, 100, 1–13. [Google Scholar] [CrossRef]
- Hibino, T.; Yamashita, Y.; Kosuge, K.; Tsunashima, A. Decarbonation behavior of Mg-Al-CO3 hydrotalcite-like compounds during heat treatment. Clays Clay Miner. 1995, 43, 427–432. [Google Scholar] [CrossRef]
- Faust, G.T. Differentiation of Aragonite from Calcite by Differential Thermal Analysis. Science 1949, 110, 402–403. [Google Scholar] [CrossRef]
- Maciejewski, M.; Oswald, H.-R.; Reller, A. Thermal transformations of vaterite and calcite. Thermochim. Acta 1994, 234, 315–328. [Google Scholar] [CrossRef]
- Ke, X.; Criado, M.; Provis, J.L.; Bernal, S.A. Slag-Based Cements That Resist Damage Induced by Carbon Dioxide. ACS Sustain. Chem. Eng. 2018, 6, 5067–5075. [Google Scholar] [CrossRef]
- Zheng, D.; Ji, T.; Wang, G. Effect of CaO on the Autogenous Shrinkage of Alkali-Activated Slag Mortar. Adv. Mater. Sci. Eng. 2021, 2021, 9918834. [Google Scholar] [CrossRef]
- Barzgar, S.; Tarik, M.; Ludwig, C.; Lothenbach, B. The effect of equilibration time on Al uptake in C-S-H. Cem. Concr. Res. 2021, 144, 106438. [Google Scholar] [CrossRef]
- Gruskovnjak, A.; Lothenbach, B.; Holzer, L.; Figi, R.; Winnefeld, F. Hydration of alkali-activated slag: Comparison with ordinary Portland cement. Adv. Cem. Res. 2006, 18, 119–128. [Google Scholar] [CrossRef]
- Kim, M.S.; Jun, Y.; Lee, C.; Oh, J.E. Use of CaO as an activator for producing a price-competitive non-cement structural binder using ground granulated blast furnace slag. Cem. Concr. Res. 2013, 54, 208–214. [Google Scholar] [CrossRef]
- L’Hôpital, E.; Lothenbach, B.; Kulik, D.; Scrivener, K. Influence of calcium to silica ratio on aluminium uptake in calcium silicate hydrate. Cem. Concr. Res. 2016, 85, 111–121. [Google Scholar] [CrossRef]
- Scrivener, K.; Snellings, R.; Lothenbach, B. A Practical Guide to Microstructural Analysis of Cementitious Materials; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Gardeh, M.G.; Kistanov, A.A.; Nguyen, H.; Manzano, H.; Cao, W.; Kinnunen, P. Exploring Mechanisms of Hydration and Carbonation of MgO and Mg(OH)2 in Reactive Magnesium Oxide-Based Cements. J. Phys. Chem. C 2022, 126, 6196–6206. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.M.; Jang, J.; Naeem, F.; Lee, H.K. Heavy Metal Leaching, CO2 Uptake and Mechanical Characteristics of Carbonated Porous Concrete with Alkali-Activated Slag and Bottom Ash. Int. J. Concr. Struct. Mater. 2015, 9, 283–294. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CaO | SiO2 | Al2O3 | Fe2O3 | MgO | Na2O | K2O | SO3 | TiO2 | P2O5 | Mn2O3 | SrO | LOI * |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 43.61 | 36.18 | 14.15 | 0.31 | 3.46 | 0.22 | 0.53 | 0.37 | 0.67 | 0.02 | 0.37 | 0.07 | 0.03 |
| Component | Slag | Q0 | Q1(I) | Q1(II) | Q2(1Al) | Q2 | Q3(1Al)/Q4(4Al) | Q3/Q4(3Al) | Q4(2Al) | Q4 |
|---|---|---|---|---|---|---|---|---|---|---|
| Position | − | −74 | −78 | −80 | −83 | −86 | −89 | −93 | −100 | −107 |
| NaOH | 49.4 | 0.0 | 1.9 | 24.1 | 14.7 | 8.8 | 1.1 | 0.0 | ||
| NaOH-sC | 10.3 | 1.7 | 2.2 | 3.7 | 7.8 | 10.8 | 26.7 | 34.9 | 1.9 | |
| Na2SiO3 | 49.3 | 0.0 | 1.9 | 6.3 | 22.7 | 12.7 | 4.0 | 3.2 | ||
| Na2SiO3-sC | 7.6 | 3.3 | 4.1 | 3.5 | 7.2 | 8.2 | 20.2 | 38.4 | 7.5 | |
| Na2SO4 | 75.4 | 0.0 | 0.4 | 1.7 | 4.8 | 12.7 | 5.0 | 0.0 | ||
| Na2SO4-sC | 21.5 | 2.6 | 0.7 | 5.0 | 6.2 | 7.4 | 25.2 | 27.7 | 3.7 | |
| CaO | 65.8 | 0.0 | 0.6 | 4.5 | 17.9 | 6.9 | 2.5 | 1.8 | ||
| CaO-sC | 20.7 | 2.0 | 1.1 | 3.2 | 8.0 | 7.9 | 15.4 | 35.4 | 6.3 | |
| MgO | 63.8 | 0.5 | 0.9 | 2.7 | 15.8 | 8.0 | 5.4 | 3.1 | ||
| MgO-sC | 27.9 | 3.3 | 1.3 | 5.6 | 9.0 | 7.1 | 18.3 | 21.1 | 6.4 |
| NaOH | Na2SiO3 | Na2SO4 | CaO | MgO | |
|---|---|---|---|---|---|
| CO2/slag | 23.49 | 18.62 | 14.88 | 20.26 | 17.36 |
| CO2/reacted slag | 27.99 | 21.03 | 21.26 | 29.99 | 29.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reddy, K.C.; Seo, J.; Yoon, H.N.; Kim, S.; Kim, G.M.; Son, H.M.; Park, S.; Park, S. Supercritical CO2-Induced Evolution of Alkali-Activated Slag Cements. Materials 2022, 15, 5873. https://doi.org/10.3390/ma15175873
Reddy KC, Seo J, Yoon HN, Kim S, Kim GM, Son HM, Park S, Park S. Supercritical CO2-Induced Evolution of Alkali-Activated Slag Cements. Materials. 2022; 15(17):5873. https://doi.org/10.3390/ma15175873
Chicago/Turabian StyleReddy, Kamasani Chiranjeevi, Joonho Seo, H. N. Yoon, Seonhyeok Kim, G. M. Kim, H. M. Son, Seunghee Park, and Solmoi Park. 2022. "Supercritical CO2-Induced Evolution of Alkali-Activated Slag Cements" Materials 15, no. 17: 5873. https://doi.org/10.3390/ma15175873
APA StyleReddy, K. C., Seo, J., Yoon, H. N., Kim, S., Kim, G. M., Son, H. M., Park, S., & Park, S. (2022). Supercritical CO2-Induced Evolution of Alkali-Activated Slag Cements. Materials, 15(17), 5873. https://doi.org/10.3390/ma15175873