
First-Principles Density Functional Theory Study of Modified Germanene-Based Electrode Materials



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
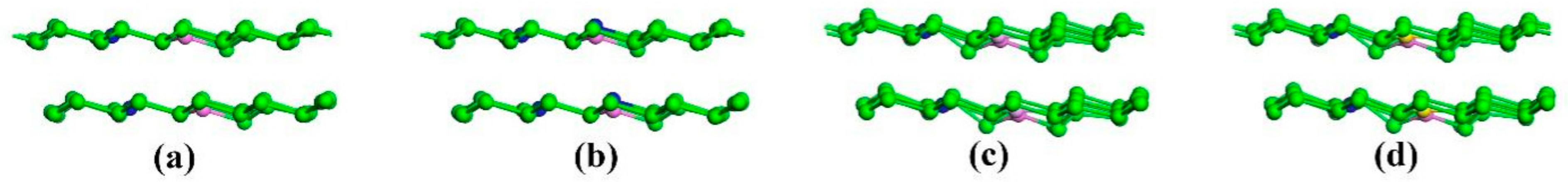
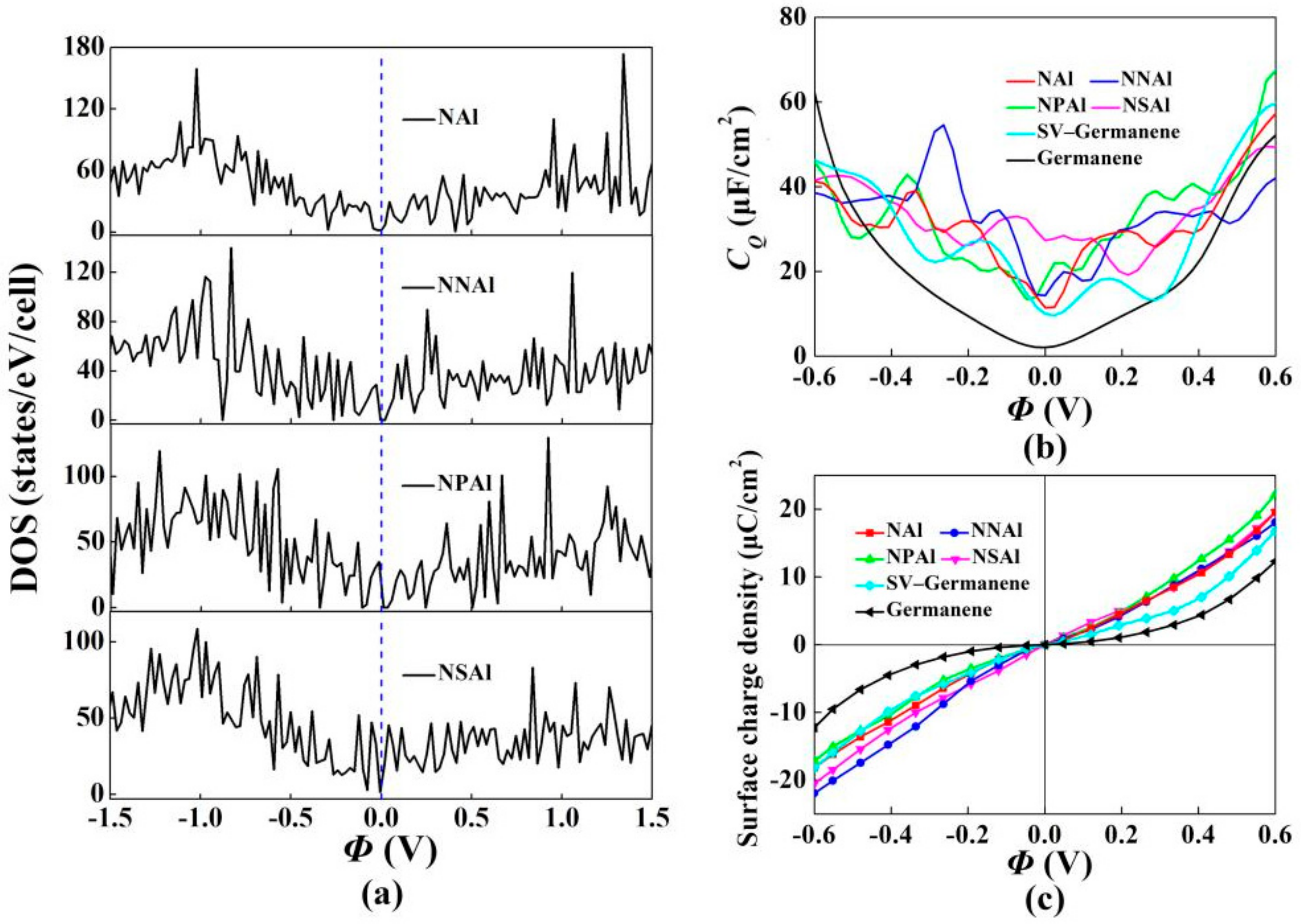
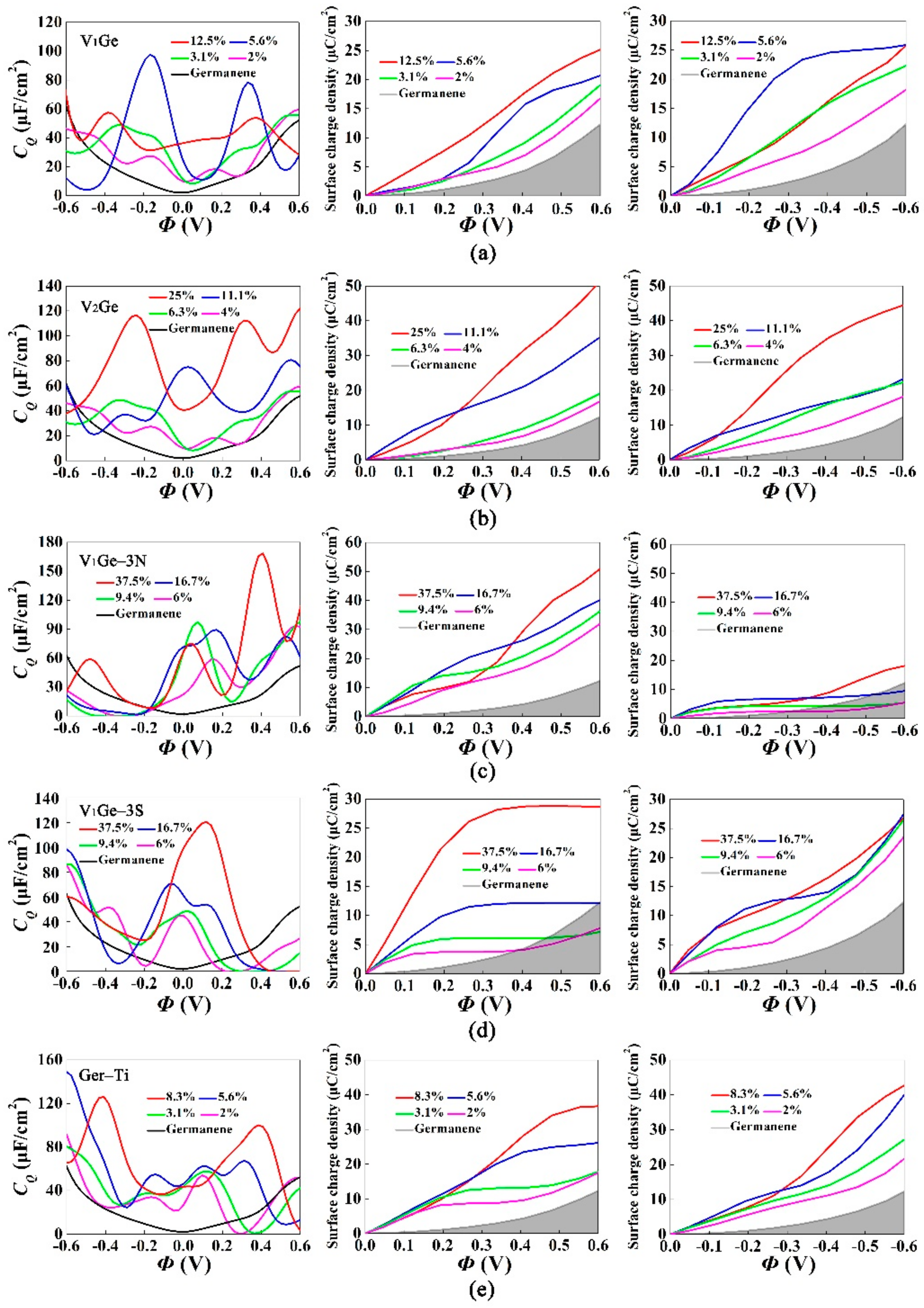
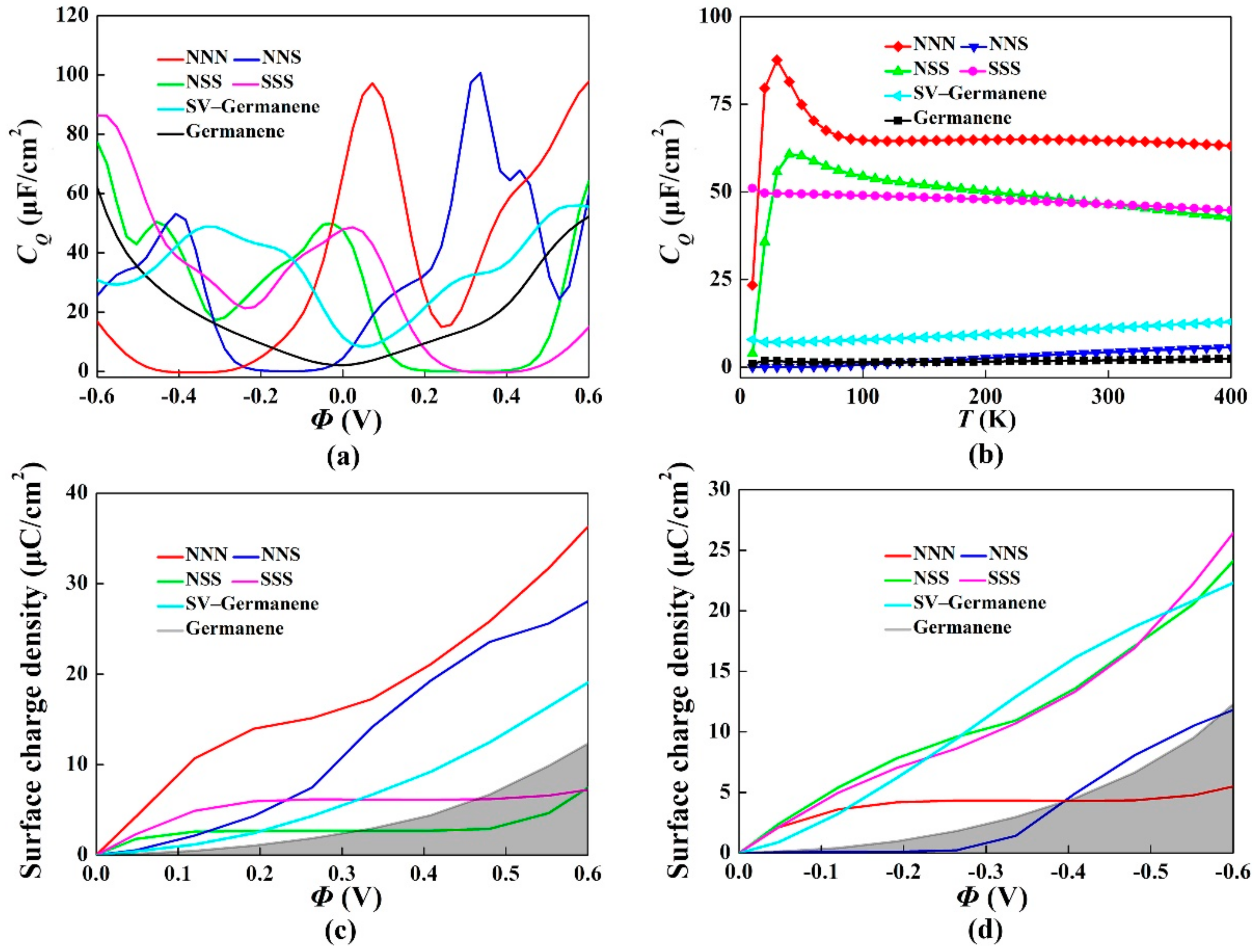
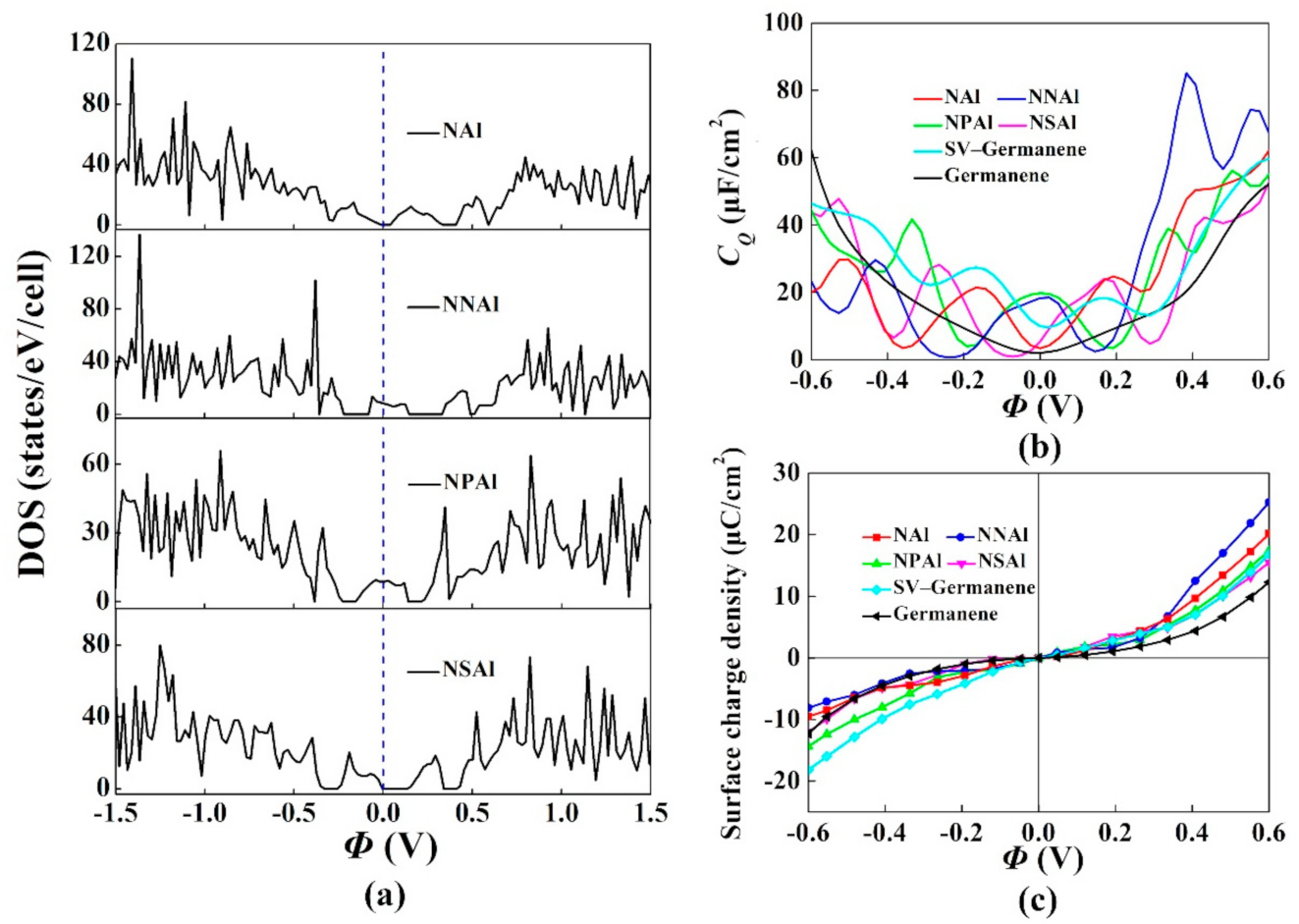
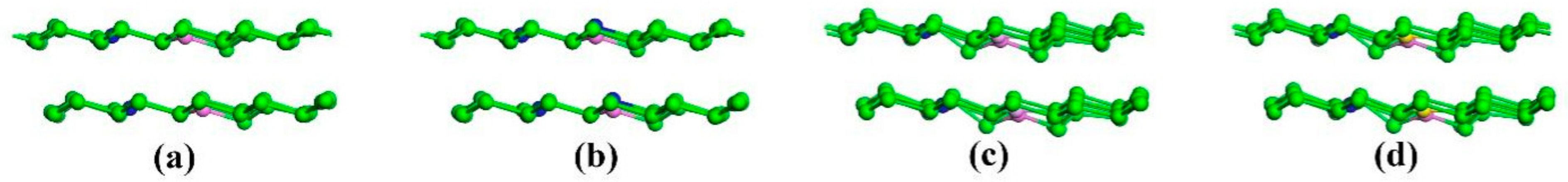
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simon, P.; Gogotsi, Y. Materials for electrochemical capacitors. Nat. Mater. 2008, 7, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Koenig, G.M. Review article: Flow battery systems with solid electroactive materials. J. Vac. Sci. Technol. B 2017, 35, 040801. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.; Qin, Z. A novel kind of activated carbon foam electrode for electric double layer capacitors. Int. J. Electrochem. Sci. 2017, 12, 1846–1862. [Google Scholar] [CrossRef]
- Chaudoy, V.; Tran Van, F.; Deschamps, M.; Ghamouss, F. Ionic liquids in a polyethylene oxide cross-linked gel polymer as an electrolyte for electrical double layer capacitor. J. Power Sources 2017, 342, 872–878. [Google Scholar] [CrossRef]
- Gaboriau, D.; Boniface, M.; Valero, A.; Aldakov, D.; Brousse, T.; Gentile, P.; Sadki, S. Atomic layer deposition alumina-passivated silicon nanowires: Probing the transition from electrochemical double-layer capacitor to electrolytic capacitor. ACS Appl. Mater. Interfaces 2017, 9, 13761–13769. [Google Scholar] [CrossRef]
- Murashko, K.; Nevstrueva, D.; Pihlajamäki, A.; Koiranen, T.; Pyrhönen, J. Cellulose and activated carbon based flexible electrical double-layer capacitor electrode: Preparation and characterization. Energy 2017, 119, 435–441. [Google Scholar] [CrossRef]
- Parida, K.; Bhavanasi, V.; Kumar, V.; Wang, J.; Lee, P.S. Fast charging self-powered electric double layer capacitor. J. Power Sources 2017, 342, 70–78. [Google Scholar] [CrossRef]
- Premathilake, D.; Outlaw, R.A.; Parler, S.G.; Butler, S.M.; Miller, J.R. Electric double layer capacitors for ac filtering made from vertically oriented graphene nanosheets on aluminum. Carbon 2017, 111, 231–237. [Google Scholar] [CrossRef]
- Xu, Y.; Chang, L.; Hu, Y. KOH-assisted microwave post-treatment of activated carbon for efficient symmetrical double-layer capacitors. Int. J. Energy Res. 2017, 41, 728–735. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, X.; Yang, J.; Bo, Z.; Hu, M.; Yan, J.; Cen, K. Molecular origin of electric double-layer capacitance at multilayer graphene edges. J. Phys. Chem. Lett. 2017, 8, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, L.; Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Sun, D.; Tan, C.; Tian, X.; Huang, Y. Al-doped ZnO monolayer as a promising transparent electrode material: A first-principles study. Materials 2017, 10, 359. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Ji, W.; Li, F.; Wang, P.; Hu, S.; Yan, S.; Liu, Y. Tunable electronic and magnetic properties in germanene by alkali, alkaline-earth, group III and 3rd transition metal atom adsorption. Phys. Chem. Chem. Phys. 2014, 16, 15968–15978. [Google Scholar] [CrossRef]
- Zhao, Z.; Fang, F.; Wu, J.; Tong, X.; Zhou, Y.; Lv, Z.; Wang, J.; Sawtell, D. Interfacial chemical effects of amorphous zinc oxide/graphene. Materials 2021, 14, 2481. [Google Scholar] [CrossRef]
- Liu, C.; Jiang, H.; Yao, Y. Low-energy effective hamiltonian involving spin-orbit coupling in silicene and two-dimensional germanium and tin. Phys. Rev. B 2011, 84, 195430. [Google Scholar] [CrossRef] [Green Version]
- Kaloni, T.P. Tuning the structural, electronic, and magnetic properties of germanene by the adsorption of 3d transition metal atoms. J. Phys. Chem. C 2014, 118, 25200–25208. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Si, X.; She, W.; Yang, G.; Fan, X.; Zheng, W. First-principles calculation of optimizing the performance of germanene-based supercapacitors by vacancies and metal atoms. J. Phys. Chem. C 2020, 124, 12346–12358. [Google Scholar] [CrossRef]
- Nair, R.R.; Blake, P.; Grigorenko, A.N.; Novoselov, K.S.; Booth, T.J.; Stauber, T.; Peres, N.M.R.; Geim, A.K. Fine structure constant defines visual transparency of graphene. Science 2008, 320, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Bampoulis, P.; Rudenko, A.N.; Yao, Q.; Van Houselt, A.; Poelsema, B.; Katsnelson, M.I.; Zandvliet, H.J.W. Structural and electronic properties of germanene on MoS2. Phys. Rev. Lett. 2016, 116, 256804. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Bo, G.; Liu, Y.; Xu, X.; Du, Y.; Dou, S. Recent progress on germanene and functionalized germanene: Preparation, characterizations, applications, and challenges. Nano. Micro Small 2019, 15, 1805147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Lu, S.; Pan, J.; Qin, Z.; Wang, Y.; Wang, Y.; Cao, G.; Du, S.; Gao, H. Buckled germanene formation on Pt(111). Adv. Mater. 2014, 26, 4820–4824. [Google Scholar] [CrossRef]
- Zhuang, J.; Gao, N.; Li, Z.; Xu, X.; Wang, J.; Zhao, J.; Dou, S.; Du, Y. Cooperative electron-phonon coupling and buckled structure in germanene on Au(111). ACS Nano 2017, 11, 3553–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derivaz, M.; Dentel, D.; Stephan, R.G.; Hanf, M.C.; Mehdaoui, A.; Sonnet, P.; Pirri, C. Continuous germanene layer on Al(111). Nano Lett. 2015, 15, 2510–2516. [Google Scholar] [CrossRef]
- Qin, Z.; Pan, J.; Lu, S.; Shao, Y.; Wang, Y.; Du, S.; Gao, H.; Cao, G. Direct evidence of dirac signature in bilayer germanene islands on Cu(111). Adv. Mater. 2017, 29, 1606046. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Uhrberg, R.I.G. Investigation of the atomic and electronic structures of highly ordered two-dimensional germanium on Au(111). Phys. Rev. Mater. 2017, 1, 074002. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.; Liu, C.; Zhou, Z.; Casillas, G.; Feng, H.; Xu, X.; Wang, J.; Hao, W.; Wang, X.; Dou, S.; et al. Dirac signature in germanene on semiconducting substrate. Adv. Sci. 2018, 5, 1800207. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, H.; Fan, X.; Zheng, W. Density functional theory calculations for the quantum capacitance performance of graphene-based electrode material. J. Phys. Chem. C 2015, 119, 6464–6470. [Google Scholar] [CrossRef]
- Paek, E.; Pak, A.J.; Hwang, G.S. Curvature effects on the interfacial capacitance of carbon nanotubes in an ionic liquid. J. Phys. Chem. C 2013, 117, 23539–23546. [Google Scholar] [CrossRef]
- Pak, A.J.; Paek, E.; Hwang, G.S. Relative contributions of quantum and double layer capacitance to the supercapacitor performance of carbon nanotubes in an ionic liquid. Phys. Chem. Chem. Phys. 2013, 15, 19741–19747. [Google Scholar] [CrossRef]
- Faizan, M.; Bhamu, K.C.; Murtaza, G.; He, X.; Kulhari, N.; Al-Anazy, M.M.; Khan, S.H. Electronic and optical properties of vacancy ordered double perovskites A2BX6 (A = Rb, Cs; B = Sn, Pd, Pt; and X = Cl, Br, I): A first principles study. Sci. Rep. 2021, 11, 6965. [Google Scholar] [CrossRef]
- Momeni, M.J.; Mousavi-Khoshdel, M.; Leisegang, T. Exploring the performance of pristine and defective silicene and silicene-like XSi3 (X = Al, B, C, N, P) sheets as supercapacitor electrodes: A density functional theory calculation of quantum capacitance. Phys. E 2020, 124, 114290. [Google Scholar] [CrossRef]
- Zhou, Q.; Ju, W.; Yong, Y.; Liu, Y.; Li, J. Quantum capacitance of supercapacitor electrodes based on germanene influenced by vacancy and co-doping: A first-principles study. Comput. Mater. Sci. 2021, 188, 110131. [Google Scholar] [CrossRef]
- Tizroespeli, F.; Parhizgar, S.S.; Beheshtian, J.; Boochani, A. The first principle study of magnetic, electronic, and optical properties of Co-and Mn-doped boron nitride nanosheets. Indian J. Phys. 2021, 33, 1–13. [Google Scholar] [CrossRef]
- Xu, Q.; Yang, G.; Fan, X.; Zheng, W. Improving the quantum capacitance of graphene-based supercapacitors by the doping and co-doping: First-principles calculations. ACS Omega 2019, 4, 13209–13217. [Google Scholar] [CrossRef] [Green Version]
- Lahourpour, F.; Boochani, A.; Parhizgar, S.S.; Elahi, S.M. Structural, electronic and optical properties of graphene-like nano-layers MoX2(X: S,Se,Te): DFT study. J. Theor. Appl. Phys. 2019, 13, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Ju, W.; Yong, Y.; Zhang, Q.; Liu, Y.; Li, J. Effect of the N/P/S, and transition-metal co-doping on the quantum capacita nce of supercapacitor electrodes based on mono-and multilayer graphene. Carbon 2020, 170, 368–379. [Google Scholar] [CrossRef]
- Zhou, Q.; Ju, W.; Liu, Y.; Li, J.; Zhang, Q. Effect of coexistence of defect and dopant on the quantum capacitance of graphene-based supercapacitors electrodes. Appl. Surf. Sci. 2020, 510, 145448. [Google Scholar] [CrossRef]
- Mohammadi, S.; Targholi, E.; Mousavi-Khoshdel, S.M. Metal—Organic framework-derived cobalt hydroxide microparticles as supercapacitor electrode materials. J. Iran. Chem. Soc. 2021, 18, 2115–2122. [Google Scholar] [CrossRef]
- Gopalsamy, K.; Balamurugan, J.; Thanh, T.D.; Kim, N.H.; Lee, J.H. Fabrication of nitrogen and sulfur co-doped graphene nanoribbons with porous architecture for high-performance supercapacitors. Chem. Eng. J. 2017, 312, 180–190. [Google Scholar] [CrossRef]
- Faizan, M.; Xie, J.; Murtaza, G.; Echeverría-Arrondo, C.; Alshahrani, T.; Bhamu, K.C.; Laref, A.; Mora-Sero, I.; Haidar Khan, S. A first-principles study of the stability, electronic structure, and optical properties of halide double perovskite Rb2Sn1−xTexI6 for solar cell applications. Phys. Chem. Chem. Phys. 2021, 23, 4646–4657. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wang, P.; Hua, C.; Yang, Y.; Zhang, Z. The impact of iron adsorption on the electronic and photocatalytic properties of the zinc oxide (0001) surface: A first-principles study. Materials 2018, 11, 417. [Google Scholar] [CrossRef] [Green Version]
- Poljak, M.; Song, E.B.; Wang, M.; Suligoj, T.; Wang, K.L. Influence of edge defects, vacancies, and potential fluctuations on transport properties of extremely scaled graphene nanoribbons. IEEE Trans. Electron. Dev. 2012, 59, 3231–3238. [Google Scholar] [CrossRef]
- Poljak, M. Electron mobility in defective nanoribbons of monoelemental 2D materials. IEEE Electron. Dev. Lett. 2020, 41, 151–154. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Yang, G.; Fan, X.; Shi, S.; Huang, H.; Zheng, W. Stability of Ptn cluster on free/defective graphene: A first-principles study. Appl. Surf. Sci. 2017, 392, 936–941. [Google Scholar] [CrossRef]
- Zhu, B.; Zhang, J.; Jiang, C.; Cheng, B.; Yu, J. First principal investigation of halogen-doped monolayer g-C3N4 photocatalyst. Appl. Catal. B Environ. 2017, 207, 27–34. [Google Scholar] [CrossRef]
- Xu, Q.; Yang, G.; Fan, X.; Zheng, W. Adsorption of metal atoms on silicene: Stability and quantum capacitance of silicene-based electrode materials. Phys. Chem. Chem. Phys. 2019, 21, 4276–4285. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.C.; de Lima, D.B.; Assali, L.V.C.; Justo, J.F. Group IV graphene-and graphene-like nanosheets. J. Phys. Chem. C 2011, 115, 13242–13246. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, S.; Srivastava, A.; Kurchania, R. Silicene and germanene: A first principle study of electronic structure and effect of hydrogenation-passivation. J. Comput. Theor. Nanosci. 2014, 11, 781–788. [Google Scholar] [CrossRef]
- John, D.L.; Castro, L.C.; Pulfrey, D.L. Quantum capacitance in nanoscale device modeling. J. Appl. Phys. 2004, 96, 5180–5184. [Google Scholar] [CrossRef]
- Paek, E.; Pak, A.J.; Kweon, K.E.; Hwang, G.S. On the origin of the enhanced supercapacitor performance of nitrogen-doped graphene. J. Phys. Chem. C 2013, 117, 5610–5616. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Zhou, J.; Xu, L.; Li, X.; Li, L.; Li, X. First-principles calculation of quantum capacitance of metals doped graphenes and nitrogen/metals co-doped graphenes: Designing strategies for supercapacitor electrodes. J. Mater. Sci. 2019, 54, 483–492. [Google Scholar] [CrossRef]
- Yang, G.; Xu, Q.; Fan, X.; Zheng, W. Quantum capacitance of silicene-based electrodes from first-principles calculations. J. Phys. Chem. C 2018, 122, 1903–1912. [Google Scholar] [CrossRef]
- Hirunsit, P.; Liangruksa, M.; Khanchaitit, P. Electronic structures and quantum capacitance of monolayer and multilayer graphenes influenced by Al, B, N and P doping, and monovacancy: Theoretical study. Carbon 2016, 108, 7–20. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Si, X.; She, W.; Xu, Q.; Yang, G.; Li, Z.; Wang, S.; Luan, J. First-Principles Density Functional Theory Study of Modified Germanene-Based Electrode Materials. Materials 2022, 15, 103. https://doi.org/10.3390/ma15010103
Si X, She W, Xu Q, Yang G, Li Z, Wang S, Luan J. First-Principles Density Functional Theory Study of Modified Germanene-Based Electrode Materials. Materials. 2022; 15(1):103. https://doi.org/10.3390/ma15010103
Chicago/Turabian StyleSi, Xue, Weihan She, Qiang Xu, Guangmin Yang, Zhuo Li, Siqi Wang, and Jingfei Luan. 2022. "First-Principles Density Functional Theory Study of Modified Germanene-Based Electrode Materials" Materials 15, no. 1: 103. https://doi.org/10.3390/ma15010103
APA StyleSi, X., She, W., Xu, Q., Yang, G., Li, Z., Wang, S., & Luan, J. (2022). First-Principles Density Functional Theory Study of Modified Germanene-Based Electrode Materials. Materials, 15(1), 103. https://doi.org/10.3390/ma15010103