
Effect of the Microstructure of the Semiconductor Support on the Photocatalytic Performance of the Pt-PtOx/TiO2 Catalyst System

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
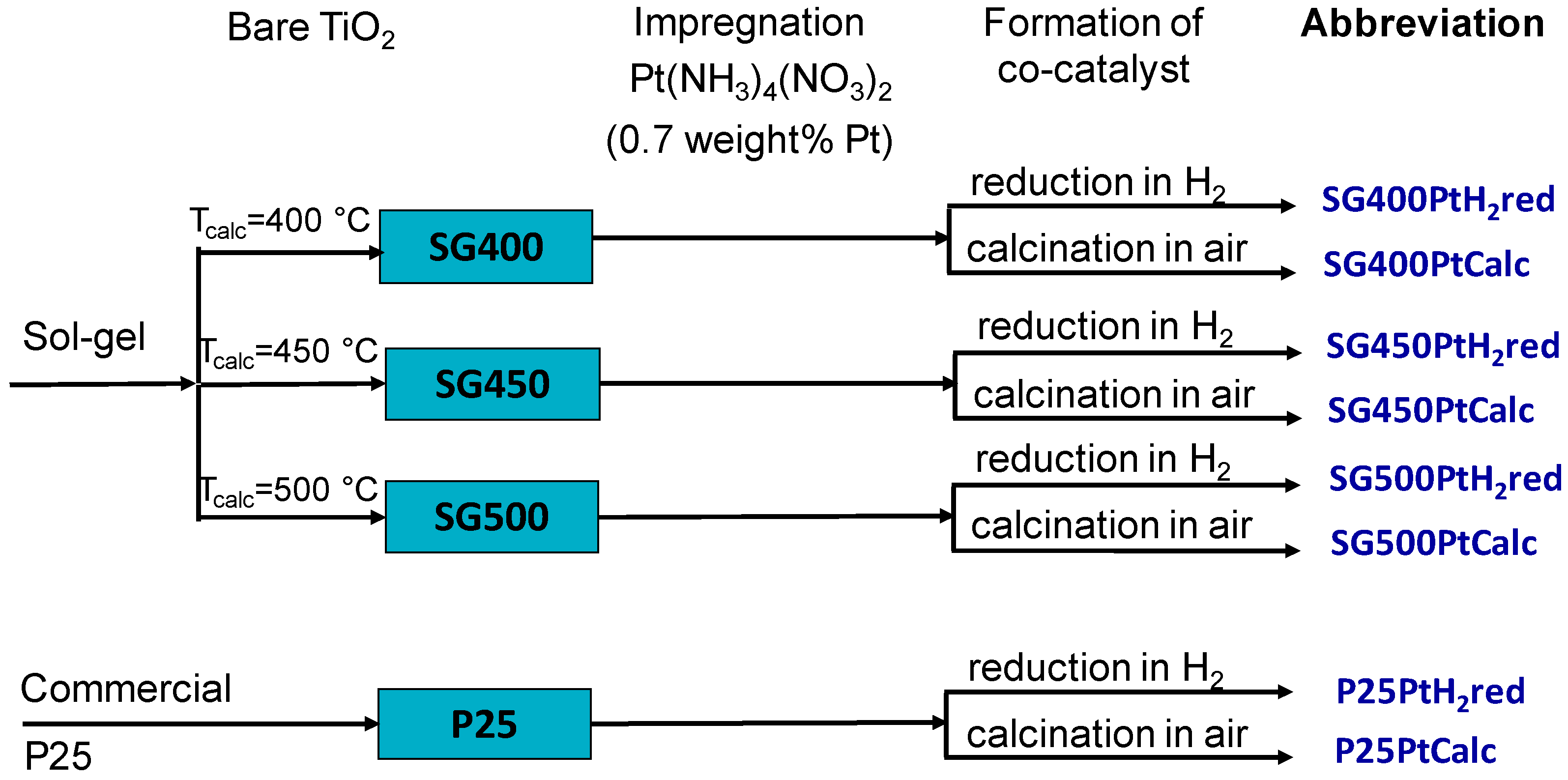
2.2. Synthesis of Photocatalysts
2.3. Physical–Chemical Characterization of Photocatalysts
2.4. Photocatalytic Hydrogen Generation
3. Results
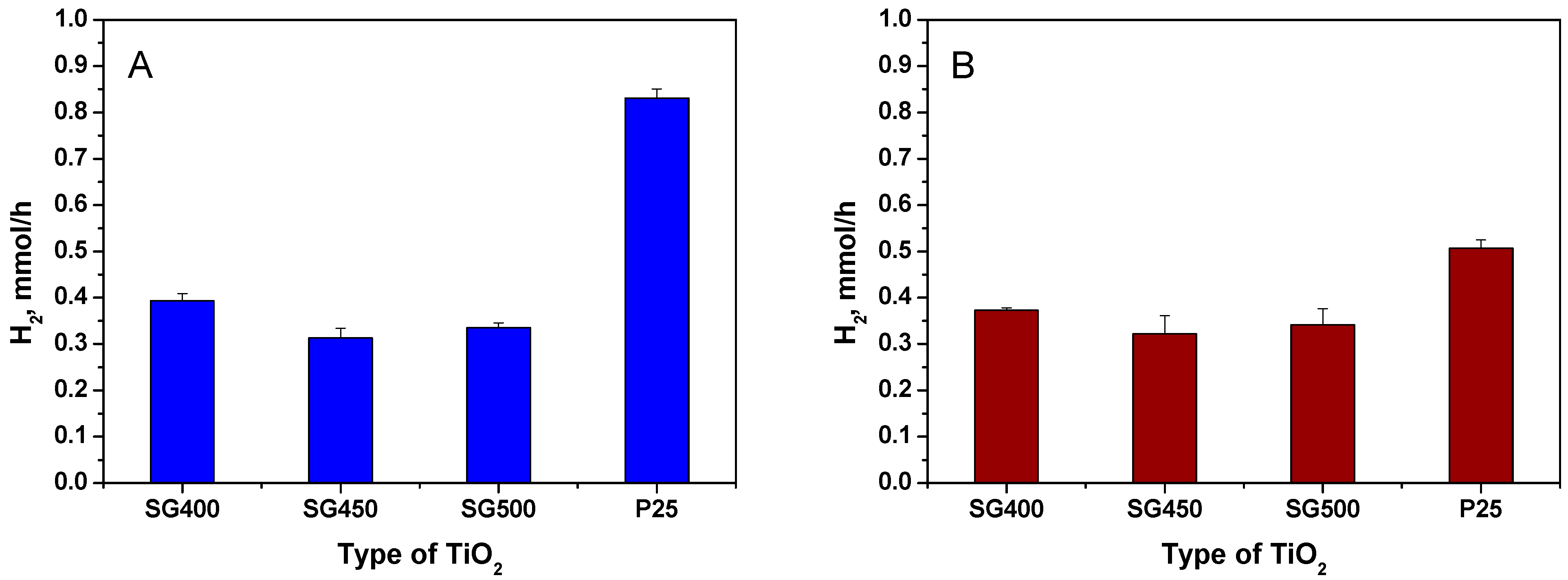
3.1. Photocatalytic Hydrogen Generation
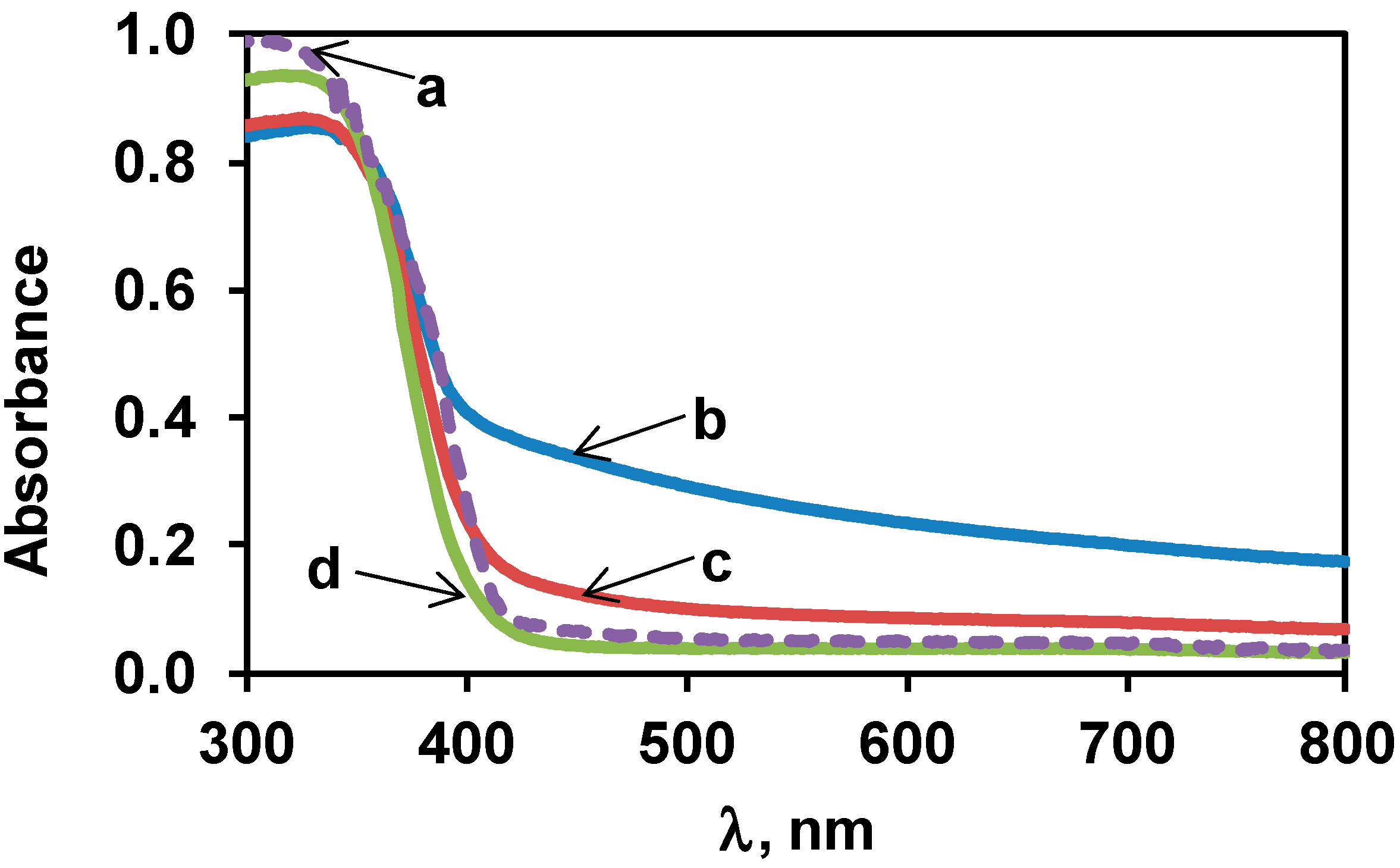
3.2. Characterization of the Bare TiO2 Materials
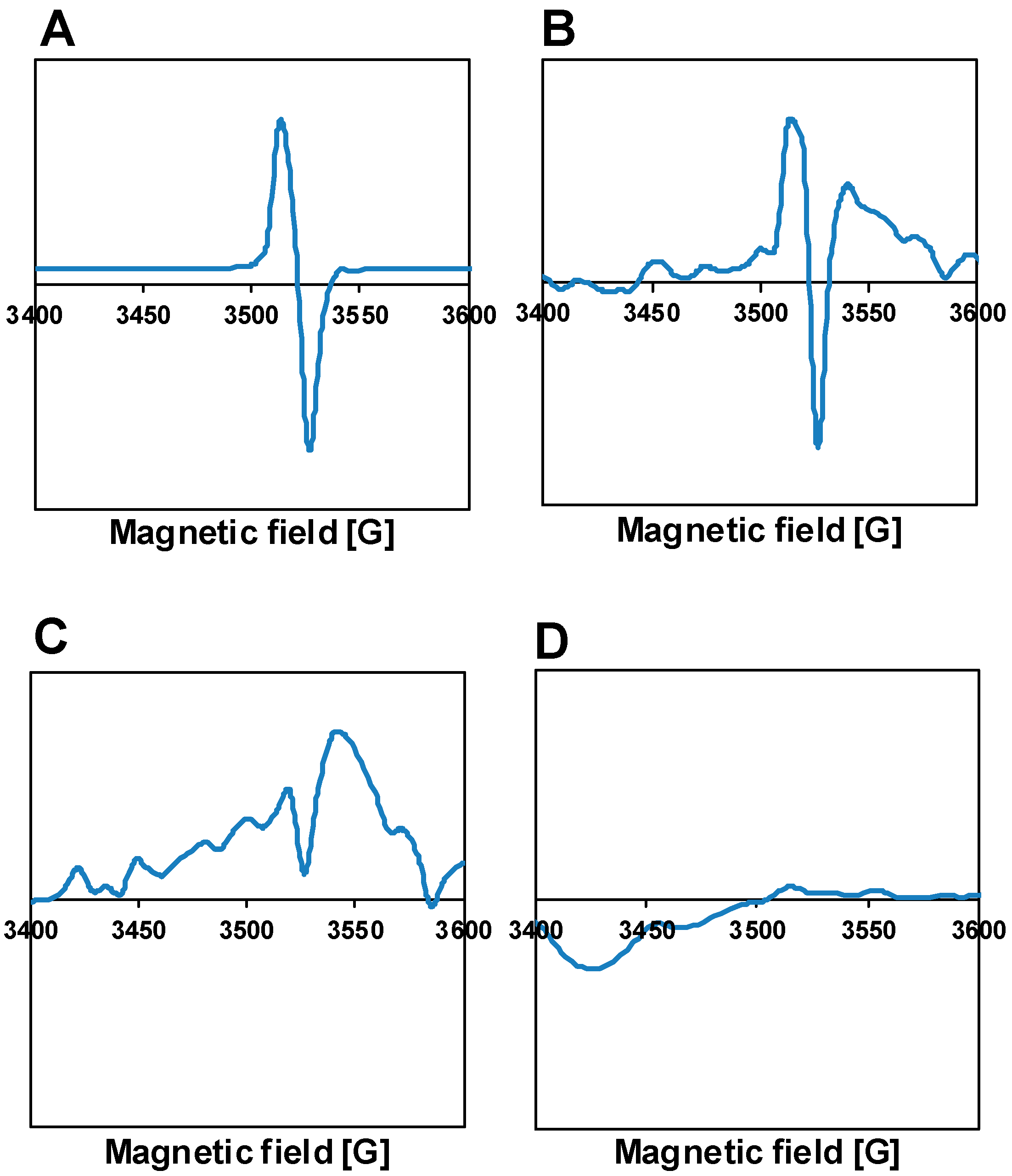
3.3. Characterization of the Fresh Pt-PtOx/TiO2 Samples
3.4. Characterization of the Catalysts Recovered after the Reaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Acar, C.; Dincer, I.; Naterer, G.F. Review of photocatalytic water-splitting methods for sustainable hydrogen production. Int. J. Energy Res. 2016, 40, 1449–1473. [Google Scholar] [CrossRef]
- Fajrina, N.; Tahir, M. A critical review in strategies to improve photocatalytic water splitting towards hydrogen production. Int. J. Hydrogen Energy 2019, 44, 540–577. [Google Scholar] [CrossRef]
- Mazloomi, K.; Gomes, C. Hydrogen as an energy carrier: Prospects and challenges. Renew. Sustain. Energy Rev. 2012, 16, 3024–3033. [Google Scholar] [CrossRef]
- Cipriani, G.; Di Dio, V.; Genduso, F.; La Cascia, D.; Liga, R.; Miceli, R.; Galluzzo, G.R. Perspective on hydrogen energy carrier and its automotive applications. Int. J. Hydrogen Energy 2014, 39, 8482–8494. [Google Scholar] [CrossRef]
- Sharma, S.; Ghoshal, S.K. Hydrogen the future transportation fuel: From production to applications. Renew. Sustain. Energy Rev. 2015, 43, 1151–1158. [Google Scholar] [CrossRef]
- Peraldobicelli, L. Hydrogen: A clean energy source. Int. J. Hydrogen Energy 1986, 11, 555–562. [Google Scholar] [CrossRef]
- Montini, T.; Monai, M.; Beltram, A.; Romero-Ocaña, I.; Fornasiero, P. H2 production by photocatalytic reforming of oxygenated compounds using TiO2-based materials. Mater. Sci. Semicond. Process. 2016, 42, 122–130. [Google Scholar] [CrossRef]
- Puga, A.V. Photocatalytic production of hydrogen from biomass-derived feedstocks. Co-ord. Chem. Rev. 2016, 315, 1–66. [Google Scholar] [CrossRef]
- Al-Mazroai, L.S.; Bowker, M.; Davies, P.; Dickinson, A.; Greaves, J.; James, D.; Millard, L. The photocatalytic reforming of methanol. Catal. Today 2007, 122, 46–50. [Google Scholar] [CrossRef]
- Wu, G.; Chen, T.; Zong, X.; Yan, H.; Ma, G.; Wang, X.; Xu, Q.; Wang, D.; Lei, Z.; Li, C. Suppressing CO formation by anion adsorption and Pt deposition on TiO2 in H2 production from photocatalytic reforming of methanol. J. Catal. 2008, 253, 225–227. [Google Scholar] [CrossRef]
- Chiarello, G.L.; Aguirre, M.H.; Selli, E. Hydrogen production by photocatalytic steam reforming of methanol on noble metal-modified TiO2. J. Catal. 2010, 273, 182–190. [Google Scholar] [CrossRef]
- Kandiel, T.A.; Dillert, R.; Robben, L.; Bahnemann, D.W. Photonic efficiency and mechanism of photocatalytic molecular hydrogen production over platinized titanium dioxide from aqueous methanol solutions. Catal. Today 2011, 161, 196–201. [Google Scholar] [CrossRef]
- Yao, Y.; Gao, X.; Li, Z.; Meng, X. Photocatalytic Reforming for Hydrogen Evolution: A Review. Catalysts 2020, 10, 335. [Google Scholar] [CrossRef]
- Melián, E.P.; López, C.R.; Santiago, D.E.; Quesada-Cabrera, R.; Méndez, J.O.; Rodríguez, J.D.; Díaz, O.G. Study of the photocatalytic activity of Pt-modified commercial TiO2 for hydrogen production in the presence of common organic sacrificial agents. Appl. Catal. A: Gen. 2016, 518, 189–197. [Google Scholar] [CrossRef]
- Gu, Q.; Long, J.; Fan, L.; Chen, L.; Zhao, L.; Lin, H.; Wang, X. Single-site Sn-grafted Ru/TiO2 photocatalysts for biomass reforming: Synergistic effect of dual co-catalysts and molecular mechanism. J. Catal. 2013, 303, 141–155. [Google Scholar] [CrossRef]
- Rosseler, O.; Shankar, M.V.; Du, M.K.-L.; Schmidlin, L.; Keller, N.; Keller, V. Solar light photocatalytic hydrogen production from water over Pt and Au/TiO2(anatase/rutile) photocatalysts: Influence of noble metal and porogen promotion. J. Catal. 2010, 269, 179–190. [Google Scholar] [CrossRef]
- Yang, J.; Wang, D.; Han, H.; Li, C. Roles of Cocatalysts in Photocatalysis and Photoelectrocatalysis. Accounts Chem. Res. 2013, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Wagner, T. Interaction of nanostructured metal overlayers with oxide surfaces. Surf. Sci. Rep. 2007, 62, 431–498. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Fernández, M.; Mendizábal, I.; Korili, S.; Soto-Armañanzas, J.; Crespo-Durante, A.; Gómez-Polo, C. Fabrication of TiO2 coated metallic wires by the sol–gel technique as a humidity sensor. Ceram. Int. 2016, 42, 9292–9298. [Google Scholar] [CrossRef]
- Arconada, N.; Durán, A.; Suárez, S.; Portela, R.; Coronado, J.M.; Sanchez, B.S.; Castro, Y. Synthesis and photocatalytic properties of dense and porous TiO2-anatase thin films prepared by sol–gel. Appl. Catal. B Environ. 2009, 86, 1–7. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. TiO2 photocatalyst for water treatment applications. J. Ind. Eng. Chem. 2013, 19, 1761–1769. [Google Scholar] [CrossRef]
- Asahi, R.; Morikawa, T.; Irie, H.; Ohwaki, T. Nitrogen-Doped Titanium Dioxide as Visible-Light-Sensitive Photocatalyst: Designs, Developments, and Prospects. Chem. Rev. 2014, 114, 9824–9852. [Google Scholar] [CrossRef]
- Wang, C.-C.; Ying, J.Y. Sol−Gel Synthesis and Hydrothermal Processing of Anatase and Rutile Titania Nanocrystals. Chem. Mater. 1999, 11, 3113–3120. [Google Scholar] [CrossRef]
- Akpan, U.; Hameed, B. The advancements in sol–gel method of doped-TiO2 photocatalysts. Appl. Catal. A: Gen. 2010, 375, 1–11. [Google Scholar] [CrossRef]
- Ali, I.; Suhail, M.; Alothman, Z.A.; Alwarthan, A. Recent advances in syntheses, properties and applications of TiO2 nanostructures. RSC Adv. 2018, 8, 30125–30147. [Google Scholar] [CrossRef]
- Livage, J.; Henry, M.; Sanchez, C. Sol-gel chemistry of transition metal oxides. Prog. Solid State Chem. 1988, 18, 259–341. [Google Scholar] [CrossRef]
- Melián, E.P.; Díaz, O.G.; Rodríguez, J.D.; Colón, G.; Navío, J.A.; Pena, J.P. Effect of hydrothermal treatment on structural and photocatalytic properties of TiO2 synthesized by sol–gel method. Appl. Catal. A: Gen. 2012, 411–412, 153–159. [Google Scholar] [CrossRef]
- Pourmand, M.; Mohammadizadeh, M.P.A.M.R. Influence of Temperature on TiO2 Nanoparticles. Curr. Nanosci. 2008, 4, 151–156. [Google Scholar] [CrossRef]
- Leyva-Porras, C.; Toxqui-Teran, A.; Vega-Becerra, O.; Miki-Yoshida, M.; Rojas-Villalobos, M.; García-Guaderrama, M.; Aguilar-Martínez, J. Low-temperature synthesis and characterization of anatase TiO2 nanoparticles by an acid assisted sol–gel method. J. Alloy. Compd. 2015, 647, 627–636. [Google Scholar] [CrossRef]
- Sheikhnejad-Bishe, O.; Zhao, F.; Rajabtabar-Darvishi, A.; Khodadad, E.; Huang, Y. Precursor and reaction time effects in evaluation of photocatalytic properties of TiO2 nanoparticles synthesized via low temperature. Int. J. Electrochem. Sci. 2014, 9, 3068–3077. Available online: http://www.electrochemsci.org/papers/vol9/90603068.pdf.
- Yang, S.; Tang, W.; Ishikawa, Y.; Feng, Q. Synthesis of titanium dioxide with oxygen vacancy and its visible-light sensitive photocatalytic activity. Mater. Res. Bull. 2011, 46, 531–537. [Google Scholar] [CrossRef]
- Mao, L.; Li, Q.; Dang, H.; Zhang, Z. Synthesis of nanocrystalline TiO2 with high photoactivity and large specific surface area by sol–gel method. Mater. Res. Bull. 2005, 40, 201–208. [Google Scholar] [CrossRef]
- Kaur, K.; Singh, C.V. Amorphous TiO2 as a Photocatalyst for Hydrogen Production: A DFT Study of Structural and Electronic Properties. Energy Procedia 2012, 29, 291–299. [Google Scholar] [CrossRef]
- Wang, J.; Li, H.; Li, H.; Zou, C.; Wang, H.; Li, D. Mesoporous TiO2 Thin Films Exhibiting Enhanced Thermal Stability and Controllable Pore Size: Preparation and Photocatalyzed Destruction of Cationic Dyes. ACS Appl. Mater. Interfaces 2014, 6, 1623–1631. [Google Scholar] [CrossRef]
- Ikeda, S.; Sugiyama, N.; Murakami, S.-Y.; Kominami, H.; Kera, Y.; Noguchi, H.; Uosaki, K.; Torimoto, T.; Ohtani, B. Quantitative analysis of defective sites in titanium(IV) oxide photocatalyst powders. Phys. Chem. Chem. Phys. 2003, 5, 778–783. [Google Scholar] [CrossRef]
- Yin, Q.; Xiang, J.; Wang, X.; Guo, X.; Zhang, T. Preparation of highly crystalline mesoporous TiO2by sol–gel method combined with two-step calcining process. J. Exp. Nanosci. 2016, 11, 1127–1137. [Google Scholar] [CrossRef]
- Qingge, F.; Huidong, C.; Haiying, L.; Siying, Q.; Zheng, L.; Dachao, M.; Yuyang, Y.; Feng, Q.; Cai, H.; Lin, H.; et al. Synthesis and structural characteristics of high surface area TiO2 aerogels by ultrasonic-assisted sol–gel method. Nanotechnology 2017, 29, 075702. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; He, T.; Zhao, L.; Wang, E.; Yang, W.; Cao, Y. Structure and Phase Transition Behavior of Sn4+-Doped TiO2 Nanoparticles. J. Phys. Chem. C 2009, 113, 18121–18124. [Google Scholar] [CrossRef]
- Shankar, K.; Basham, J.I.; Allam, N.K.; Varghese, O.K.; Mor, G.K.; Feng, X.; Paulose, M.; Seabold, J.A.; Choi, K.-S.; Grimes, C.A. Recent Advances in the Use of TiO2 Nanotube and Nanowire Arrays for Oxidative Photoelectrochemistry. J. Phys. Chem. C 2009, 113, 6327–6359. [Google Scholar] [CrossRef]
- Jiang, Z.; Shangguan, W. Rational removal of stabilizer-ligands from platinum nanoparticles supported on photocatalysts by self-photocatalysis degradation. Catal. Today 2015, 242, 372–380. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Tsubota, S.; Nakamura, T.; Haruta, M. Photoassisted hydrogen production from a water-ethanol solution: A comparison of activities of Au TiO2 and Pt TiO2. J. Photochem. Photobiol. A Chem. 1995, 89, 177–189. [Google Scholar] [CrossRef]
- Wenderich, K.; Mul, G. Methods, Mechanism, and Applications of Photodeposition in Photocatalysis: A Review. Chem. Rev. 2016, 116, 14587–14619. [Google Scholar] [CrossRef]
- Majrik, K.; Pászti, Z.; Korecz, L.; Trif, L.; Domján, A.; Bonura, G.; Cannilla, C.; Frusteri, F.; Tompos, A.; Tálas, E. Study of PtOx/TiO2 Photocatalysts in the Photocatalytic Reforming of Glycerol: The Role of Co-Catalyst Formation. Materials 2018, 11, 1927. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Fu, X.; Zhang, L.; Meng, S.; Chen, S. Photocatalytic reforming of glycerol for H2 evolution on Pt/TiO2: Fundamental understanding the effect of co-catalyst Pt and the Pt deposition route. J. Mater. Chem. A 2014, 3, 2271–2282. [Google Scholar] [CrossRef]
- Song, S.; Sheng, Z.; Liu, Y.; Wang, H.; Wu, Z. Influences of pH value in deposition-precipitation synthesis process on Pt-doped TiO2 catalysts for photocatalytic oxidation of NO. J. Environ. Sci. 2012, 24, 1519–1524. [Google Scholar] [CrossRef]
- Xing, J.; Li, Y.H.; Jiang, H.B.; Wang, Y.; Yang, H.G. The size and valence state effect of Pt on photocatalytic H2 evolution over platinized TiO2 photocatalyst. Int. J. Hydrogen Energy 2014, 39, 1237–1242. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Zhou, X.; Jiang, H.; Yang, H.G.; Li, C. Positively charged Pt-based cocatalysts: An orientation for achieving efficient photocatalytic water splitting. J. Mater. Chem. A 2020, 8, 17–26. [Google Scholar] [CrossRef]
- Wang, D.; Subban, C.V.; Wang, H.; Rus, E.; Disalvo, F.J.; Abrunña, H.D. Highly Stable and CO-Tolerant Pt/Ti0.7W0.3O2Electrocatalyst for Proton-Exchange Membrane Fuel Cells. J. Am. Chem. Soc. 2010, 132, 10218–10220. [Google Scholar] [CrossRef]
- Majrik, K.; Tálas, E.; Pászti, Z.; Sajó, I.; Mihály, J.; Korecz, L.; Drotár, E.; Tompos, A. Enhanced activity of sol–gel prepared SnOx–TiO2 in photocatalytic methanol reforming. Appl. Catal. A: Gen. 2013, 466, 169–178. [Google Scholar] [CrossRef]
- Fernandes, S.; Da Silva, J.C.E.; Da Silva, L.P. Life Cycle Assessment of the Sustainability of Enhancing the Photodegradation Activity of TiO2 with Metal-Doping. Materials 2020, 13, 1487. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-Ray Photoelectron Spectroscopy; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Wagner, C.D.; Naumkin, A.V.; Kraut-Vass, A.; Allison, J.W.; Powell, C.J.; Rumble, J.R., Jr. NIST X-ray Photoelectron Spectroscopy Database, Version 3.4, (the most comprehensive online data set for photoelectron- and Auger electron spectroscopy). National Institute of Standards and Technology: Gaithersburg, MD, USA, 2003. Available online: http://srdata.nist.gov/xps/ (accessed on 5 February 2021).
- Fairley, N. CasaXPS: Spectrum Processing Software for XPS, AES and SIMS, Version 2.3.13. Casa Software Ltd.: Cheshire, UK, 2006; Available online: http://www.casaxps.com (accessed on 5 February 2021).
- Mohai, M. XPS MultiQuant: Multimodel XPS quantification software. Surf. Interface Anal. 2004, 36, 828–832. [Google Scholar] [CrossRef]
- Mohai, M. XPS MultiQuant: Multi-Model X-ray Photoelectron Spectroscopy Quantification Program. Version 7.00.92. 2011. (Available at the Author’s Website without Charge). Available online: http://aki.ttk.mta.hu/XMQpages/XMQhome.php (accessed on 5 February 2021).
- Hou, L.; Zhang, M.; Guan, Z.; Li, Q.; Yang, J. Effect of platinum dispersion on photocatalytic performance of Pt-TiO2. J. Nanoparticle Res. 2018, 20, 60. [Google Scholar] [CrossRef]
- Bingham, M.; Mills, A. Photonic efficiency and selectivity study of M (M = Pt, Pd, Au and Ag)/TiO2 photocatalysts for methanol reforming in the gas phase. J. Photochem. Photobiol. A Chem. 2020, 389, 112257. [Google Scholar] [CrossRef]
- Jiang, Z.; Qi, R.; Huang, Z.; Shangguan, W.; Wong, R.J.; Lee, A.F. Impact of Methanol Photomediated Surface Defects on Photocatalytic H2 Production Over Pt/TiO2. Energy Environ. Mater. 2020, 3, 202–208. [Google Scholar] [CrossRef]
- Tálas, E.; Pászti, Z.; Korecz, L.; Domján, A.; Németh, P.; Szíjjártó, G.; Mihály, J.; Tompos, A. PtOx-SnOx-TiO2 catalyst system for methanol photocatalytic reforming: Influence of cocatalysts on the hydrogen production. Catal. Today 2018, 306, 71–80. [Google Scholar] [CrossRef]
- Su, C.; Lin, K.-F.; Lin, Y.-H.; You, B.-H. Preparation and characterization of high-surface-area titanium dioxide by sol-gel process. J. Porous Mater. 2006, 13, 251–258. [Google Scholar] [CrossRef]
- Ohtani, B.; Prieto-Mahaney, O.O.; Li, D.; Abe, R. What is Degussa (Evonik) P25? Crystalline composition analysis, reconstruction from isolated pure particles and photocatalytic activity test. J. Photochem. Photobiol. A Chem. 2010, 216, 179–182. [Google Scholar] [CrossRef]
- Mistrik, J.; Kasap, S.; Ruda, H.E.; Koughia, C.; Singh, J. Optical Properties of Electronic Materials: Fundamentals and Characterization. In Springer Handbook of Electronic and Photonic Materials; Kasap, S., Capper, P., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Reddy, K.M.; Manorama, S.V.; Reddy, A.R. Bandgap studies on anatase titanium dioxide nanoparticles. Mater. Chem. Phys. 2003, 78, 239–245. [Google Scholar] [CrossRef]
- Han, H.; Bai, R. Highly effective buoyant photocatalyst prepared with a novel layered-TiO2 configuration on polypropylene fabric and the degradation performance for methyl orange dye under UV–Vis and Vis lights. Sep. Purif. Technol. 2010, 73, 142–150. [Google Scholar] [CrossRef]
- Samet, L.; March, K.; Brun, N.; Hosni, F.; Stephan, O.; Chtourou, R. Effect of gamma radiation on the photocatalytic properties of Cu doped titania nanoparticles. Mater. Res. Bull. 2018, 107, 1–7. [Google Scholar] [CrossRef]
- Liu, H.; Ma, H.; Li, X.; Li, W.; Wu, M.; Bao, X. The enhancement of TiO2 photocatalytic activity by hydrogen thermal treatment. Chemosphere 2003, 50, 39–46. [Google Scholar] [CrossRef]
- Rajaraman, T.; Parikh, S.P.; Gandhi, V.G. Black TiO2: A review of its properties and conflicting trends. Chem. Eng. J. 2020, 389, 123918. [Google Scholar] [CrossRef]
- Maqbool, Q.; Srivastava, A. Benign Synthesis of Black Microspheres of Anatase TiO2 with Paramagnetic Oxygen Vacancies through NH3 Treatment. Chem. A Eur. J. 2017, 23, 13864–13868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Xing, C.; Guo, M.; Xu, F.; Wang, X.; Gruber, H.J.; Zhang, B.; Tang, J. Sodium citrate: A universal reducing agent for reduction / decoration of graphene oxide with au nanoparticles. Nano Res. 2011, 4, 599–611. [Google Scholar] [CrossRef]
- Hilaire, L.; Guerrero, G.; Légaré, P.; Maire, G.; Krill, G. A photoemission study of the oxidation of platinum in Pt-based alloys: Pt-Pd, Pt-Ru, Pt-Ir. Surf. Sci. 1984, 146, 569–582. [Google Scholar] [CrossRef]
- Oudenhuijzen, M.K.; Kooyman, P.J.; Tappel, B.; Van Bokhoven, J.A.; Koningsberger, D.C. Understanding the Influence of the Pretreatment Procedure on Platinum Particle Size and Particle-Size Distribution for SiO2 Impregnated with [Pt2+(NH3)4](NO3−)2: A Combination of HRTEM, Mass Spectrometry, and Quick EXAFS. J. Catal. 2002, 205, 135–146. [Google Scholar] [CrossRef]
- Pan, X.; Yang, M.-Q.; Fu, X.; Zhang, N.; Xu, Y.-J. Defective TiO2 with oxygen vacancies: Synthesis, properties and photocatalytic applications. Nanoscale 2013, 5, 3601–3614. [Google Scholar] [CrossRef]
- Hu, H.; Qian, D.; Lin, P.; Ding, Z.; Cui, C. Oxygen vacancies mediated in-situ growth of noble-metal (Ag, Au, Pt) nanoparticles on 3D TiO2 hierarchical spheres for efficient photocatalytic hydrogen evolution from water splitting. Int. J. Hydrogen Energy 2020, 45, 629–639. [Google Scholar] [CrossRef]
- Xu, L.; Wang, S.; Zhang, T.; Chen, F. Photo-induced re-modulation of Pt particles loaded on V-TiO2 for enhanced CO photocatalytic oxidation. Catal. Sci. Technol. 2017, 7, 3698–3701. [Google Scholar] [CrossRef]
- Wu, J.; Ma, X.; Xu, L.; Zhao, B.; Chen, F. Fluorination promoted photoinduced modulation of Pt clusters on oxygen vacancy enriched TiO2/Pt photocatalyst for superior photocatalytic performance. Appl. Surf. Sci. 2019, 489, 510–518. [Google Scholar] [CrossRef]
- Qiu, H.; Ma, X.; Sun, C.; Zhao, B.; Chen, F. Surface oxygen vacancies enriched Pt/TiO2 synthesized with a defect migration strategy for superior photocatalytic activity. Appl. Surf. Sci. 2020, 506, 145021. [Google Scholar] [CrossRef]
- Ma, X.; Wu, J.; Xu, L.; Zhao, B.; Chen, F. Modulation of Pt species on oxygen vacancies enriched TiO2 via UV illumination for photocatalytic performance optimization. Colloids Surf. A Physicochem. Eng. Asp. 2020, 586, 124243. [Google Scholar] [CrossRef]
- Kydd, R.; Scott, J.; Teoh, W.Y.; Chiang, K.; Amal, R. Understanding Photocatalytic Metallization of Preadsorbed Ionic Gold on Titania, Ceria, and Zirconia. Langmuir 2010, 26, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Li, Y.; Chen, X.; Tian, T.; Fang, P.; Zheng, F.; Zhao, X. Tuning the Relative Concentration Ratio of Bulk Defects to Surface Defects in TiO2 Nanocrystals Leads to High Photocatalytic Efficiency. J. Am. Chem. Soc. 2011, 133, 16414–16417. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of TiO2 | SSA, m2/g | Crystalline Phases b, % | Grain Size, nm | ||
|---|---|---|---|---|---|
| Anatase | Rutile | Anatase | Rutile | ||
| SG400 | 45 ± 10 a | 100 | - | 15–20 c | - |
| SG450 | 47 ± 10 a | 100 | - | 15–30 c | - |
| SG500 | 28 ± 10 a | 100 | - | 20–30 c | - |
| P25 | 50 ± 10 | 82 | 18 | 24 b | 45 b |
| Catalyst | Fresh | Recovered | ||
|---|---|---|---|---|
| Pt Content (Weight%) | Pt/Ti Ratio | Pt Content (Weight%) | Pt/Ti Ratio | |
| SG400PtH2red | 2.2 | 1:95 = 0.011 | 1.5 | 1:150 = 0.007 |
| SG400PtCalc | 3.0 | 1:74 = 0.014 | 1.9 | 1:117 = 0.009 |
| SG450PtH2red | 2.1 | 1:104 = 0.010 | 1.5 | 1:146 = 0.007 |
| SG450PtCalc | 3.6 | 1:62 = 0.016 | 2.0 | 1:108 = 0.009 |
| SG500PtH2red | 2.4 | 1:89 = 0.011 | 1.5 | 1:140 = 0.007 |
| SG500PtCalc | 4.2 | 1:50 = 0.020 | 1.9 | 1:115 = 0.009 |
| P25PtH2red | 2.0 | 1:120 = 0.008 | 1.5 | 1:139 = 0.007 |
| P25PtCalc | 2.2 | 1:102 = 0.010 | 1.8 | 1:150 = 0.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majrik, K.; Pászti, Z.; Korecz, L.; Mihály, J.; May, Z.; Németh, P.; Cannilla, C.; Bonura, G.; Frusteri, F.; Tompos, A.; et al. Effect of the Microstructure of the Semiconductor Support on the Photocatalytic Performance of the Pt-PtOx/TiO2 Catalyst System. Materials 2021, 14, 943. https://doi.org/10.3390/ma14040943
Majrik K, Pászti Z, Korecz L, Mihály J, May Z, Németh P, Cannilla C, Bonura G, Frusteri F, Tompos A, et al. Effect of the Microstructure of the Semiconductor Support on the Photocatalytic Performance of the Pt-PtOx/TiO2 Catalyst System. Materials. 2021; 14(4):943. https://doi.org/10.3390/ma14040943
Chicago/Turabian StyleMajrik, Katalin, Zoltán Pászti, László Korecz, Judith Mihály, Zoltán May, Péter Németh, Catia Cannilla, Giuseppe Bonura, Francesco Frusteri, András Tompos, and et al. 2021. "Effect of the Microstructure of the Semiconductor Support on the Photocatalytic Performance of the Pt-PtOx/TiO2 Catalyst System" Materials 14, no. 4: 943. https://doi.org/10.3390/ma14040943
APA StyleMajrik, K., Pászti, Z., Korecz, L., Mihály, J., May, Z., Németh, P., Cannilla, C., Bonura, G., Frusteri, F., Tompos, A., & Tálas, E. (2021). Effect of the Microstructure of the Semiconductor Support on the Photocatalytic Performance of the Pt-PtOx/TiO2 Catalyst System. Materials, 14(4), 943. https://doi.org/10.3390/ma14040943