
Crystal Structure Determination of 4-[(Di-p-tolyl-amino)-benzylidene]-(5-pyridin-4-yl-[1,3,4]thiadiazol-2-yl)-imine along with Selected Properties of Imine in Neutral and Protonated Form with Camforosulphonic Acid: Theoretical and Experimental Studies

 , , , , and
, , , , and
Abstract
1. Introduction
2. Experimental
2.1. Materials
2.1.1. Synthesis of 4-[(Di-p-tolyl-amino)-benzylidene]-(5-pyridin-4-yl-[1,3,4]thiadiazol-2-yl)-imine (PPL9)
2.1.2. Protonation of Imine PPL9
2.2. Methods
2.2.1. X-ray Crystallography
2.2.2. Hirshfeld Surface Analysis
2.2.3. NMR Spectroscopy
2.2.4. Vibrational Spectroscopy
2.2.5. Thermal Analysis
2.2.6. UV Spectroscopy
2.2.7. Electrochemical Analysis
2.2.8. Thermal Imaging
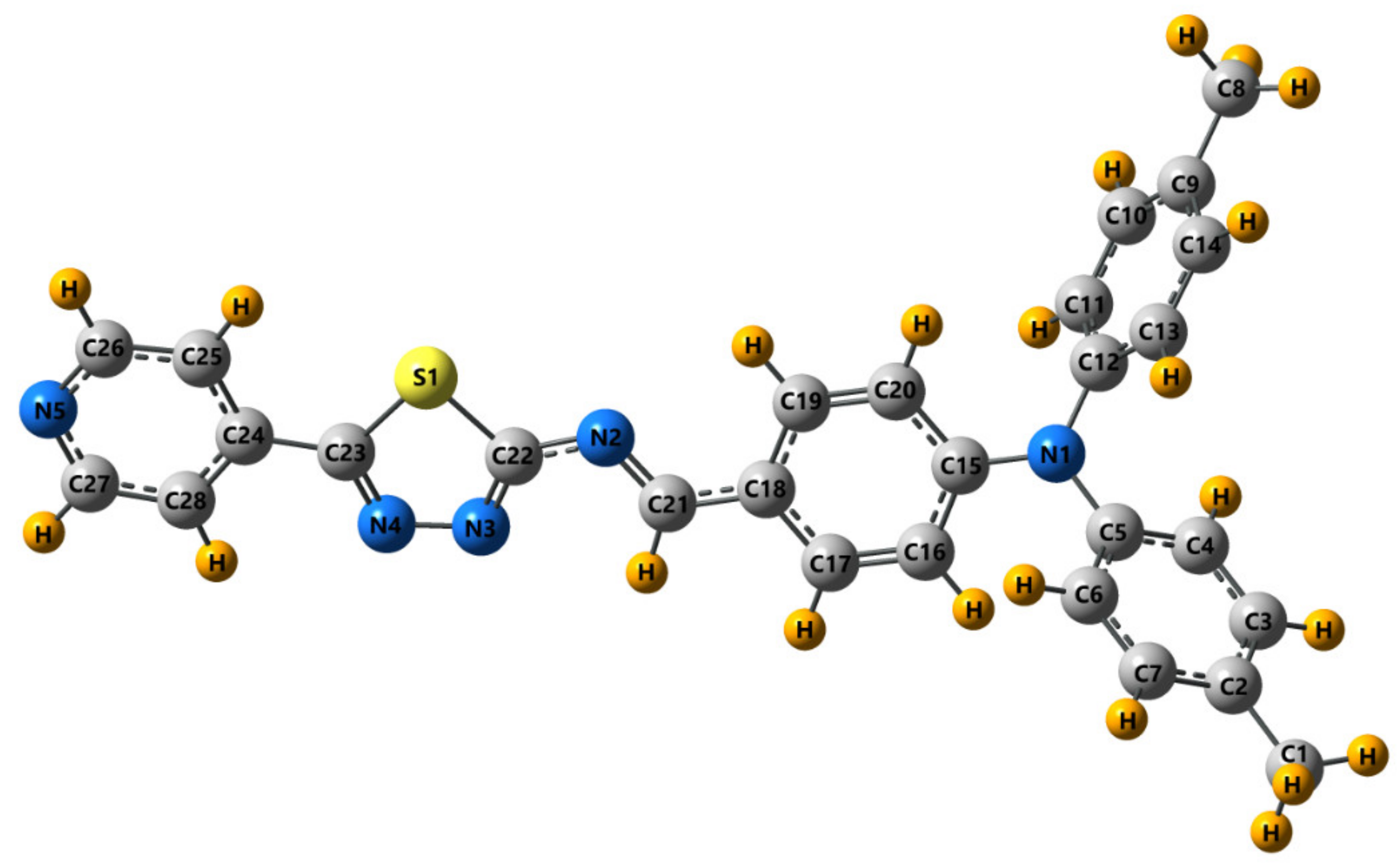
2.2.9. Theoretical Calculations
3. Results and Discussion
3.1. Synthesis of PPL9
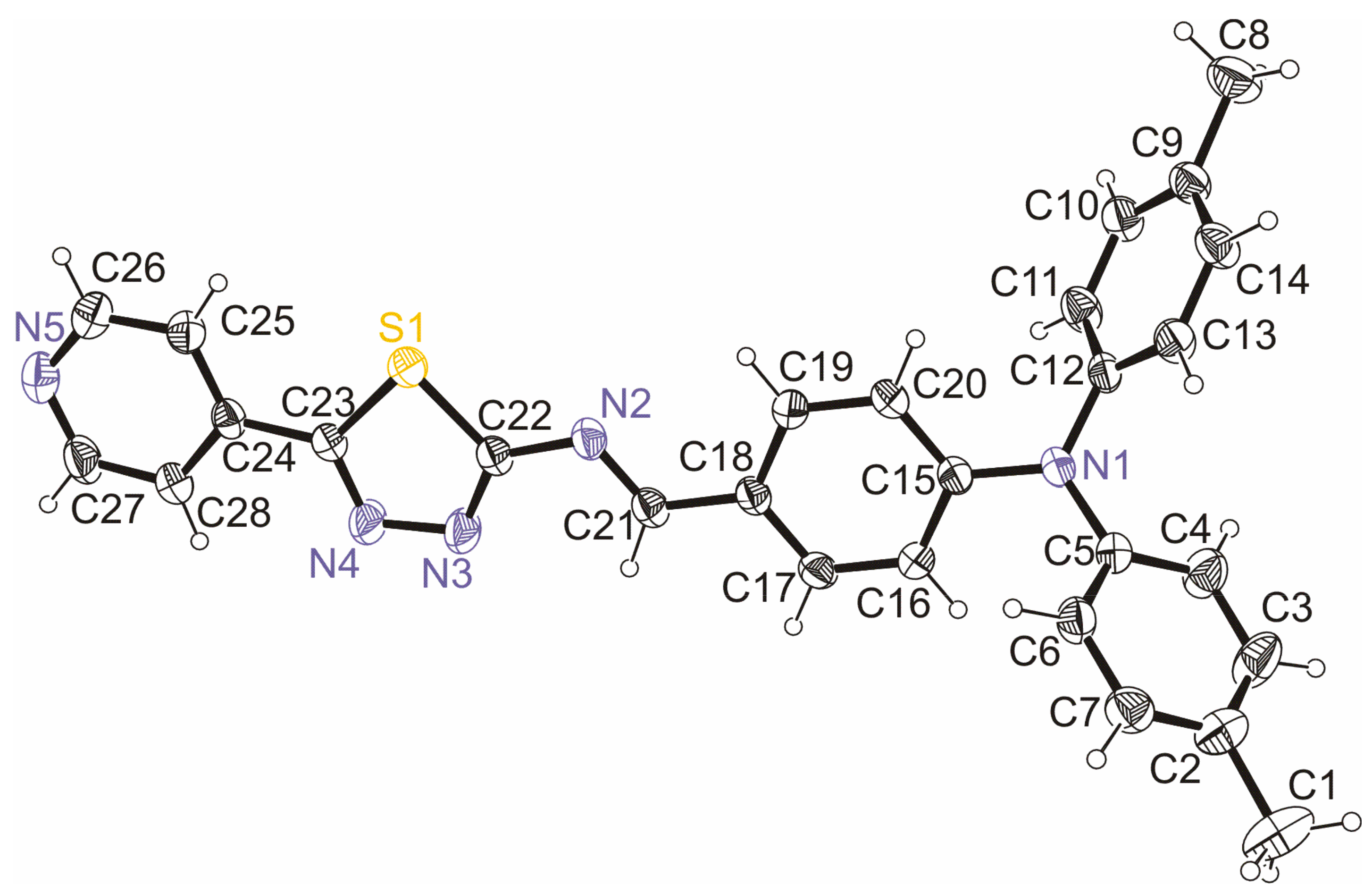
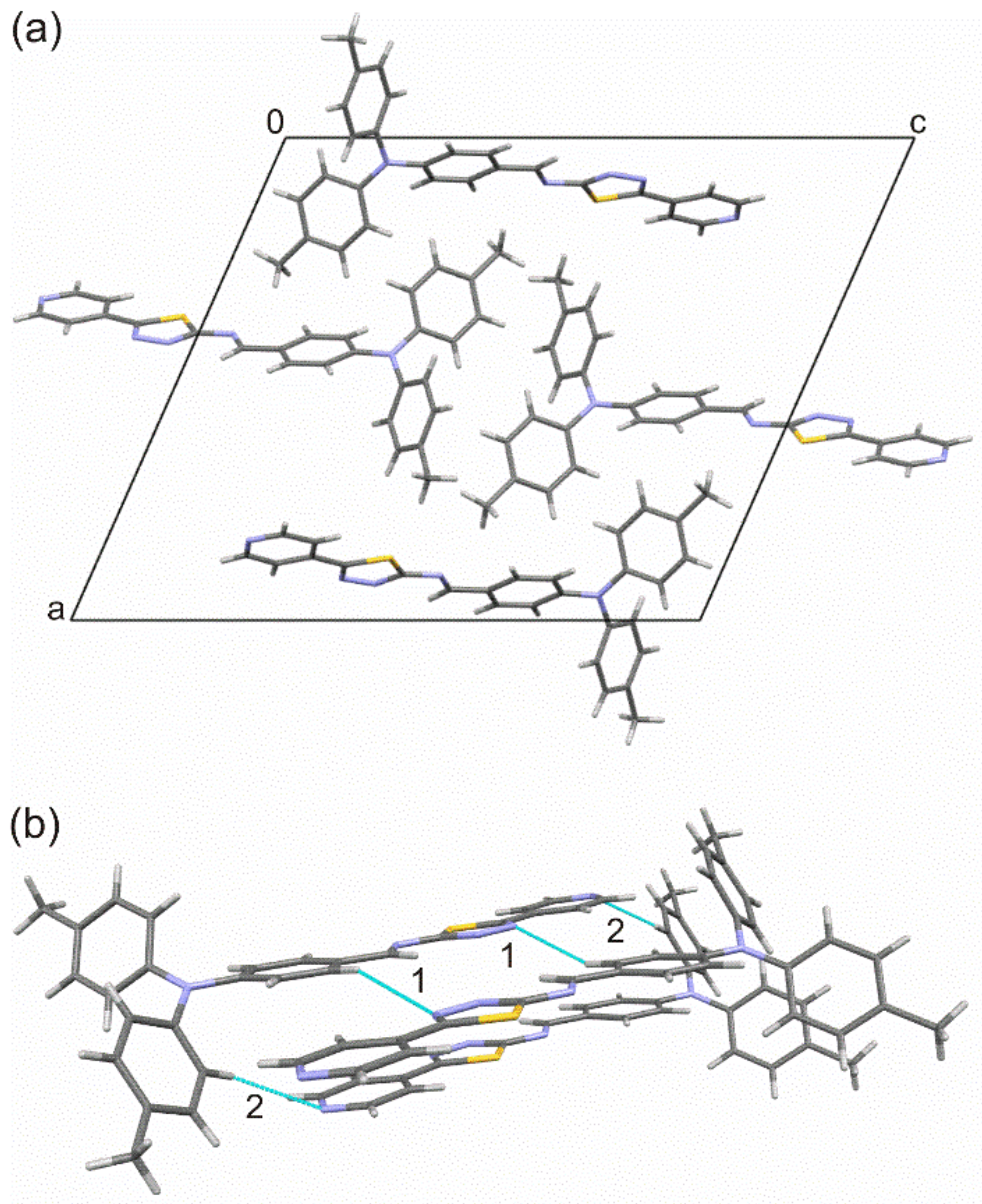
3.2. Crystal and Molecular Structure of Trans-PPL9
3.3. Spectroscopy
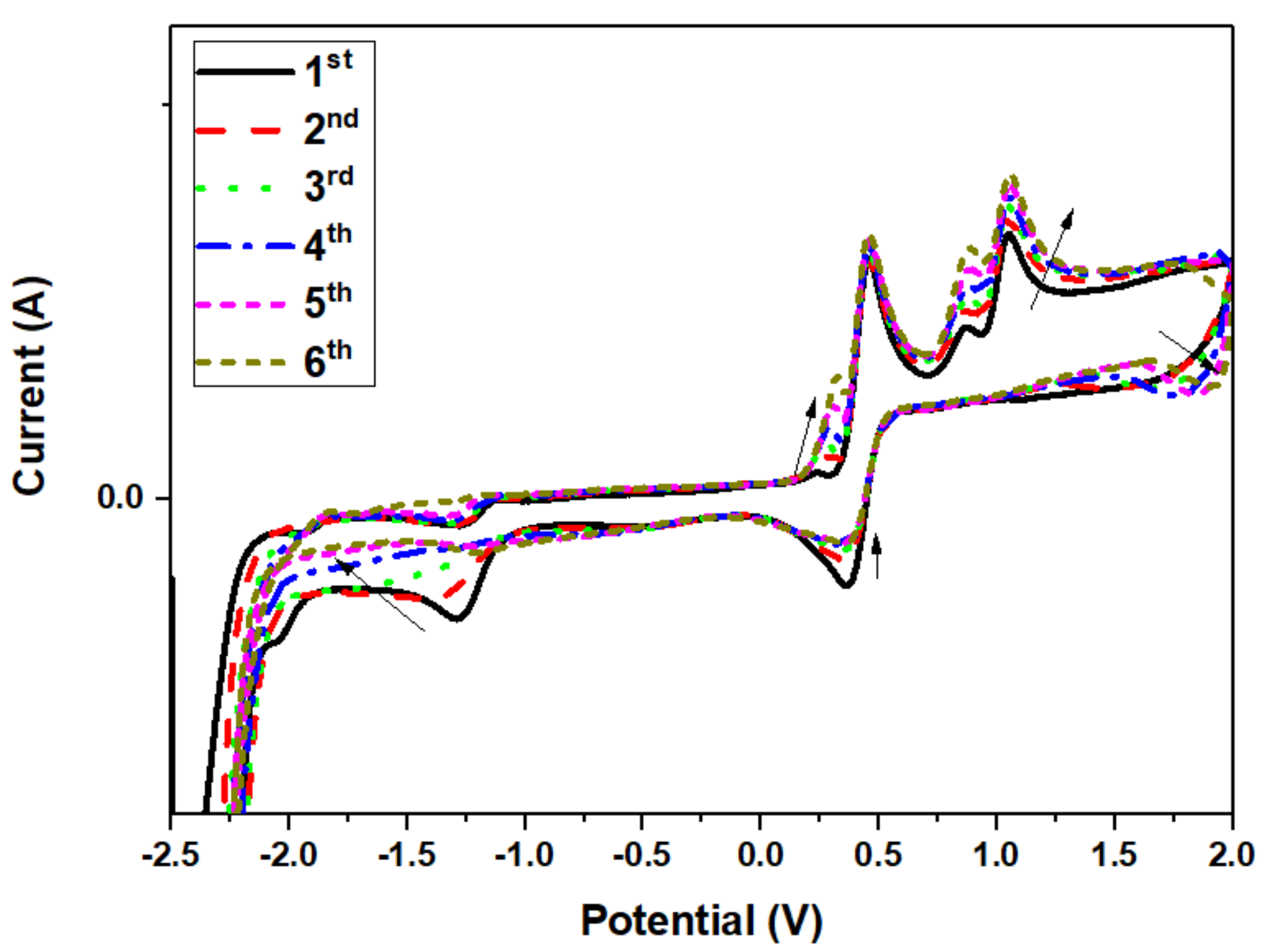
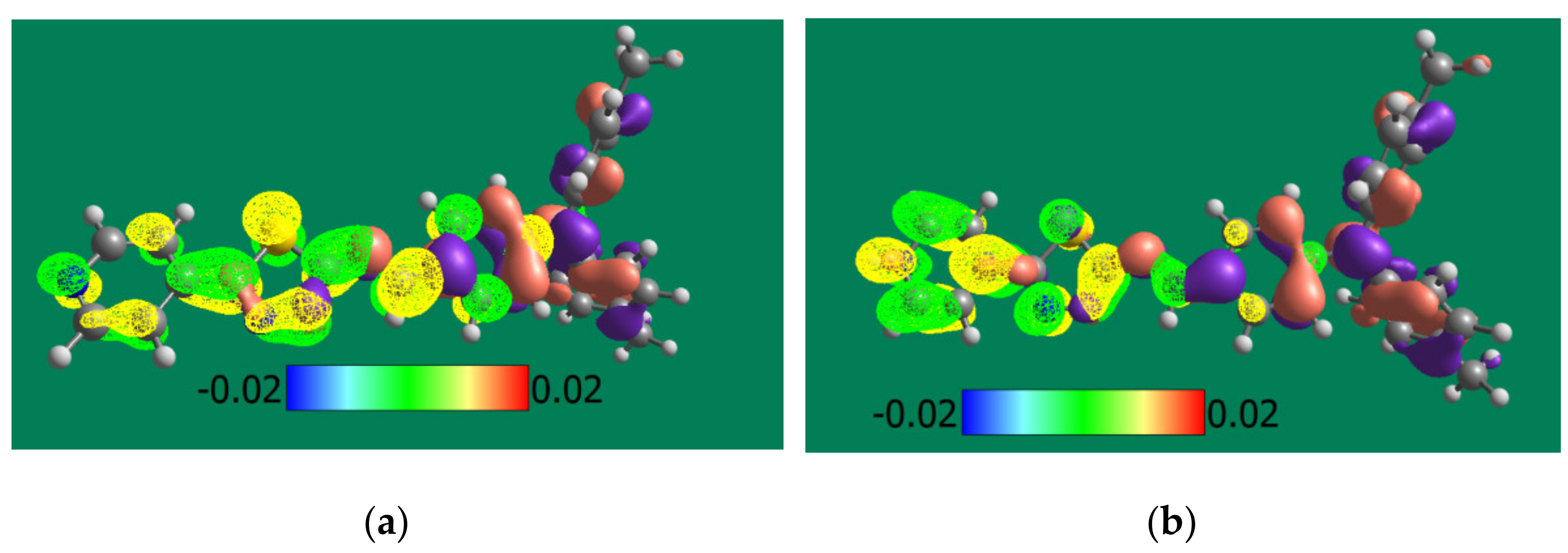
3.4. Theoretical and Experimental Cyclic Voltammetry Study of PPL9 and Their Protonated Form
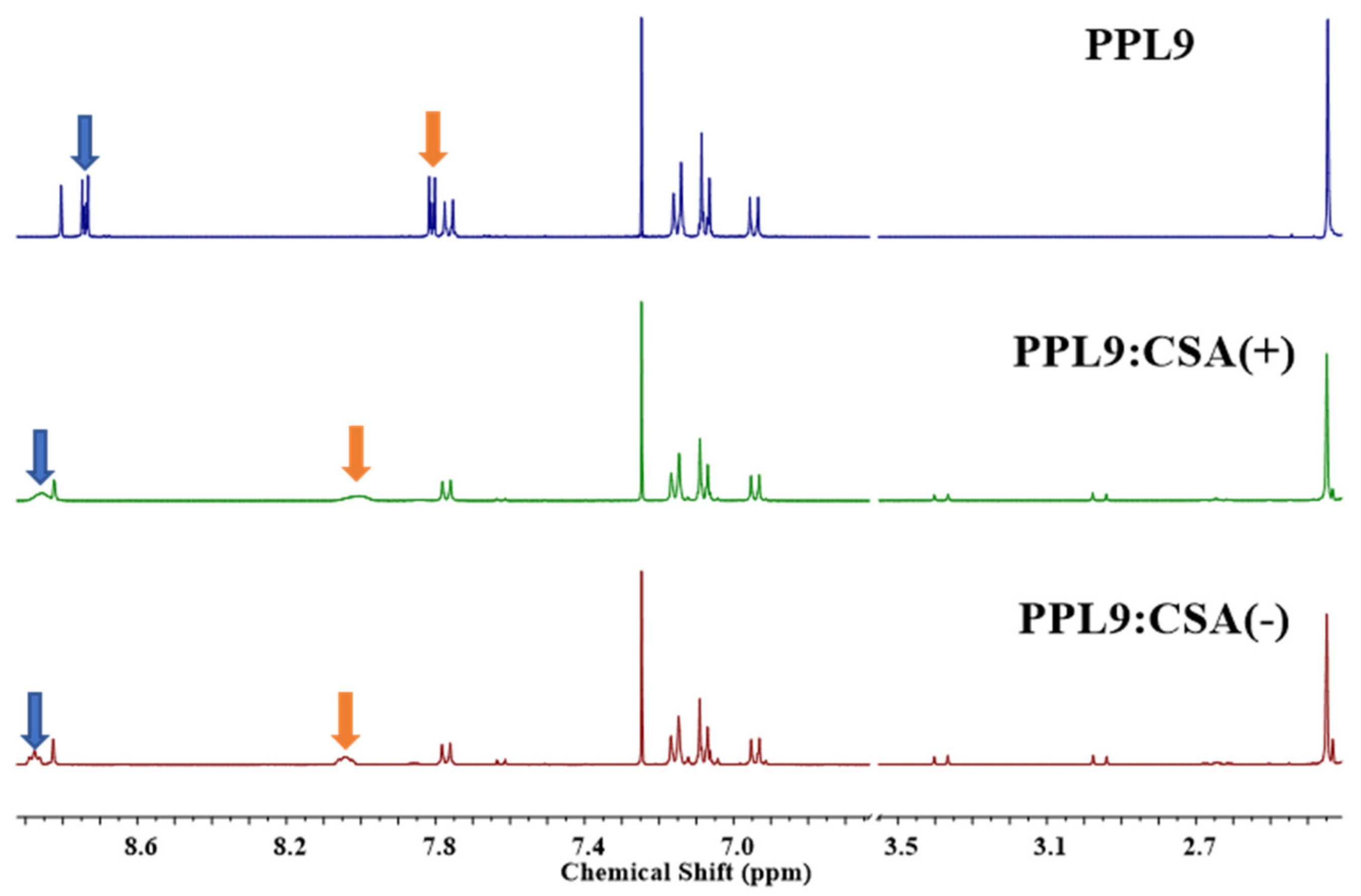
3.5. Proton NMR Study of PPL9:CSA(+) and PPL9:CSA(−)
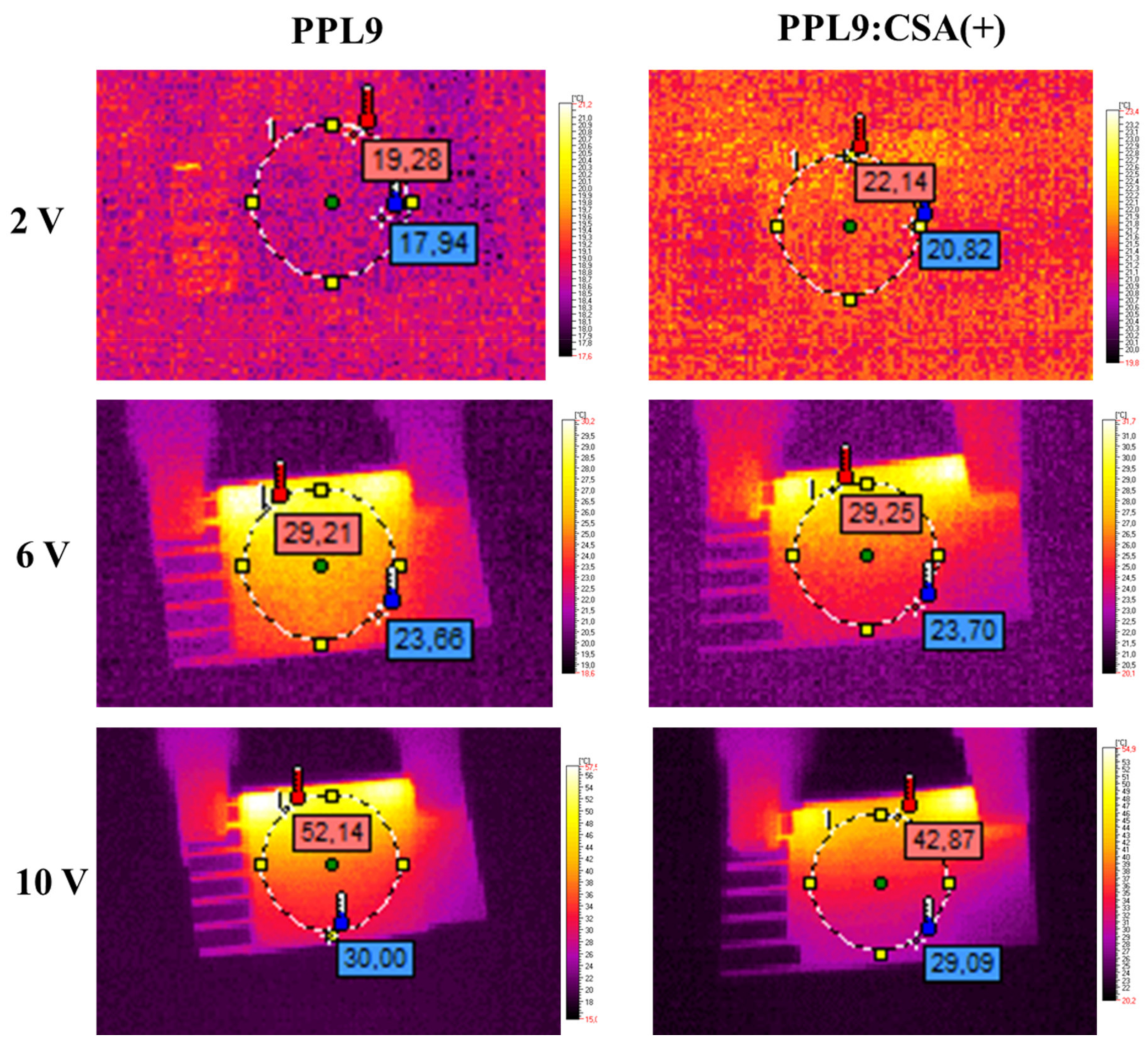
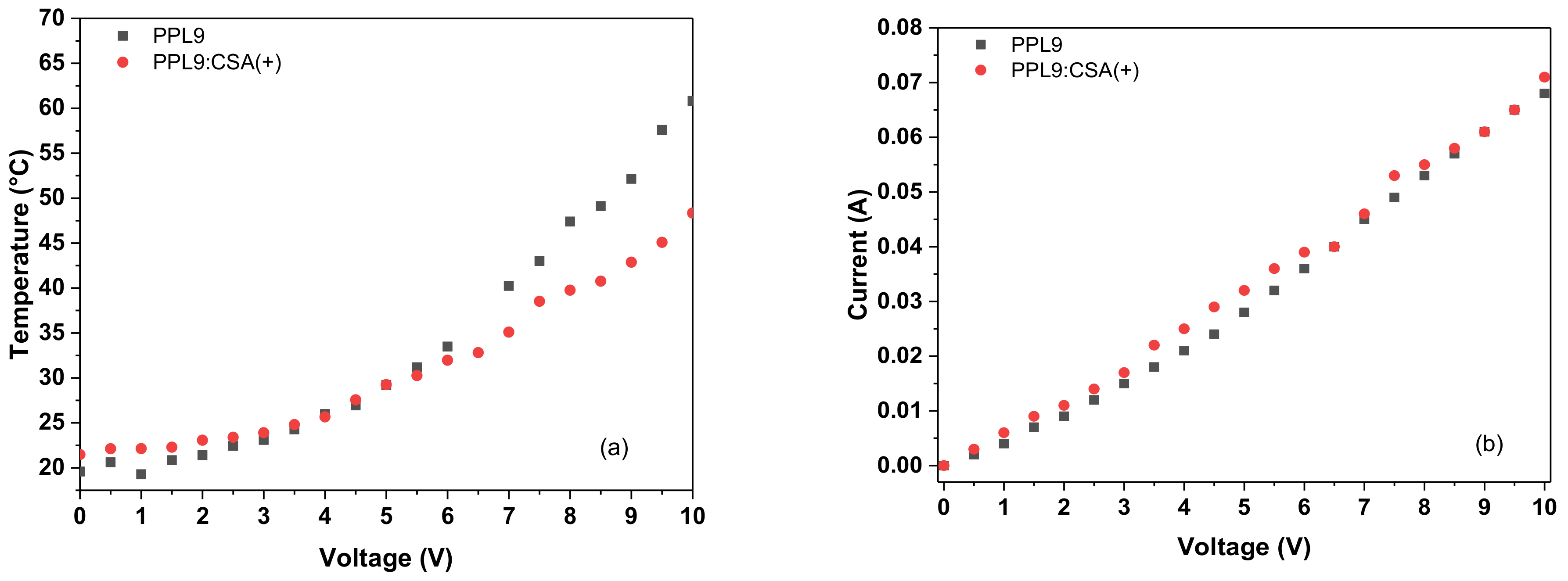
3.6. Thermal Imaging of PPL9 and PPL9:CSA
3.7. Thermal Stability PPL9
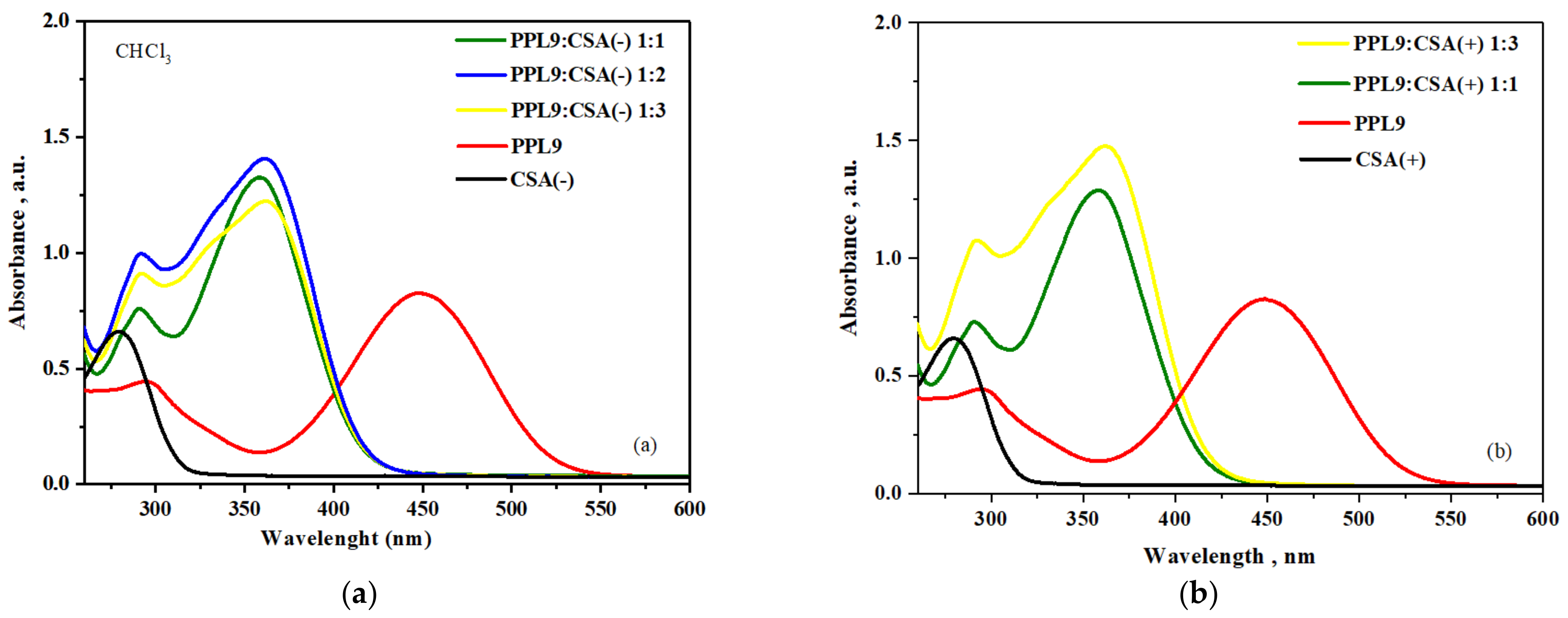
3.8. Protonation of PPL9 by CSA: UV–Vis Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iwan, A.; Sek, D. Processible polyazomethines and polyketanils: From aerospace to light-emitting diodes and other advanced applications. Prog. Polym. Sci. 2008, 33, 289–345. [Google Scholar] [CrossRef]
- Wei, L.; Tan, W.; Zhang, J.; Mi, Y.; Dong, F.; Li, Q.; Guo, Z. Synthesis, characterization, and antifungal activity of schiff bases of inulin bearing pyridine ring. Polymers 2019, 11, 371. [Google Scholar] [CrossRef]
- Shamim, S.; Murtaza, S.; Nazar, M.F. Synthesis of Schiff Bases of Pyridine-4-Carbaldehyde and their Antioxidant and DNA Binding Studies. J. Chem. Soc. Pak. 2016, 38, 03. [Google Scholar]
- Petrus, M.L.; Bouwer, R.K.; Lafont, U.; Athanasopoulos, S.; Greenham, N.C.; Dingemans, T.J. Small-molecule azomethines: Organic photovoltaics via Schiff base condensation chemistry. J. Mater. Chem. A 2014, 2, 9474–9477. [Google Scholar] [CrossRef]
- Jewloszewicz, B.; Bogdanowicz, K.A.; Przybyl, W.; Dysz, K.; Dylong, A.; Gonciarz, A.; Pich, R.; Mech, W.; Korona, K.P.; Kamińska, M.; et al. A comprehensive optical and electrical study of unsymmetrical imine with four thiophene rings and their binary and ternary compositions with PTB7 and PC70BM towards organic photovoltaics. RSC Adv. 2020, 10, 44958–44972. [Google Scholar] [CrossRef]
- Wenzel, M.; Wichmann, K.; Gloe, K.; Gloe, K.; Buschmann, H.J.; Otho, K.; Schröder, M.; Blake, A.J.; Wilson, C.; Mills, A.M.; et al. Interaction of tripodal Schiff-base ligands with silver (I): Structural and solution studies. CrystEngComm 2010, 12, 4176–4183. [Google Scholar] [CrossRef]
- Koskinen, L.; Jääskeläinen, S.; Oresmaa, L.; Haukka, M. Argentophilic interactions in multinuclear Ag complexes of imidazole containing Schiff bases. CrystEngComm 2012, 14, 3509–3514. [Google Scholar] [CrossRef]
- Jeevadason, A.W.; Murugavel, K.K.; Neelakantan, M.A. Review on Schiff bases and their metal complexes as organic photovoltaic materials. Renew. Sustain. Energy Rev. 2014, 36, 220–227. [Google Scholar] [CrossRef]
- Barik, S.; Bletzacker, T.; Skene, W.G. π-Conjugated fluorescent azomethine copolymers: Opto-electronic, halochromic, and doping properties. Macromolecules 2012, 45, 1165–1173. [Google Scholar] [CrossRef]
- Wen, H.; Niu, H.; Li, B.; Ma, X.; Bai, X.; Zhang, Y.; Wang, W. Synthesis and acidochromic, electrochromic properties of Schiff bases containing furan and triphenylamine units. Synth. Met. 2015, 202, 89–97. [Google Scholar] [CrossRef]
- Wu, X.; Wang, W.; Li, B.; Hou, Y.; Niu, H.; Zhang, Y.; Wang, S.; Bai, X. Synthesis and electrochromic, acidochromic properties of Schiff bases containing triphenylamine and thiophene units. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 140, 398–406. [Google Scholar] [CrossRef]
- Iwan, A.; Mazurak, Z.; Kaczmarczyk, B.; Jarzabek, B.; Sek, D. Synthesis and characterization of polyketanils with 3,8-diamino-6-phenylphenanthridine moieties exhibiting light emitting properties. Molecular and supramolecular engineering concept. Spectrochim. Acta Part A 2008, 69, 291–303. [Google Scholar] [CrossRef]
- Mathivanan, M.; Tharmalingam, B.; Lin, C.-H.; Pandiyan, B.V.; Thiagarajan, V.; Murugesapandian, B. Synthesis and characterization of polyketanils with 3, 8-diamino-6-phenylphenanthridine moieties exhibiting light emitting properties: Molecular and supramolecular engineering concept. CrystEngComm 2020, 22, 213–228. [Google Scholar] [CrossRef]
- Ha, S.-T.; Ong, L.-K.; Win, Y.-F.; Yeap, G.-Y.; Bonde, N.L.; Boey, P.-L. New Schiff bases with pyridine core: Spectra, thermal and optical characterizations. Am. J. Appl. Sci. 2010, 7, 656–660. [Google Scholar] [CrossRef]
- Iwan, A.; Kaczmarczyk, B.; Janeczek, H.; Sek, D.; Ostrowski, S. Similarities and differences between azomethines and ketimines: Synthesis, materials characterization and structure of novel imines compounds. Spectrochim Acta Part A Mol. Biomol. Spectrosc. 2007, 66, 1030–1041. [Google Scholar] [CrossRef]
- Sek, D.; Iwan, A.; Jarzabek, B.; Kaczmarczyk, B.; Kasperczyk, J.; Mazurak, Z.; Domanski, M.; Karon, K.; Lapkowski, M. Hole transport triphenylamine-azomethine conjugated system: Synthesis and optical, photoluminescence and electrochemical properties. Macromolecules 2008, 41, 6653–6663. [Google Scholar] [CrossRef]
- Sek, D.; Iwan, A.; Jarzabek, B.; Kaczmarczyk, B.; Kasperczyk, J.; Janeczek, H.; Mazurak, Z. Characterization and optical properties of oligoazomethines with triphenylamine moieties exhibiting blue, blue-green and green light. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 72, 1–10. [Google Scholar] [CrossRef]
- Sęk, D.; Grabiec, E.; Janeczek, H.; Jarząbek, B.; Kaczmarczyk, B.; Domański, M.; Iwan, A. Structure-properties relationship of linear and star-shaped imines with triphenylamine moieties as hole-transporting materials. Opt. Mater. 2010, 32, 1514–1525. [Google Scholar]
- Marin, L.; van der Lee, A.; Shova, S.; Arvinte, A.; Barboiu, M. Molecular amorphous glasses toward large azomethine crystals with aggregation-induced emission. New J. Chem. 2015, 39, 6404. [Google Scholar] [CrossRef]
- Sudhakar, S.; Narasimhaswamy, T.; Srinivasan, K.S.V. Synthesis, characterization and thermal properties of 4, 4′-bis (4-n-alkoxybenzoyloxy) benzylideneanilines and bis (4-benzylidene-4’-n-alkoxyaniline) terephthalates. Liq. Cryst. 2000, 27, 1525–1532. [Google Scholar] [CrossRef]
- Godzwon, J.; Sienkowska, M.J.; Galewski, Z. Liquid crystalline properties of 4-hexyloxybenzylidene-4′-alkyloxyanilines. Liq. Cryst. 2007, 34, 911–917. [Google Scholar] [CrossRef]
- Henderson, P.A.; Imrie, C.T. Semiflexible liquid crystalline tetramers as models of structurally analogous copolymers. Macromolecules 2005, 38, 3307–3311. [Google Scholar] [CrossRef]
- Imrie, C.T.; Stewart, D.; Remy, C.; Christie, D.W.; Hamley, I.W.; Harding, R. Liquid crystal tetramers. J. Mater. Chem. 1999, 9, 2321–2325. [Google Scholar] [CrossRef]
- Bhowmik, P.K.; Han, H.; Nedeltchev, A.K.; Mandal, H.D.; Jimenez-Hernandez, J.A.; McGannon, P.M.; Lopez, L.; Kang, S.-W.; Kumar, S. Synthesis and characterisation of thermotropic liquid-crystalline properties of azomethine dimers. Liq. Cryst. 2009, 36, 1389–1399. [Google Scholar] [CrossRef]
- Stamatoiu, O.; Bubnov, A.; Tarcomnicu, I.; Iovu, M. Synthesis and spectral characterisation of new amido-ether Schiff bases. J. Mol. Struct. 2008, 886, 187–196. [Google Scholar] [CrossRef]
- Smith, J.A.; DiStasio, R.A.; Hannah, N.A.; Winter, R.W.; Weakley, T.J.; Gard, G.L.; Rananavare, S.B. SF5-terminated fluorinated Schiff base liquid crystals. J. Phys. Chem. B 2004, 108, 19940–19948. [Google Scholar] [CrossRef]
- Narasimhaswamy, T.; Srinivasan, K.S. Synthesis and characterization of novel thermotropic liquid crystals containing a dimethylamino group. Liq. Cryst. 2004, 31, 1457–1462. [Google Scholar] [CrossRef]
- Nesrullajev, A.; Bilgin-Eran, B. Mesomorphic, morphologic and thermotropic properties of 4-hexyl-N-(4-hexadecyloxysalicylidene)aniline. Mater. Chem. Phys. 2005, 93, 21–25. [Google Scholar] [CrossRef]
- Eran, B.B.; Nesrullajev, A.; Canlı, N.Y. Characterization and investigation of the mesogenic, thermo-morphologic and thermotropic properties of new chiral (S)-5-octyloxy-2-[{4-(2-methylbuthoxy)-phenylimino (methyl] phenol liquid crystalline compound. Mater. Chem. Phys. 2008, 111, 555–558. [Google Scholar] [CrossRef]
- Thaker, B.T.; Patel, P.H.; Vansadiya, A.D.; Kanojiya, J.B. Substitution effects on the liquid crystalline properties of thermotropic liquid crystals containing Schiff base chalcone linkages. Mol. Cryst. Liq. Cryst. 2009, 515, 135–147. [Google Scholar] [CrossRef]
- Marin, L.; Destri, S.; Porzio, W.; Bertini, F. Synthesis and characterization of new azomethine derivatives exhibiting liquid crystalline properties. Liq. Cryst. 2009, 36, 21–32. [Google Scholar] [CrossRef]
- Ha, S.-T.; Koh, T.-M.; Lee, S.-L.; Yeap, G.-Y.; Lin, H.-C.; Ong, S.-T. Synthesis of new schiff base ester liquid crystals with a benzothiazole core. Liq. Cryst. 2010, 37, 547–554. [Google Scholar] [CrossRef]
- Bilgin-Eran, B.; Yorur, C.; Tschierske, C.; Prehm, M.; Baumeister, U. Liquid crystals based on semiperfluorinated imines and salicylaldimato metal complexes. A comparative study of alkyl, alkoxy and polyether substituents. J. Mater. Chem. 2007, 17, 2319–2328. [Google Scholar] [CrossRef]
- Kadkin, O.N.; Han, H.; Galyametdinov, Y.G. Synthesis, computational modelling and liquid crystalline properties of some [3] ferrocenophane-containing Schiff’s bases and β-aminovinylketone: Molecular geometry–phase behaviour relationship. J. Organomet. Chem. 2007, 692, 5571–5582. [Google Scholar] [CrossRef]
- Deun, R.V.; Binnemans, K. Mesomorphic lanthanide complexes with azomethine ligands. J. All. Comp. 2000, 303–304, 146–150. [Google Scholar] [CrossRef]
- Paschke, R.; Liebsch, S.; Tschierske, C.; Oakley, M.A.; Sinn, E. Synthesis and mesogenic properties of binuclear copper (II) complexes derived from salicylaldimine Schiff bases. Inorg. Chem. 2003, 42, 8230–8240. [Google Scholar] [CrossRef]
- Iwan, A.; Janeczek, H.; Jarząbek, B.; Domański, M.; Rannou, P. Characterization, optical and thermal properties of new azomethines based on heptadecafluoroundecyloxy benzaldehyde. Liq. Cryst. 2009, 36, 873–883. [Google Scholar] [CrossRef]
- Iwan, A.; Bilski, P.; Janeczek, H.; Jarząbek, B.; Domański, M.; Rannou, P.; Sikora, A.; Pociecha, D.; Kaczmarczyk, B. Thermal, optical, electrical and structural study of new symmetrical azomethine based on poly (1, 4-butanediol) bis (4-aminobenzoate). J. Mol. Struct. 2010, 963, 175–182. [Google Scholar] [CrossRef]
- Iwan, A.; Sęk, D.; Pociecha, D.; Sikora, A.; Palewicz, M.; Janeczek, H. New discotic-shaped azomethines with triphenylamine moieties: Thermal, structural behaviors and opto-electrical properties. J. Mol. Struct. 2010, 981, 120–129. [Google Scholar] [CrossRef]
- Pickering, A.L.; Seeber, G.; Long, D.-L.; Cronin, L. The importance of π–π, π–CH and N–CH interactions in the crystal packing of Schiff-base derivatives of cis, cis-and cis, trans-1, 3, 5-triaminocyclohexane. CrystEngComm 2005, 7, 504–510. [Google Scholar] [CrossRef]
- Lai, C.S.; Mohr, F.; Tiekink, E.R.T. The importance of C–H⋯N, C–H⋯π and π⋯π interactions in the crystal packing of the isomeric N1,N4-bis((pyridine-n-yl)methylene)-cyclohexane-1,4-diamines, n = 2, 3 and 4. CrystEngComm 2006, 8, 909–915. [Google Scholar] [CrossRef]
- Bolduc, A.; Dufresne, S.; Skene, W.G. EDOT-containing azomethine: An easily prepared electrochromically active material with tuneable colours. J. Mater. Chem. 2010, 20, 4820–4826. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A.; Rodríguez, M.; Métivier, R.; Ramos-Ortíz, G.; Maldonado, J.L.; Réboles, N.; Farfán, N.; Nakatani, K.; Santillan, R. Synthesis and crystal structures of a series of Schiff bases: A photo-, solvato-and acidochromic compound. New J. Chem. 2014, 38, 730. [Google Scholar] [CrossRef]
- Gumbau-Brisa, R.; Hayward, J.J.; Wallis, J.D.; Rawsonc, J.M.; Pilkington, M. Structural insights into the coordination chemistry and reactivity of a 3, 3′-bis-imine-2, 2′-bipyridine ligand. CrystEngComm 2016, 18, 1892–1903. [Google Scholar] [CrossRef]
- Mainsah, E.N.; Ntum, S.-J.E.; Conde, M.A.; Chi, G.T.; Raftery, J.; Ndifon, P.T. Synthesis, Characterization and Crystal Structure of Cobalt (II) Complex of a Schiff Base Derived from Isoniazid and Pyridine-4-Carboxaldehyde. Cryst. Struct. Theor. Appl. 2019, 8, 45–56. [Google Scholar] [CrossRef]
- Zhang, W.; Saraei, N.; Nie, H.; Vaughn, J.R.; Jones, A.S.; Mashuta, M.S.; Buchanan, R.M.; Grapperhaus, C.A. Reversible methanol addition to copper Schiff base complexes: A kinetic, structural and spectroscopic study of reactions at azomethine C [double bond, length as m-dash] N bonds. Dalton Trans. 2016, 45, 15791–15799. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry; Blackwell Publishing: Hoboken, NJ, USA, 2000; Volume 4. [Google Scholar]
- Caradonna, J.P.; Lippard, S.J.; Gait, M.J.; Singh, M. The antitumor drug cis-dichlorodiammineplatinum forms an intrastrand d (GpG) crosslink upon reaction with [d (ApGpGpCpCpT)] 2. J. Am. Chem. Soc. 1982, 104, 5793. [Google Scholar] [CrossRef]
- Baik, M.; Friesner, R.A.; Lippard, S.J. Theoretical study of cisplatin binding to purine bases: Why does cisplatin prefer guanine over adenine. J. Am. Chem. Soc. 2003, 125, 14082. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, H.; Chen, C.; Deng, H.; Li, T.; Ji, D.L. Interaction of macrocyclic copper (II) complexes with calf thymus DNA: Effects of the side chains of the ligands on the DNA-binding behaviors. Dalton Trans. 2003, 1, 114–119. [Google Scholar] [CrossRef]
- Jain, A.K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1, 3, 4-Thiadiazole and its derivatives: A review on recent progress in biological activities. Chem. Biol. Drug. Des. 2013, 81, 557–576. [Google Scholar] [CrossRef]
- Iwan, A.; Sek, D. Polymers with Triphenylamine Units: Photonic and Electroactive Materials. Prog. Polym. Sci. 2011, 36, 1277–1325. [Google Scholar] [CrossRef]
- Bogdanowicz, K.A.; Jewłoszewicz, B.; Iwan, A.; Dysz, K.; Przybyl, W.; Januszko, A.; Marzec, M.; Cichy, K.; Świerczek, K.; Kavan, L.; et al. Selected Electrochemical Properties of 4,4’-((1E,1’E)-((1,2,4-thiadiazole-3,5-diyl)bis(azaneylylidene))bis(methaneylylidene))bis(N,N-di-p-tolylaniline) towards Perovskite Solar Cells with 14.4% Efficiency. Materials 2020, 13, 2440. [Google Scholar] [CrossRef]
- Petrus, M.L.; Bein, T.; Dingemans, T.; Docampo, P. A low cost azomethine-based hole transporting material for perovskite photovoltaics. J. Mater. Chem. A 2015, 3, 12159–12162. [Google Scholar] [CrossRef]
- Safin, D.A.; Robeyns, K.; Garcia, Y. 1, 2, 4-Triazole-based molecular switches: Crystal structures, Hirshfeld surface analysis and optical properties. CrystEngComm 2016, 18, 7284–7296. [Google Scholar] [CrossRef]
- Safin, A.; Robeyns, K.; Babashkina, M.G.; Filinchuk, Y.; Rotaru, A.; Jureschi, C.; Mitoraj, M.P.; Hooper, J.; Brela, M.; Garcia, Y. Polymorphism driven optical properties of an anil dye. CrystEngComm 2016, 18, 7249–7259. [Google Scholar] [CrossRef]
- Dylong, A.; Dysz, K.; Iwan, A.; Bogdanowicz, K.A. Krystaliczna forma PPL9 i sposób jej wytwarzania. Polish Patent Application P.430631, 2019. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017; Available online: http://hirshfeldsurface.net (accessed on 10 October 2019).
- Bogdanowicz, K.A.; Jewloszewicz, B.; Dysz, K.; Przybyl, W.; Dylong, A.; Mech, W.; Korona, K.P.; Skompska, M.; Kaim, A.; Kamińska, M.; et al. Electrochemical and optical studies of new symmetrical and unsymmetrical imines with thiazole and thiophene moieties. Electrochim. Acta 2020, 332, 135476. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Effects Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Michalska, D.; Wysokiński, R. The prediction of Raman spectra of platinum(II) anticancer drugs by density functional theory. Chem. Phys. Lett. 2005, 403, 211–217. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frish, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-functional theory for time-dependent systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Luo, Y.; Utecht, M.; Dokić, J.; Korchak, S.; Vieth, H.; Haag, R.; Saalfrank, P. Cis–trans isomerisation of substituted aromatic imines: A comparative experimental and theoretical study. ChemPhysChem 2011, 12, 2311–2321. [Google Scholar] [CrossRef]
- Lucio, A.J.; Shaw, S.K. Pyridine and pyridinium electrochemistry on polycrystalline gold electrodes and implications for CO2 reduction. J. Phys. Chem. C 2015, 119, 12523–12530. [Google Scholar] [CrossRef]
- Chiu, K.Y.; Su, T.X.; Li, J.H.; Lin, T.-H.; Liou, G.-S.; Cheng, S.-H. Novel trends of electrochemical oxidation of amino-substituted triphenylamine derivatives. J. Electroanal. Chem. 2005, 575, 95–101. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contact | D−H [Å] | H⋯A [Å] | D⋯A [Å] | <DHA [°] |
|---|---|---|---|---|
| C6−H6⋯N5i | 0.92(2) | 2.68(2) | 3.498(3) | 148(2) |
| C17−H17⋯N4ii | 0.96(2) | 2.64(2) | 3.509(3) | 152(2) |
| Fragments | Cg1⋯Cg2 [Å] | D1 [Å] | D2 [Å] | |
| C22→S1, C22i→S1i | 3.701 | 3.538 | 1.086 | |
| N1→N5, N1i→N5i | 4.090 | 3.614 | 1.915 |
| Chemical Formula | C28H23N5S |
| Formula weight | 461.57 |
| Crystal dimensions/mm | 0.44 × 0.16 × 0.05 |
| Temperature/K | 298 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 18.9567(7) |
| b/Å | 6.18597(17) |
| c/Å | 22.5897(7) |
| β/° | 114.009(4) |
| V/Å3 | 2419.81(15) |
| Z | 4 |
| Dx/Mg m−3 | 1.267 |
| μ/mm−1 | 0.160 |
| Reflections collected | 17048 |
| Reflections independent | 4762 |
| Reflections ind. observed | 3343 |
| Rint | 0.032 |
| R, wR (F2 > 2σ(F2)), S | 0.054, 0.113, 1.03 |
| Δρmax, Δρmin/eÅ−3 | 0.29, −0.36 |
| FT-IR | FT-R | νa | Assignments |
|---|---|---|---|
| n.o. | n.o. | 3233 | ν(CH)pyridine |
| n.o. | n.o. | 3226 | ν(CH)sphe |
| n.o. | n.o. | 3225 | ν(CH)sphe |
| n.o. | n.o. | 3212 | ν(CH)asphe |
| n.o. | n.o. | 3210 | ν(CH)sarom |
| n.o. | n.o. | 3209 | ν(CH)sarom |
| n.o. | n.o. | 3208 | ν(CH)asarom |
| n.o. | n.o. | 3207 | ν(CH)asarom |
| n.o. | n.o. | 3199 | ν(CH)spyridine |
| n.o. | n.o. | 3182 | ν(CH)asphe |
| n.o. | n.o. | 3180 | ν(CH)asarom |
| n.o. | n.o. | 3179 | ν(CH)asarom |
| n.o. | n.o. | 3179 | ν(CH)asarom |
| 3027vw | 3064vw | 3178 | ν(CH)asarom |
| n.o. | n.o. | 3166 | ν(CH)aspyridine |
| n.o. | n.o. | 3127 | ν(CH3)as |
| 2919vw | 2922vw | 3126 | ν(CH3)as |
| n.o. | n.o. | 3098 | ν(CH3) |
| 2856vw | n.o. | 3038 | ν(CH3)s |
| 1604vw | 1615vw 1606vw | 1664 | ν(CC)arom, ν(C=N) |
| 1573m | 1581m 1575m | 1631 | ν(C=N), ν(CC)arom |
| 1542m | 1552m 1544m | 1588 | ν(C=N), ν(CC)arom |
| 1501s | 1502m | 1540 | δ(CCH), ν(CN)pyridine |
| 1433m | n.o. | 1488 | ν(CN), ν(CC) |
| n.o. | 1432m | 1478 | ν(CN), ν(CC) |
| n.o. | 1409vw | 1459 | δ(CCH), ν(CC) |
| 1408m 1398m | 1397vs | 1455 | δ(NCH), ν(CC), δ(CCH), ν(CN)th |
| 1442 | ν(CN)th, ν(C=N), δ(CCH) | ||
| 1363w | 1366m | 1404 | ν(CN)th, δ(NCH), ν(C=N) |
| 1320m | 1327m | 1360 | ν(CN)arom, δ(CCH) |
| n.o. | 1296w | 1326 | ν(CN)arom, ν(CC) |
| 1292m | 1249m | 1297 | ν(CN), ν(CC) |
| 1274m | 1240m | 1277 | ν(CC), δ(NCH) |
| n.o. | 1214w | 1250 | δ(NCH), δ(CCH), ν(CN) |
| 1215w | 1181m | 1207 | δ(NNC), δ(CCH), ν(CN) |
| 1190w | n.o. | 1185 | δ(CCH), ν(CN) |
| 1159vs | 1166m | 1171 | ν(NN), δ(NNC) |
| 1110m | 1113m | 1144 | δ(CCH) |
| 833m | 827w | 839 | γ(H(CCC) |
| 814s | n.o. | 838 | γ(H(CCC) |
| n.o. | 803m | 821 | ν(CN), δ(NNC), ν(CS) |
| n.o. | 792w | 804 | ν(CS), ν(CC), δ(NNC) |
| 784w | n.o. | 796 | ν(CC)arom, δ(CCC) |
| 765m | 767w | 765 | ν(CS), δ(NNC), ν(CC) |
| 728w | 729w | 729 | τ(CCCC), γ(C(CCC) |
| 713w | 665w | 700 | δ(CNC), ν(CS) |
| 695m | n.o. | 662 | δ(CCC), ν(CS) |
| n.o. | 654w | 637 | τ(SCNN), τ(CNNC), τ(SCNN) |
| 653m | 638br | 624 | δ(CSC), δ(SCN) |
| 565m | n.o. | 575 | δ(CCC), ν(CC) |
| 522s | n.o. | 540 | γ(C(CNC)pyridine, γ(C(CCC)phe |
| 511s | 525vw | 524 | γ(C(CNC), τ(CCNC) |
| 497m | 490w | 495 | γ(C(CCC), ν(CS) δ(CCC) |
| 454m | 421m | 431 | τ(CCCC)phe |
| 374vw | 362w | 356 | δ(CCC) |
| 345m | 346w | 342 | γ(C(CCC), τ(CCCN) |
| n.o. | 319w | 322 | δ(NCC), τ(NCCC) |
| n.o. | 264w | 221 | τ(CCCC), τ(NCCC) |
| Frontier Orbital Energy at DFT/B3LYP/6-31G(d,p) | |||
|---|---|---|---|
| HOMO [eV] | LUMO [eV] | Eg [eV] | |
| trans-PPL9 | −5.17 | −2.46 | 2.71 |
| H+ doped trans-PPL9 | −7.07 | −5.95 | 1.12 |
| TD-DFT/B3LYP/6-31G(d,p) | |||
| trans-PPL9 | −5.26 | −2.26 | 3.00 |
| H+ doped trans-PPL9 | −5.75 | −7.35 | 1.60 |
| Cyclic Voltammetry | |||
| PPL9 | −5.41 | −2.52 | 2.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dylong, A.; Dysz, K.; Bogdanowicz, K.A.; Przybył, W.; Konieczny, K.A.; Turowska-Tyrk, I.; Kaim, A.; Iwan, A. Crystal Structure Determination of 4-[(Di-p-tolyl-amino)-benzylidene]-(5-pyridin-4-yl-[1,3,4]thiadiazol-2-yl)-imine along with Selected Properties of Imine in Neutral and Protonated Form with Camforosulphonic Acid: Theoretical and Experimental Studies. Materials 2021, 14, 1952. https://doi.org/10.3390/ma14081952
Dylong A, Dysz K, Bogdanowicz KA, Przybył W, Konieczny KA, Turowska-Tyrk I, Kaim A, Iwan A. Crystal Structure Determination of 4-[(Di-p-tolyl-amino)-benzylidene]-(5-pyridin-4-yl-[1,3,4]thiadiazol-2-yl)-imine along with Selected Properties of Imine in Neutral and Protonated Form with Camforosulphonic Acid: Theoretical and Experimental Studies. Materials. 2021; 14(8):1952. https://doi.org/10.3390/ma14081952
Chicago/Turabian StyleDylong, Agnieszka, Karolina Dysz, Krzysztof A. Bogdanowicz, Wojciech Przybył, Krzysztof A. Konieczny, Ilona Turowska-Tyrk, Andrzej Kaim, and Agnieszka Iwan. 2021. "Crystal Structure Determination of 4-[(Di-p-tolyl-amino)-benzylidene]-(5-pyridin-4-yl-[1,3,4]thiadiazol-2-yl)-imine along with Selected Properties of Imine in Neutral and Protonated Form with Camforosulphonic Acid: Theoretical and Experimental Studies" Materials 14, no. 8: 1952. https://doi.org/10.3390/ma14081952
APA StyleDylong, A., Dysz, K., Bogdanowicz, K. A., Przybył, W., Konieczny, K. A., Turowska-Tyrk, I., Kaim, A., & Iwan, A. (2021). Crystal Structure Determination of 4-[(Di-p-tolyl-amino)-benzylidene]-(5-pyridin-4-yl-[1,3,4]thiadiazol-2-yl)-imine along with Selected Properties of Imine in Neutral and Protonated Form with Camforosulphonic Acid: Theoretical and Experimental Studies. Materials, 14(8), 1952. https://doi.org/10.3390/ma14081952