
Solid-State NMR and Raman Spectroscopic Investigation of Fluoride-Substituted Apatites Obtained in Various Thermal Conditions


Abstract
:1. Introduction
2. Materials and Methods
- HAP-800: hydroxyapatite granules sintered at 800 °C;
- HAP-1000: hydroxyapatite granules sintered at 1000 °C;
- HAP-1200: hydroxyapatite granules sintered at 1200 °C;
- FAP-800: fluoride-substituted apatite granules sintered at 800 °C;
- FAP-1000: fluoride-substituted apatite granules sintered at 1000 °C;
- FAP-1200: fluoride-substituted apatite granules sintered at 1200 °C.
3. Results and Discussion
3.1. Powder X-ray Diffraction (PXRD)
3.2. Transmission Electron Microscopy (TEM)
3.3. Scaning Electron Microscopy (SEM)
3.4. Raman Spectroscopy
3.5. Fourier Transmition Infrared Spectroscopy (FT-IR)
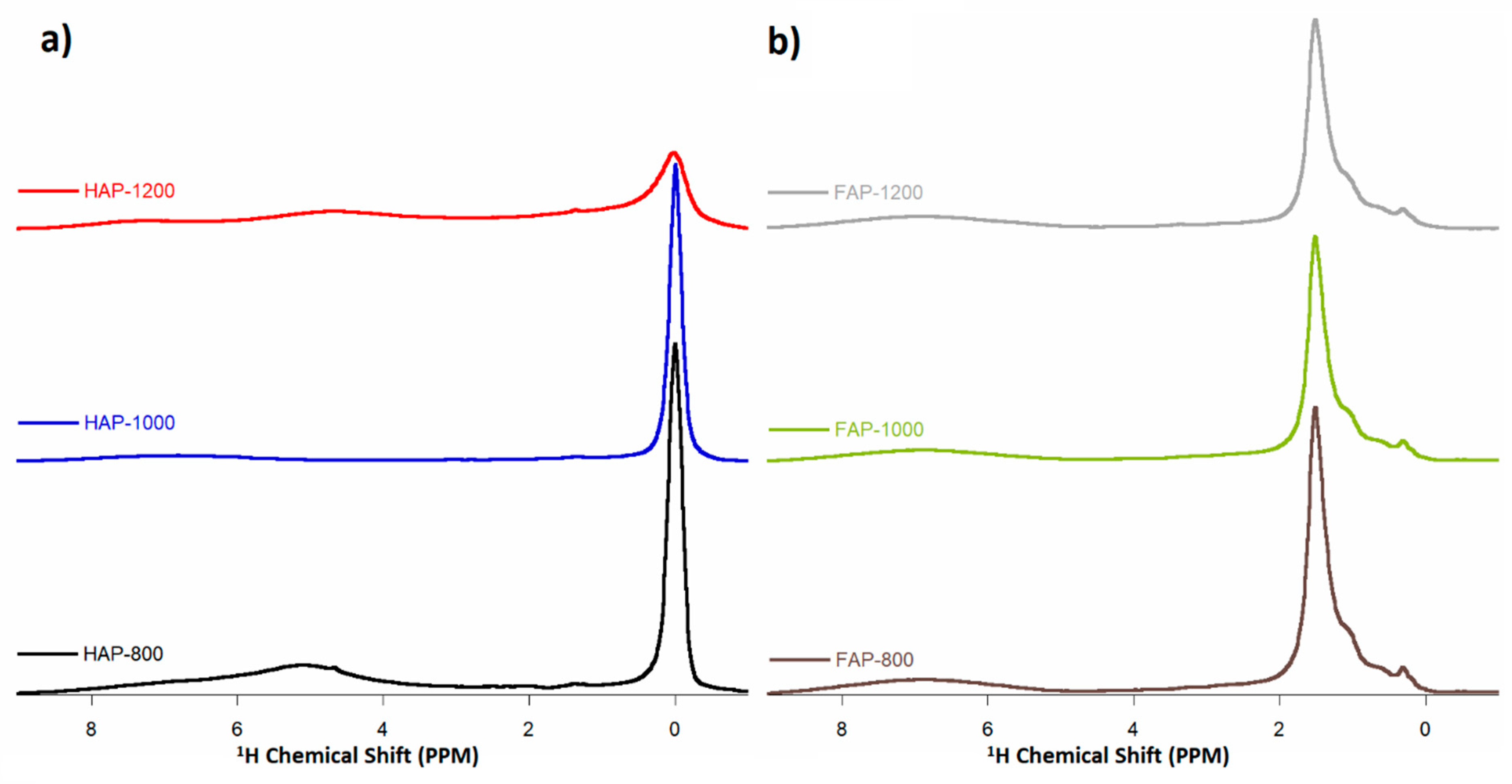
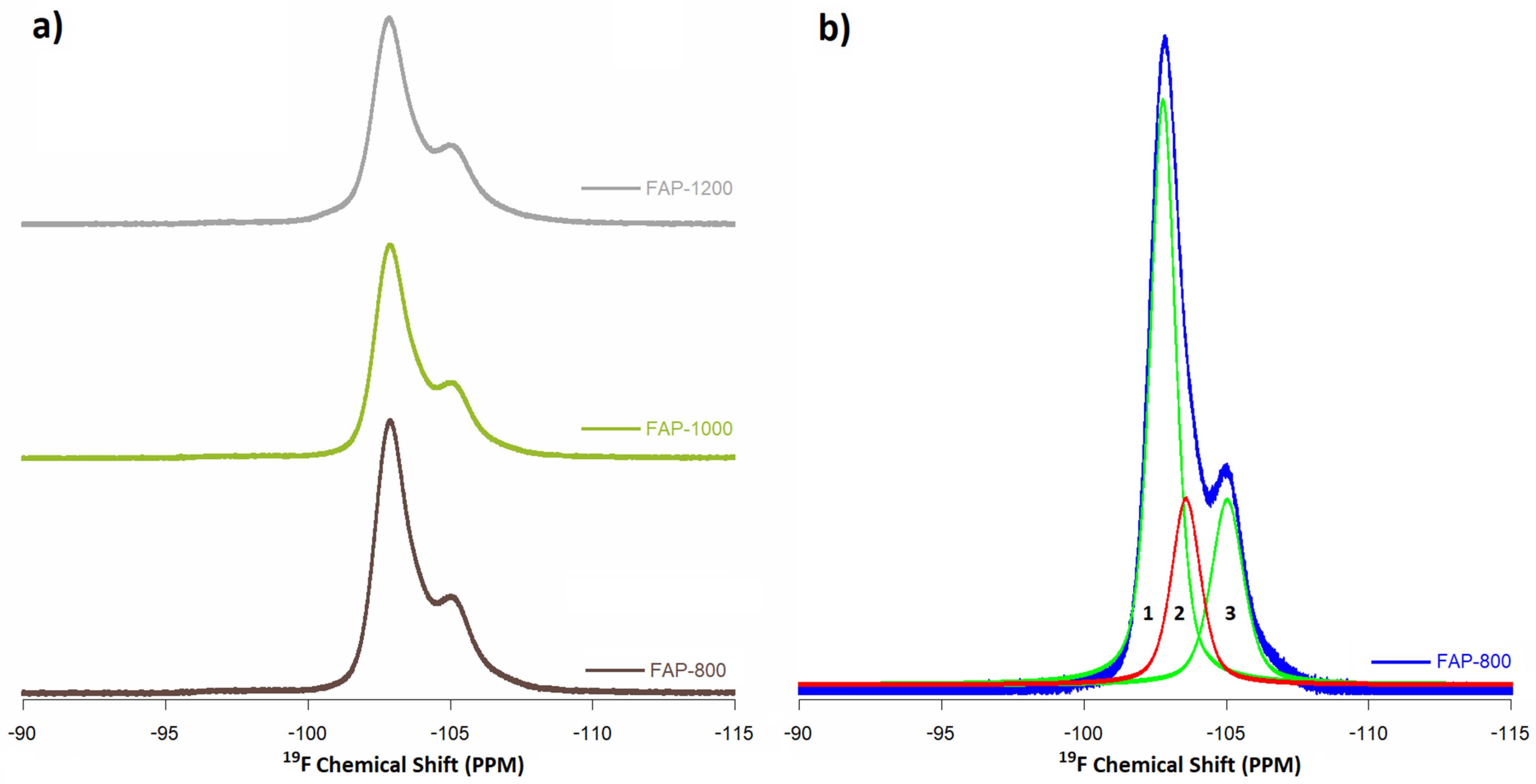
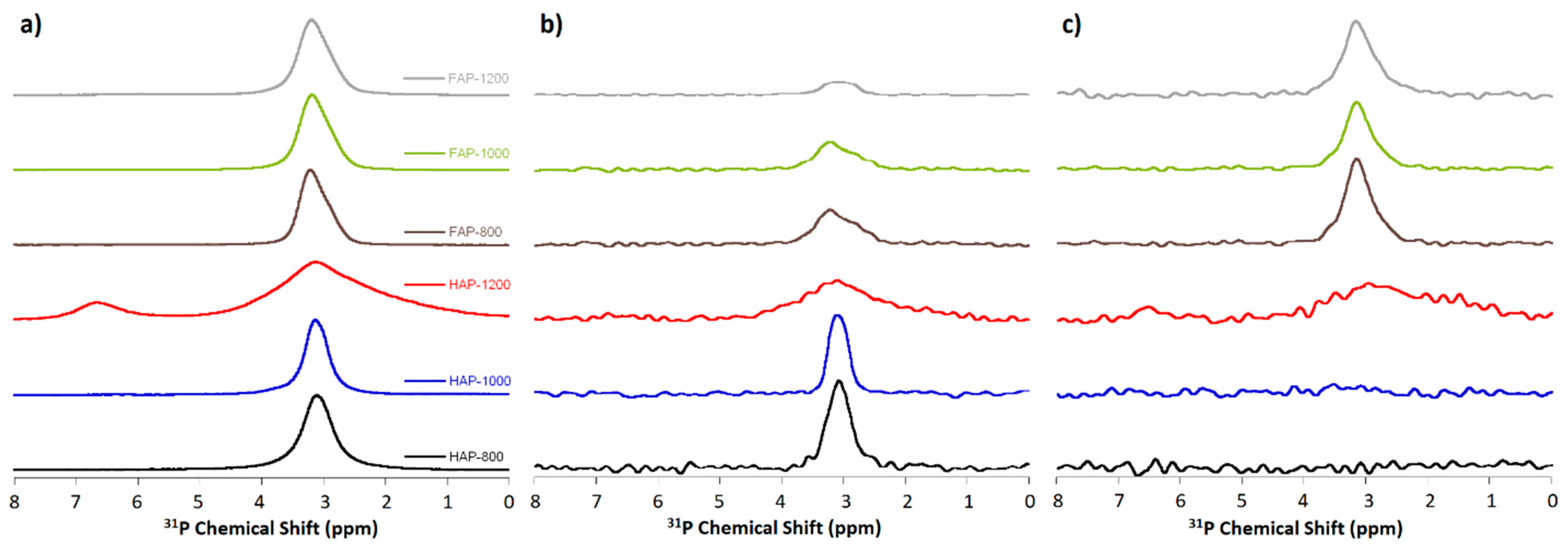
3.6. Solid-State Nuclear Magnetic Resonance Spectroscopy (ssNMR)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, K.; Chang, J. Structure and properties of hydroxyapatite for biomedical applications. In Hydroxyapatite (Hap) for Biomedical Applications; Woodhead Publishing: Cambridge, UK, 2015; pp. 3–19. [Google Scholar]
- Dorozhkin, S.V.; Epple, M. Biological and medical significance of calcium phosphates. Angew. Chem. Int. Ed. Engl. 2002, 41, 3130–3146. [Google Scholar] [CrossRef]
- Pajchel, L.; Kolodziejski, W. Solid-state MAS NMR, TEM, and TGA studies of structural hydroxyl groups and water in nanocrystalline apatites prepared by dry milling. J. Nanoparticle Res. 2013, 15, 1868. [Google Scholar] [CrossRef] [Green Version]
- Pajor, K.; Pajchel, L.; Kolmas, J. Hydroxyapatite and Fluorapatite in Conservative Dentistry and Oral Implantology—A Review. Materials 2019, 12, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph Nathanael, A.; Mangalaraj, D.; Hong, S.I.; Masuda, Y.; Rhee, Y.H.; Kim, H.W. Influence of fluorine substitution on the morphology and structure of hydroxyapatite nanocrystals prepared by hydrothermal method. Mater. Chem. Phys. 2013, 137, 967–976. [Google Scholar] [CrossRef]
- Kolmas, J.; Jabłoński, M.; Ślósarczyk, A.; Kolodziejski, W. Solid-State NMR Study of Mn2+ for Ca2+ Substitution in Thermally Processed Hydroxyapatites. J. Am. Ceram. Soc. 2015, 98, 1265–1274. [Google Scholar] [CrossRef]
- Ren, C.; Yu, Z.; Phillips, B.L.; Wang, H.; Ji, J.; Pan, B.; Li, W. Molecular-scale investigation of fluoride sorption mechanism by nanosized hydroxyapatite using 19F solid-state NMR spectroscopy. J. Colloid Interface Sci. 2019, 557, 357–366. [Google Scholar] [CrossRef]
- Nasker, P.; Samanta, A.; Rudra, S.; Sinha, A.; Mukhopadhyay, A.K.; Das, M. Effect of fluorine substitution on sintering behaviour, mechanical and bioactivity of hydroxyapatite. J. Mech. Behav. Biomed. Mater. 2019, 95, 136–142. [Google Scholar] [CrossRef]
- Suzuki, S.; Sakamura, M.; Ichiyanagi, M.; Ozawa, M. Internal friction of hydroxyapatite and fluorapatite. Ceram. Int. 2004, 30, 625–627. [Google Scholar] [CrossRef]
- Joseph Nathanael, A.; Oyane, A.; Nakamura, M.; Mahanti, M.; Koga, K.; Shitomi, K.; Miyaji, H. Rapid and area-specific coating of fluoride-incorporated apatite layers by a laser-assisted biomimetic process for tooth surface functionalization. Acta Biomater. 2018, 79, 148–157. [Google Scholar] [CrossRef]
- Sadat-Shojai, M.; Khorasani, M.T.; Dinpanah-Khoshdargi, E.; Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater. 2013, 9, 7591–7621. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, Y.; Yan, W.; Hu, Q.; Tao, J.; Zhang, M.; Shi, Z.; Tang, R. Role of hydroxyapatite nanoparticle size in bone cell proliferation. J. Mater. Chem. 2007, 17, 3780–3787. [Google Scholar] [CrossRef]
- Penel, G.; Leroy, G.; Rey, C.; Sombret, B.; Huvenne, J.P.; Bres, E. Infrared and Raman microspectrometry study of fluor-fluor-hydroxy and hydroxy-apatite powders. J. Mater. Sci. Mater. Med. 1997, 8, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Tredwin, C.J.; Young, A.M.; Georgiou, G.; Shin, S.-H.; Kim, H.-W.; Knowles, J.C. Hydroxyapatite, fluor-hydroxyapatite and fluorapatite produced via the sol–gel method. Optimisation, characterisation and rheology. Dent. Mater. 2013, 29, 166–173. [Google Scholar] [CrossRef]
- Gross, K.A.; Bhadang, K.A. Sintered hydroxyfluorapatites. Part III: Sintering and resultant mechanical properties of sintered blends of hydroxyapatite and fluorapatite. Biomaterials 2004, 25, 1395–1405. [Google Scholar] [CrossRef]
- Barinov, S.M.; Shvorneva, L.I.; Ferro, D.; Fadeeva, I.V.; Tumanov, S.V. Solid solution formation at the sintering of hydroxyapatite–fluorapatite ceramics. Sci. Technol. Adv. Mater. 2004, 5, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Gross, K.A.; Rodríguez-Lorenzo, L.M. Sintered hydroxyfluorapatites. Part I: Sintering ability of precipitated solid solution powders. Biomaterials 2004, 25, 1375–1384. [Google Scholar] [CrossRef]
- Gross, K.A.; Rodríguez-Lorenzo, L.M. Sintered hydroxyfluorapatites. Part II: Mechanical properties of solid solutions determined by microindentation. Biomaterials 2004, 25, 1385–1394. [Google Scholar] [CrossRef]
- Harrison, J.; Melville, A.J.; Forsythe, J.S.; Muddle, B.C.; Trounson, A.O.; Gross, K.A.; Mollard, R. Sintered hydroxyfluorapatites—IV: The effect of fluoride substitutions upon colonisation of hydroxyapatites by mouse embryonic stem cells. Biomaterials 2004, 25, 4977–4986. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, Z.; Zhu, P.; Wang, J.; Gan, Z.; Wei, J.; Zhao, Y.; Wei, S. Effects of fluorine on the structure of fluorohydroxyapatite: A study by XRD, solid-state NMR and Raman spectroscopy. J. Mater. Chem. B 2015, 3, 34–38. [Google Scholar] [CrossRef]
- Borkowski, L.; B, A.; Przekora, A.; Ginalska, G. Production Method for Biocompatible Implant Material. Patent no. 235803, 2020. [Google Scholar]
- Borkowski, L.; Przekora, A.; Belcarz, A.; Palka, K.; Jozefaciuk, G.; Lübek, T.; Jojczuk, M.; Nogalski, A.; Ginalska, G. Fluorapatite ceramics for bone tissue regeneration: Synthesis, characterization and assessment of biomedical potential. Mater. Sci. Eng. C 2020, 116, 111211. [Google Scholar] [CrossRef]
- Szurkowska, K.; Zgadzaj, A.; Kuras, M.; Kolmas, J. Novel hybrid material based on Mg2+ and SiO44- co-substituted nano-hydroxyapatite, alginate and chondroitin sulphate for potential use in biomaterials engineering. Ceram. Int. 2018, 44, 18551–18559. [Google Scholar] [CrossRef]
- Guggino, S.N.; Hervig, R.L. Fluorine Partitioning between Nominally Anhydrous Minerals (cpx, ol, plag) and Silicate Melt. In AGU Fall Meeting Abstracts; abstract id. V31D-2828; American Geophysical Union: Washington, DC, USA, 2012. [Google Scholar]
- Chen, Y.; Miao, X. Thermal and chemical stability of fluorohydroxyapatite ceramics with different fluorine contents. Biomaterials 2005, 26, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.D.; Hill, R.G.; Law, R.V.; Fong, S. Raman spectroscopy, 19F and 31P MAS-NMR of a series of fluorochloroapatites. J. Eur. Ceram. Soc. 2009, 29, 377–384. [Google Scholar] [CrossRef]
- Freund, F.; Knobel, R.M. Distribution of fluorine in hydroxyapatite studied by infrared spectroscopy. J. Chem. Soc. Dalton Trans. 1977, 1136–1140. [Google Scholar] [CrossRef]
- Kolmas, J.; Kolodziejski, W. Concentration of hydroxyl groups in dental apatites: A solid-state 1H MAS NMR study using inverse 31P → 1H cross-polarization. Chem. Commun. 2007, 4390–4392. [Google Scholar] [CrossRef]
- Ou, S.-F.; Chiou, S.-Y.; Ou, K.-L. Phase transformation on hydroxyapatite decomposition. Ceram. Int. 2013, 39, 3809–3816. [Google Scholar] [CrossRef]
- Mason, H.E.; McCubbin, F.M.; Smirnov, A.; Phillips, B.L. Solid-state NMR and IR spectroscopic investigation of the role of structural water and F in carbonate-rich fluorapatite. Am. Mineral. 2009, 94, 507–516. [Google Scholar] [CrossRef]
- Jäger, C.; Welzel, T.; Meyer-Zaika, W.; Epple, M. A solid-state NMR investigation of the structure of nanocrystalline hydroxyapatite. Magn. Reson. Chem. 2006, 44, 573–580. [Google Scholar] [CrossRef]
- Vyalikh, A.; Simon, P.; Rosseeva, E.; Buder, J.; Scheler, U.; Kniep, R. An NMR Study of Biomimetic Fluorapatite—Gelatine Mesocrystals. Sci. Rep. 2015, 5, 15797. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a (Å) | c (Å) | Cell Volume (Å3) | Crystallite Size Along c-Axis | Crystallite Size Along a-Axis |
|---|---|---|---|---|---|
| FAP-800 | 9.379 (3) | 6.885 (2) | 524.6 (3) | 58 ± 4 nm | 49 ± 4 nm |
| FAP-1000 | 9.376 (2) | 6.885 (3) | 524.2 (3) | 54 ± 3 nm | 47 ± 3 nm |
| FAP-1200 | 9.377 (1) | 6.887 (3) | 524.5 (2) | 74 ± 4 nm | 68 ± 4 nm |
| HAP-800 | 9.415 (2) | 6.881 (1) | 528.2 (1) | 30 ± 2 nm | 24 ± 2 nm |
| HAP-1000 | 9.419 (3) | 6.882 (3) | 528.8 (3) | 55 ± 4 nm | 44 ± 3 nm |
| HAP-1200 | 9.408 (2) | 6.885 (2) | 527.7 (1) | 54 ± 3 nm | 48 ± 3 nm |
| Material | Type of Crystals | Width | Length |
|---|---|---|---|
| HAP-800 | uniform | 32 ± 9 | 49 ± 19 |
| HAP-1000 | uniform | 154 ± 75 | 205 ± 122 |
| HAP-1200 | small crystals | 15 ± 6 | 22 ± 11 |
| sharp edges crystals | 166 ± 124 | 269 ± 179 | |
| FAP-800 | uniform | 46 ± 35 | 70 ± 46 |
| FAP-1000 | small crystals | 21 ± 13 | 32 ± 19 |
| sharp edges crystals | 121 ± 51 | 212 ± 52 | |
| FAP-1200 | small crystals | 22 ± 12 | 33 ± 20 |
| sharp edges crystals | 163 ± 56 | 264 ± 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajchel, L.; Borkowski, L. Solid-State NMR and Raman Spectroscopic Investigation of Fluoride-Substituted Apatites Obtained in Various Thermal Conditions. Materials 2021, 14, 6936. https://doi.org/10.3390/ma14226936
Pajchel L, Borkowski L. Solid-State NMR and Raman Spectroscopic Investigation of Fluoride-Substituted Apatites Obtained in Various Thermal Conditions. Materials. 2021; 14(22):6936. https://doi.org/10.3390/ma14226936
Chicago/Turabian StylePajchel, Lukasz, and Leszek Borkowski. 2021. "Solid-State NMR and Raman Spectroscopic Investigation of Fluoride-Substituted Apatites Obtained in Various Thermal Conditions" Materials 14, no. 22: 6936. https://doi.org/10.3390/ma14226936
APA StylePajchel, L., & Borkowski, L. (2021). Solid-State NMR and Raman Spectroscopic Investigation of Fluoride-Substituted Apatites Obtained in Various Thermal Conditions. Materials, 14(22), 6936. https://doi.org/10.3390/ma14226936