3.1. Hydrothermal Synthesis
Hydroxyapatite nanopowders prepared from natural sources were synthesized under hydrothermal conditions at temperatures, between 100–150 °C, and different pressures (2, 6, and 10 MPa). The resulting powders were denoted HAP-20, HAP-60, and HAP-100, with the numbers in the sample code signifying the working pressure in bar units.
The chemical reactions that lead to hydroxyapatite, starting with Rapana Thomasiana shells, are written below:
Equations (1) and (2) describe chemical reactions which take place duringthe dissolving of Rapana shells in the HCl-HNO3 mixture. Hydrothermal reactions are represented by Equations (3) and (4). Calcium solution, consisting of CaCl2 and Ca(NO3)2 aqueous species, is mixed with NH4·H2PO4 as the P precursor of HAp and precipitated with NH3 25% solution. During hydrothermal synthesis at a high temperature and pressure, crystalline HAp nanoparticles are formed.
It is well known that crystalline hydroxyapatite can be obtained using the hydrothermal method in a relatively wide temperature range (from 70 °C to 200 °C) [
4,
44,
45,
46]. An important advantage of using the hydrothermal process for HAp synthesis, besides controlled morphology and nanometer particle size, is that no hydroxyl defects are produced in the structure [
44].
Hydrothermal synthesis takes place in a perfectly sealed reaction system, in aqueous solution, at a high temperature and pressure. Usually, pressure is created by the saturated vapor phase which forms above the solution, and the most varied hydrothermal synthesis parameters, according to literature data, are temperature, time, and pH [
44,
46]. Although preliminary results on hydrothermal synthesis of HAp obtained from Rapana shells in high pressure conditions (10 MPa) were reported in our previous paper [
43], the effect of pressure on the physical-chemical properties of HAp was not investigated.
Numerous scientific papers [
47,
48,
49,
50] have developed various models for thermodynamic calculation of physicochemical processes that take place in aqueous or non-aqueous solutions during hydrothermal synthesis. Depending on the results obtained, not only can the appropriate solvent be selected, but also the pressure-temperature range that leads to the formation of the desired reaction products and allows for the control of the shape and size of the obtained particles. The behavior of the solvent under hydrothermal conditions, in terms of its structure in critical, super-critical, and sub-critical conditions, as well as the dielectric constant, pH variation, viscosity, coefficient of expansion, and density, must all be correlated with the temperature and working pressure.
In the case of high-pressure hydrothermal synthesis, an external pressure higher than the water vapor pressure at equilibrium is used. Under these conditions, remarkable results are obtained because the solubility of inorganic materials increases with increasing pressure. Thermodynamic stability also varies with pressure (at very high pressures, denser phases crystallize). Hydrothermal reactions are based on the equilibrium reactions of dissolution-reprecipitation and crystallization.
In the studied system, the pressure above the aqueous suspension in the autoclave vessel corresponds to the vapor pressure of the substances in the reaction system, but also to the external pressure of the inert gas introduced. The temperature is kept constant at values much lower than the critical water temperature (T = 100–150 °C << 374 °C) so that the liquid–gas equilibria will be neglected. In this way, due to the high pressure in the reaction system, the crystallization of hydroxyapatite under hydrothermal conditions takes place at temperatures much lower than the usual values for obtaining crystalline calcium phosphates. The influence of the pressure on the equilibrium constant for real systems is determined by the variation of the molar volumes of the participants in the reaction (reactants and reaction products), denoted ΔrV. In the case of hydrothermal reactions ΔrV < 0, the value of the equilibrium constant increases with increasing pressure, meaning the equilibrium shifts to the reaction products (HAp formation reaction is favored by the introduction of external pressure). The introduction of external pressure in the hydrothermal synthesis autoclave favors the obtaining of nanostructured crystalline HAp at relatively low temperatures.
The chemical analysis results (Ca:P ratio) are shown in
Table 2.
The Ca:P ratio is higher than theoretical value of Ca:P = 1.67, which is calculated from the HAp chemical formula Ca10(PO4)6(OH)2, which could be explained by the formation of non-stoichiometric hydroxyapatite, because of the natural Ca source used in the synthesis.
3.3. Analysis of Particle Size Distribution by DLS Technique
Dynamic light scattering (DLS) is an established measurement technique for the characterization of particle sizes in suspension, based on the Brownian motion of particles. The smaller the particles, the faster they will move in a solution. The hydrodynamic diameter represents the particle size plus the dielectric layer, which adheres to its surface during movement through the liquid medium. The movement of the particles causes intensity fluctuations in the scattered light. From these fluctuations, the diffusion coefficient can be determined, and thus the hydrodynamic diameter of the particle is obtained from the Stokes–Einstein equation:
where: d(H) = hydrodynamic diameter, k = Boltzmann’s constant (1.38 × 10
−23 NmK
−1), T = absolute temperature (K), η = solvent viscosity (N·s·m
−2), and D = diffusion coefficient (m
2·s
−1).
The results obtained for synthesized hydroxyapatite powders (hydrodynamic diameter and polydispersity index-PdI) are presented in
Table 3 and
Figure S1. The average particle size (hydrodynamic diameter in aqueous solutions) varies between 76 nm and 97 nm, with a monomodal size distribution. The low values of the polydispersity index suggest that the investigated samples are homogenous in size. The PdI is situated in the range of 0.009–0.087.
3.4. SEM Characterization
An SEM image of HAp nanopowder prepared at 10 MPa is presented in
Figure 3.
All three analyzed samples are formed of irregularly shaped microcrystalline aggregates that have dimensions on the order of microns up to tens of microns. SEM images of HAP-20 and HAP-60 samples are presented in
Figure S2. Microcrystalline aggregates are in turn formed of exceedingly small microcrystals (on the order of nanometers), whose shapes and sizes could not be highlighted. Moreover, morphological investigation revealed a porous structure of hydroxyapatite, regardless of the synthesis pressure. This porous structure, with nano-sized pores (determined by BJH method), represents an advantage for medical applications (bone tissue reconstruction) [
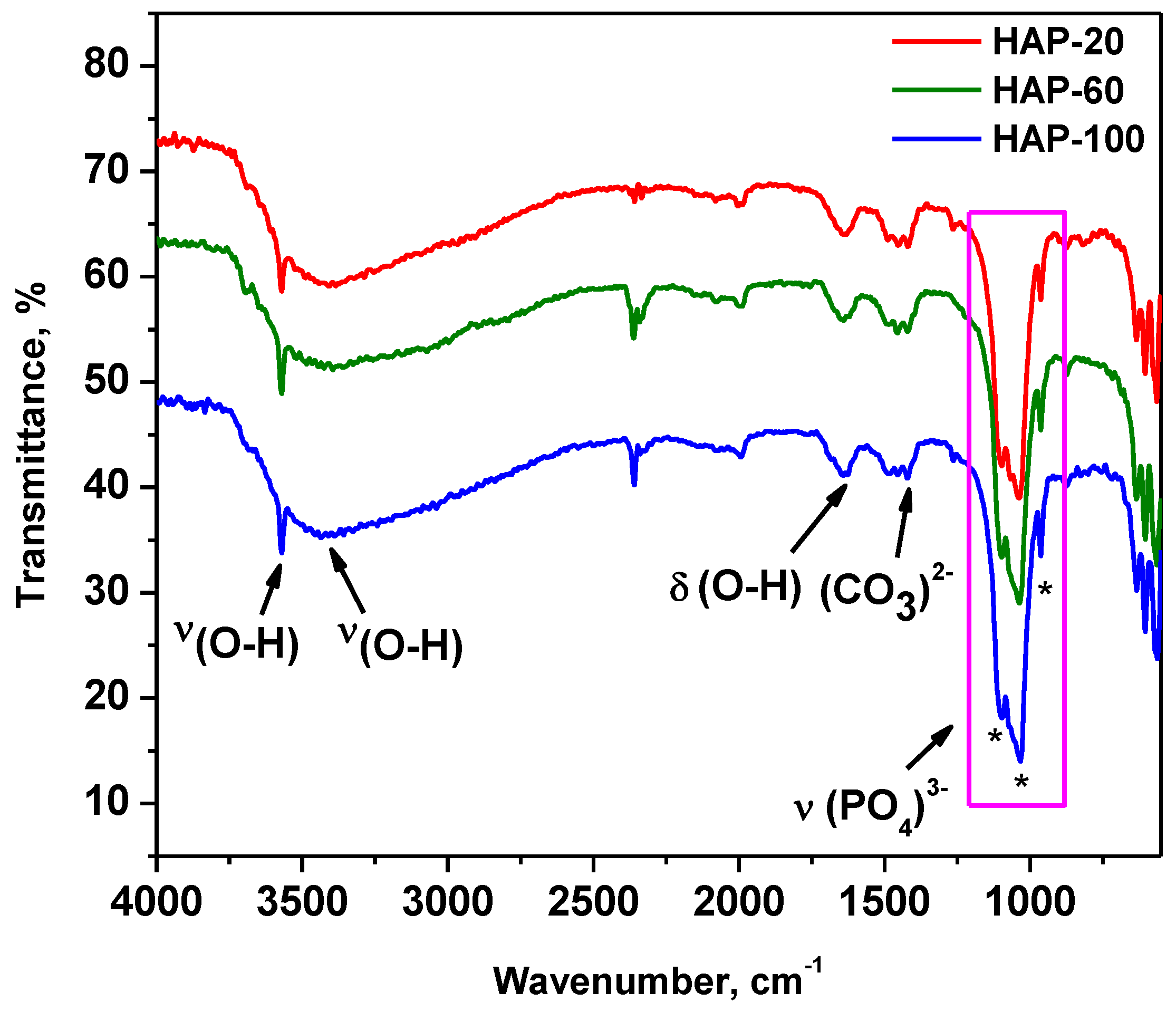
56]. Thus, the BJH adsorption average pore width is 2.75 nm for HAP-20, 2.74 nm for HAP-60, and 2.67 nm for HAP-100 powder samples.
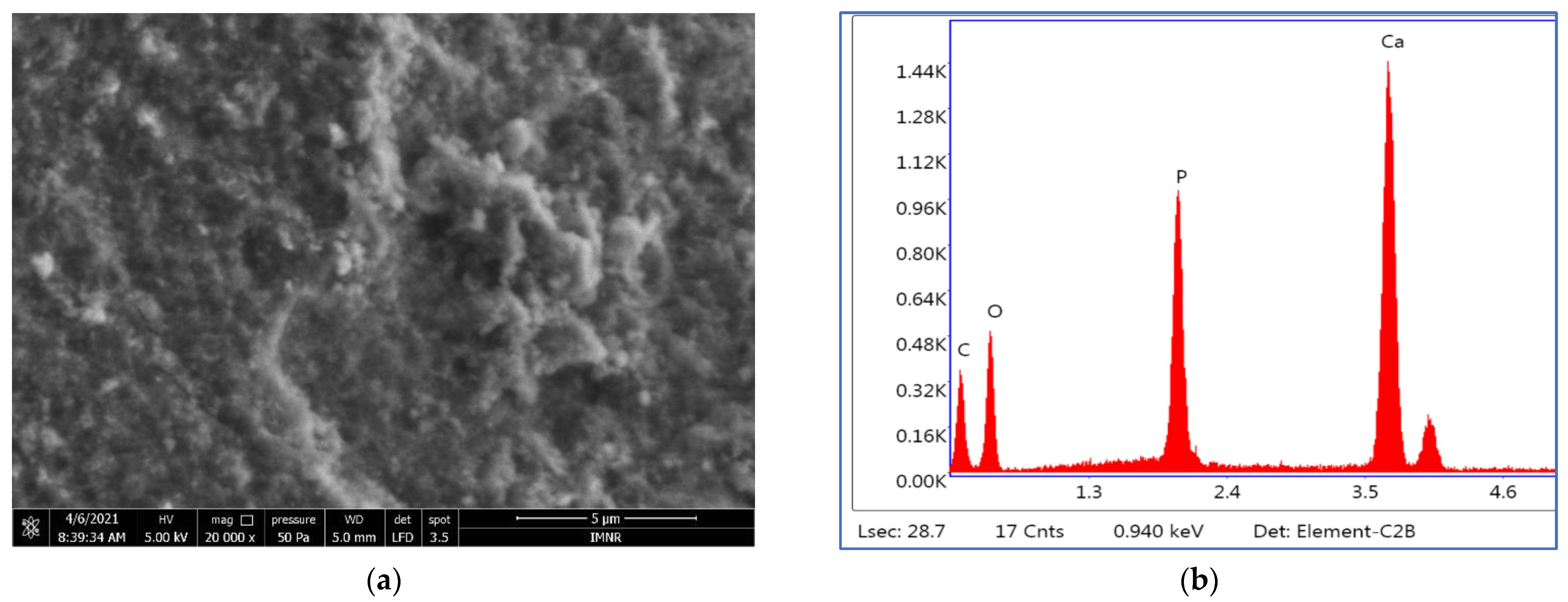
The EDS semiquantitative analysis results are presented in
Table 5.
The Ca:P ratios calculated based on EDS analysis agree with those obtained by chemical analysis.
Based on the results obtained from the physical-chemical characterization of the HAp samples, powders prepared at 10 MPa were further selected for mechanical and in vitro testing. The reason is that HAP-100 shows the highest crystallinity index, 79.4%, calculated from XRD measurements. In X-ray diffraction, it is revealed that the smaller the crystallite size, the more amorphous the material will be considered [
57]. We also assume, based on our previous results in this field, that a higher pressure positively influences the biocompatible properties of the material [
43,
58].
3.6. Biocompatibility Assessment
Biocompatibility can be broadly defined as the physical, chemical, and biological compatibility between a biomaterial and body tissues and the optimal compatibility of a biomaterial with the mechanical behavior of the body.
The biocompatibility of any biomaterial (medical device) must be evaluated using in vitro and in vivo testing before use in patients. While animal experiments are expensive and require extended periods of experimentation, cell culture methods can be performed at lower cost, are faster and easier to perform, and can be easily reproduced. In recent years, a wide range of in vitro tests have been developed to evaluate the biocompatibility of different biomaterials (powders, solutions, hydrogels, medical devices). Such an in vitro assay uses MTT {3-(4,5-dimethithiazol-2-yl)-2,5-diphenyl tetrazolium bromide} and is a sensitive, quantitative, and reliable colorimetric assay that measures cell viability, proliferation, and activation. In living cells, water-soluble yellow MTT is reduced to a dark blue formazan product by the mitochondrial dehydrogenase enzyme. The amount of formazan produced is directly proportional to the number of viable cells present. Therefore, measuring the optical density will help to determine the amount of formazan produced and thus, the number of viable cells present.
3.6.1. Cytotoxicity Test
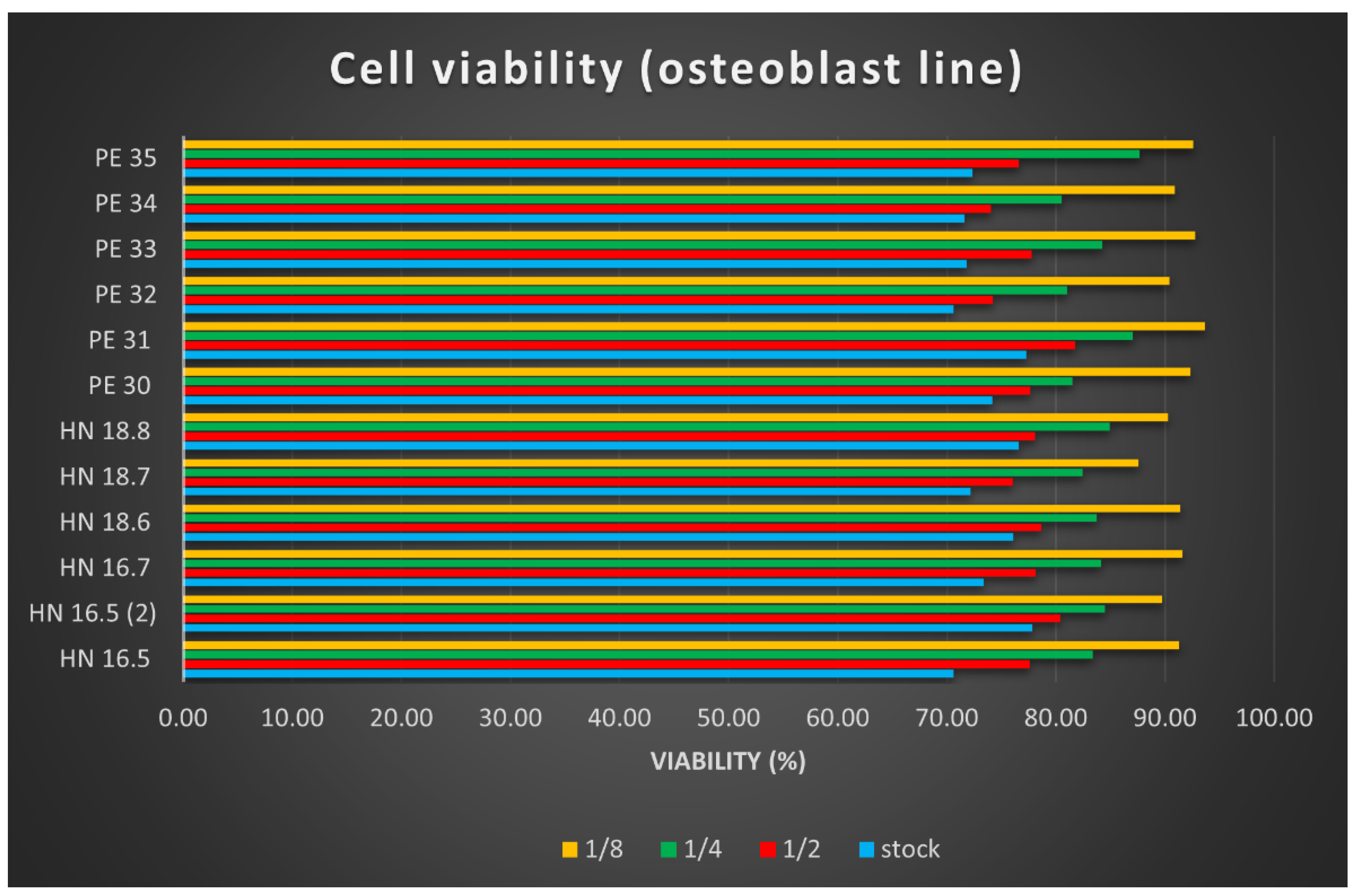
After reading the values of the optical densities for the osteoblast cell line used in testing the cytotoxicity of the 12 HAp samples, the arithmetic means of the values were calculated, and the viability calculation formula was applied. The results were plotted in
Figure 7. Samples called HN-x are square specimens (3D printed samples with dimensions of 15 × 15 × 5 mm
3), and they were studied for comparative reasons.
The cell viability results determined using the MTT test helped us to conclude the following:
The cytotoxic effect of the 12 HAp samples tested is low, with very small differences depending on their size; square (printed) samples, dried at 100 °C showed better results.
The cytotoxicity of the tested samples was dose-dependent; the lower the concentration of the tested product, the lower the cytotoxicity.
The cell viability is lowest in culture wells with an extract stock concentration, and it increases in direct proportion to the increase in dilution.
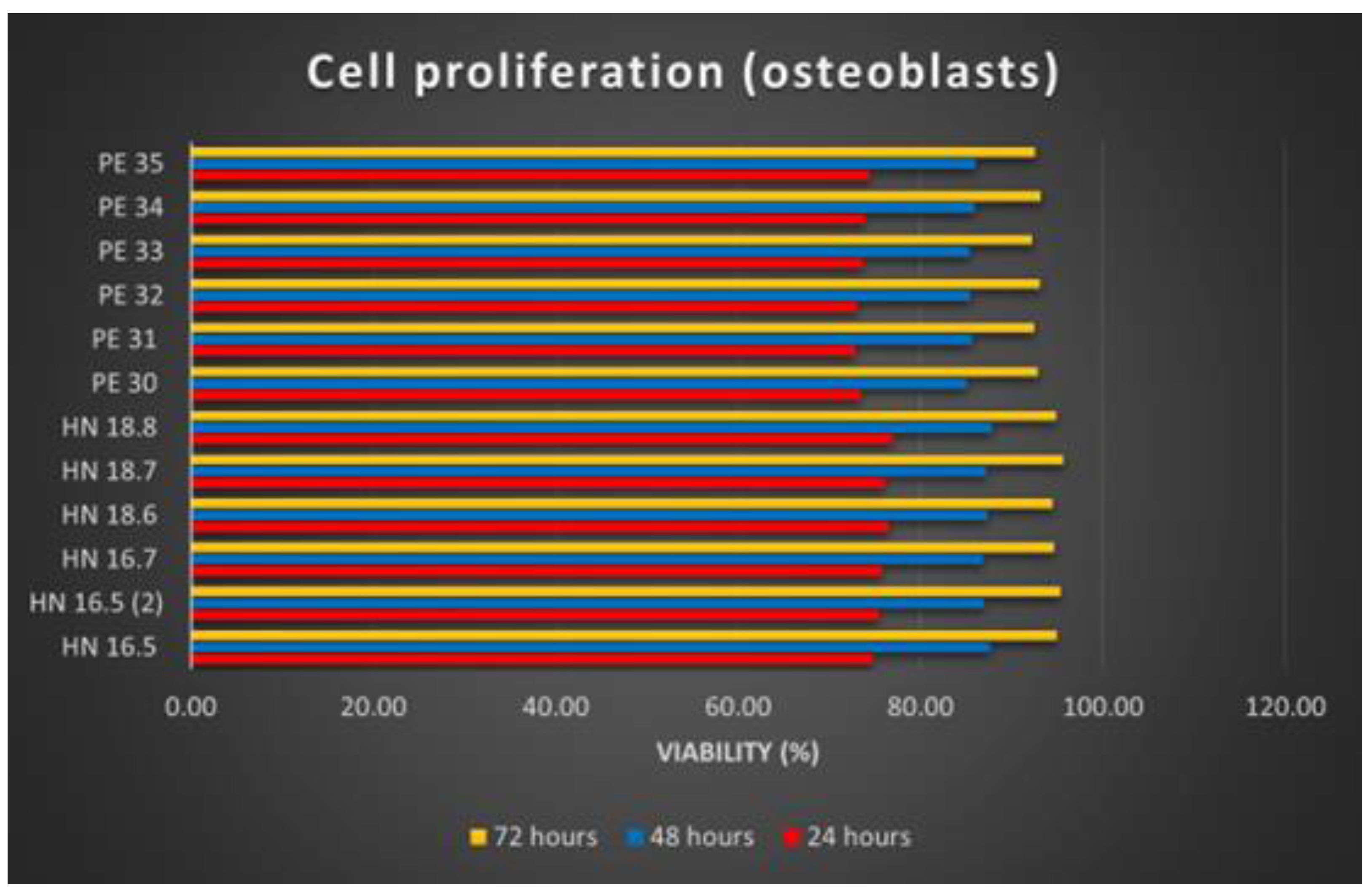
3.6.2. Cell Proliferation Study
Osteoblast proliferation on the 12 HAp samples was analyzed at 24, 48, and 72 h of substrate–cell interaction. In the case of the cell proliferation test, after reading the absorbance values, calculating the average of the values read for each sample and applying the calculation formula, the results were synthesized as graphs (
Figure 8).
The increasing number of osteoblasts used for the proliferation tests of the 12 samples shows the proliferation production on all these samples. Comparing the 12 HAp samples, it is noticeable that the differences are insignificant. Better proliferation has been observed for osteoblasts grown on square (3D printed) samples. Regarding the time dynamics of cell proliferation, it increases in direct proportion to the increase in the substrate–cell contact period.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}