
Accessing Structural, Electronic, Transport and Mesoscale Properties of Li-GICs via a Complete DFTB Model with Machine-Learned Repulsion Potential

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational
2.2. Experimental
3. Results and Discussion
3.1. Structural Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPrep-DFTB | Experiment | DFT | ML | |||||
|---|---|---|---|---|---|---|---|---|
| Method: | (a) | (b) | (c) | (d) | (e) | (f) | (g) | |
| Å] | ||||||||
| Å] | ||||||||
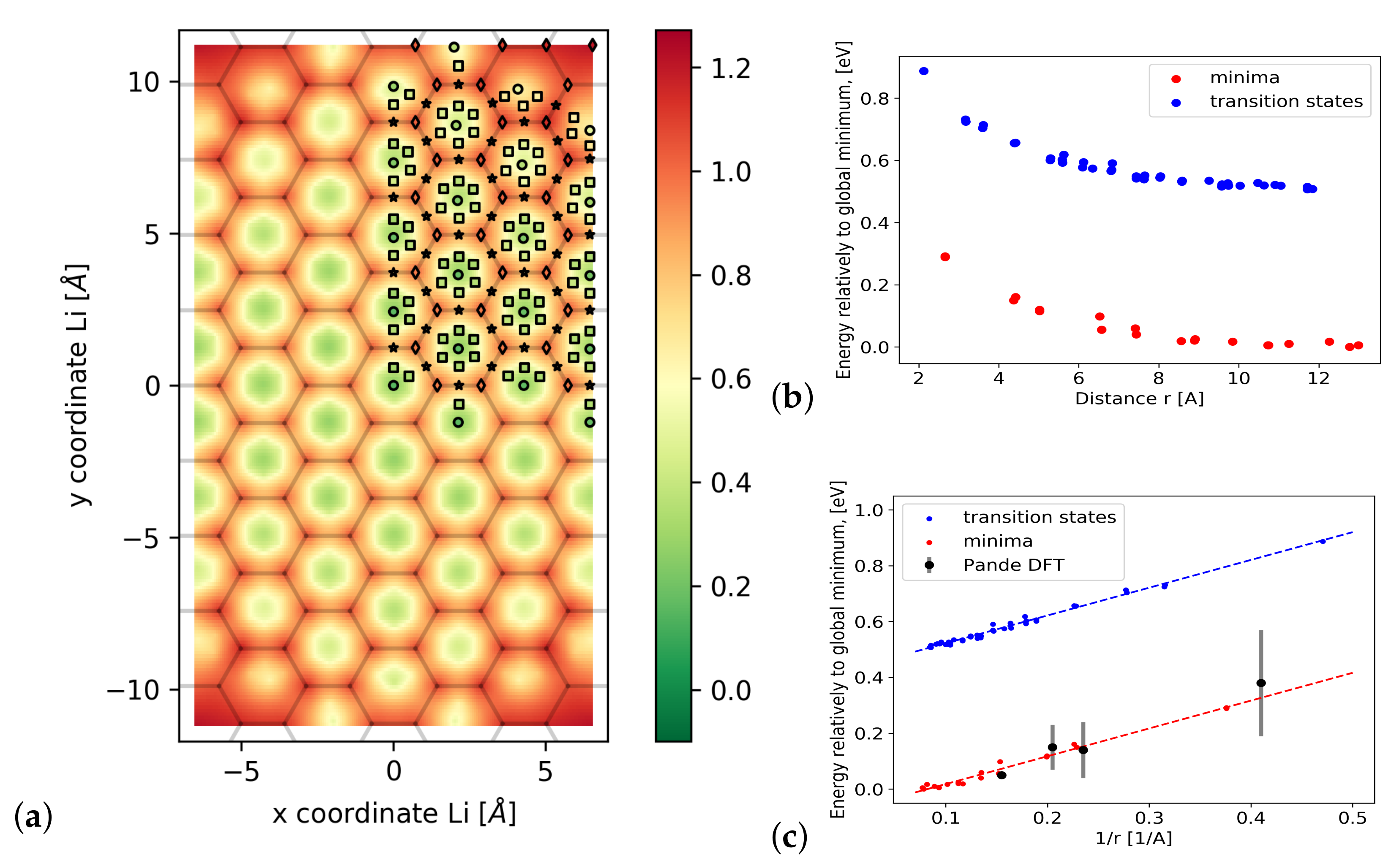
3.2. Diffusion Barriers
3.3. Formation Energetics
3.4. Long-Range Interactions
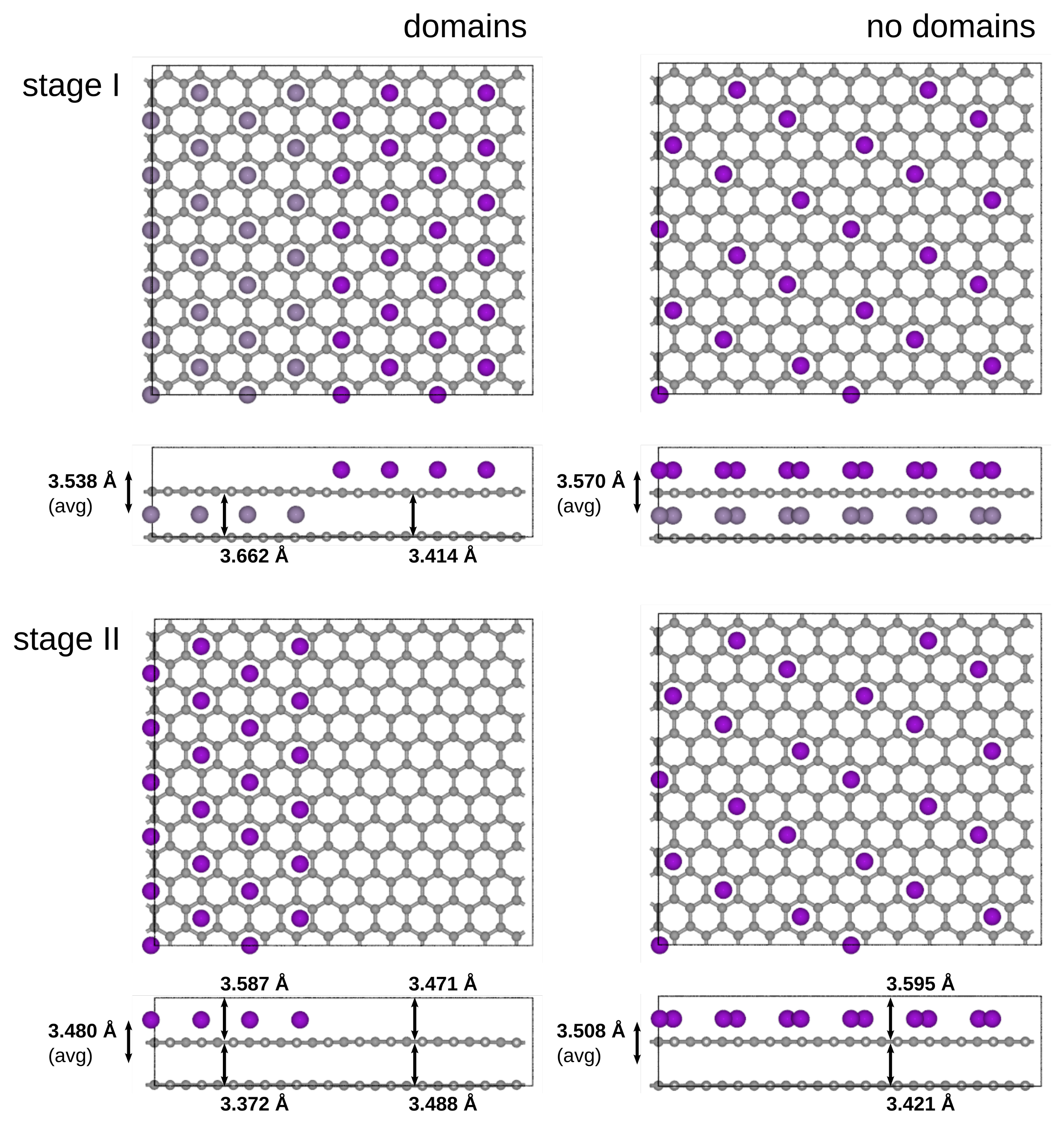
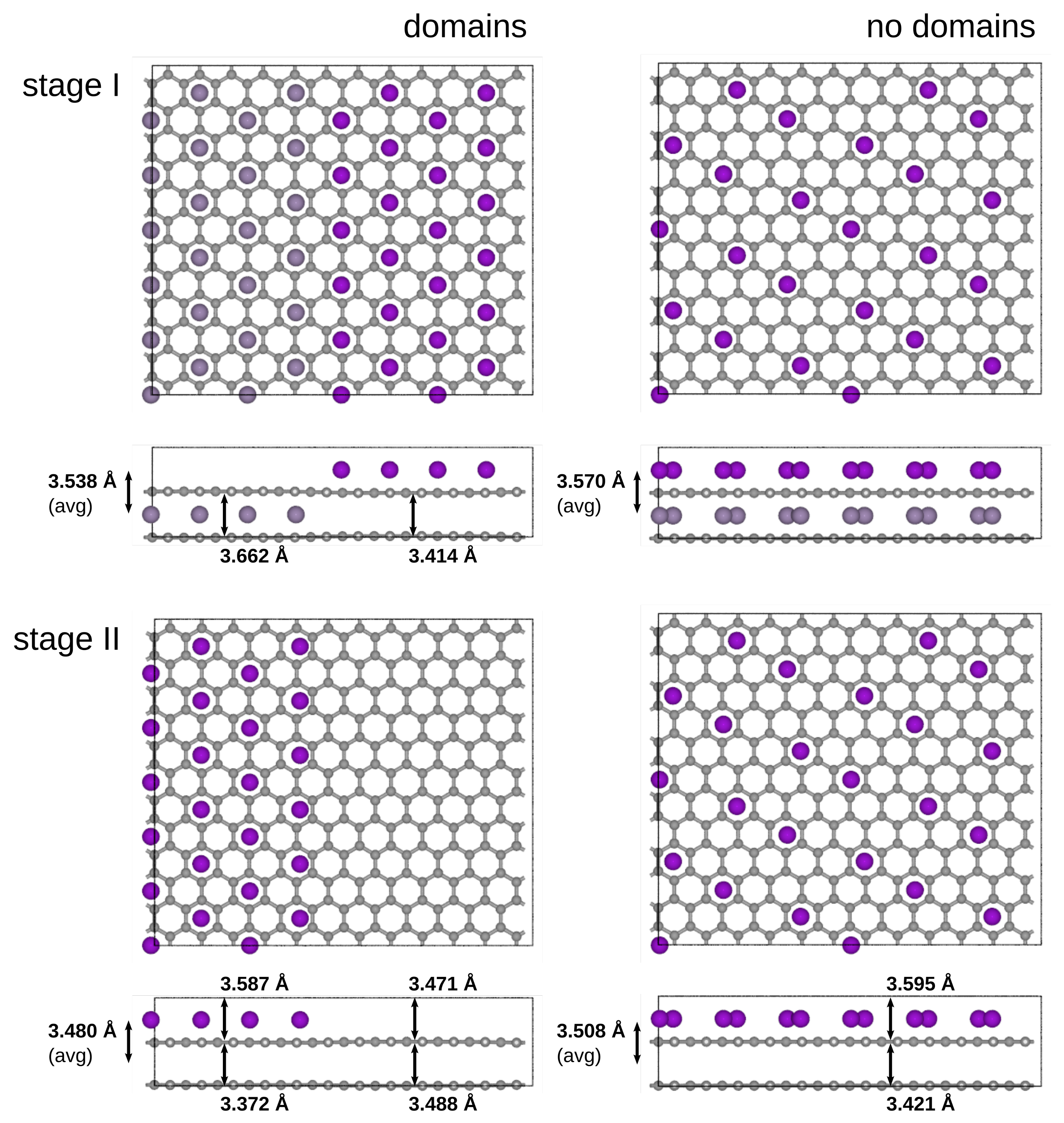
3.5. Domains vs. Dilute
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, H.; Yang, Y.; Ren, D.; Wang, L.; He, X. Graphite as anode materials: Fundamental mechanism, recent progress and advances. Energy Storage Mater. 2020, 36, 147–170. [Google Scholar] [CrossRef]
- Asenbauer, J.; Eisenmann, T.; Kuenzel, M.; Kazzazi, A.; Chen, Z.; Bresser, D. The success story of graphite as a lithium-ion anode material–fundamentals, remaining challenges, and recent developments including silicon (oxide) composites. Sustain. Energy Fuels 2020, 4, 5387. [Google Scholar] [CrossRef]
- Grosu, C.; Panosetti, C.; Merz, S.; Jakes, P.; Matera, S.; Eichel, R.A.; Granwehr, J.; Scheurer, C. Lithium intercalation into graphite beyond LiC6 at ambient pressure. arXiv 2021, arXiv:2107.11137. [Google Scholar]
- Zhang, C.; Ma, J.; Han, F.; Liu, H.; Zhang, F.; Fan, C.; Liu, J.; Li, X. Strong anchoring effect of ferric chloride-graphite intercalation compounds (FeCl 3-GICs) with tailored epoxy groups for high-capacity and stable lithium storage. J. Mater. Chem. A 2018, 6, 17982. [Google Scholar] [CrossRef]
- Dahn, J.; Fong, R.; Spoon, M. Suppression of staging in lithium-intercalated carbon by disorder in the host. Phys. Rev. B 1990, 42, 6424. [Google Scholar] [CrossRef] [Green Version]
- Reynier, Y.; Yazami, R.; Fultz, B. The entropy and enthalpy of lithium intercalation into graphite. J. Power Sources 2003, 119, 850. [Google Scholar] [CrossRef]
- Daumas, N.; Heróld, A. Relations between phase concept and reaction mechanics in graphite insertion compounds. C. R. Acad. Sci. C 1969, 268, 373. [Google Scholar]
- Vadlamani, B.; An, K.; Jagannathan, M.; Chandran, K.R. An in situ electrochemical cell for neutron diffraction studies of phase transitions in small volume electrodes of Li-ion batteries. J. Electrochem. Soc. 2014, 161, A1731. [Google Scholar] [CrossRef]
- Kambe, N.; Dresselhaus, M.; Dresselhaus, G.; Basu, S.; McGhie, A.; Fischer, J. Intercalate ordering in first stage graphite-lithium. Mater. Sci. Eng. 1979, 40, 1. [Google Scholar] [CrossRef]
- Babar, M.; Parks, H.L.; Houchins, G.; Viswanathan, V. An accurate machine learning calculator for the lithium-graphite system. J. Phys. Energy 2020, 3, 014005. [Google Scholar] [CrossRef]
- Imai, Y.; Watanabe, A. Energetic evaluation of possible stacking structures of Li-intercalation in graphite using a first-principle pseudopotential calculation. J. Alloy. Compd. 2007, 439, 258. [Google Scholar] [CrossRef]
- Lenchuk, O.; Adelhelm, P.; Mollenhauer, D. Comparative study of density functionals for the description of lithium-graphite intercalation compounds. J. Comput. Chem. 2019, 40, 2400. [Google Scholar] [CrossRef]
- Pande, V.; Viswanathan, V. Robust high-fidelity DFT study of the lithium-graphite phase diagram. Phys. Rev. Mater. 2018, 2, 125401. [Google Scholar] [CrossRef] [Green Version]
- Thinius, S.; Islam, M.M.; Heitjans, P.; Bredow, T. Theoretical study of Li migration in lithium–graphite intercalation compounds with dispersion-corrected DFT methods. J. Phys. Chem. C 2014, 118, 2273. [Google Scholar] [CrossRef]
- Fujikake, S.; Deringer, V.L.; Lee, T.H.; Krynski, M.; Elliott, S.R.; Csányi, G. Gaussian approximation potential modeling of lithium intercalation in carbon nanostructures. J. Chem. Phys. 2018, 148, 241714. [Google Scholar] [CrossRef] [PubMed]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef]
- Trucano, P.; Chen, R. Structure of graphite by neutron diffraction. Nature 1975, 258, 136. [Google Scholar] [CrossRef]
- Yin, M.; Cohen, M.L. Structural theory of graphite and graphitic silicon. Phys. Rev. B 1984, 29, 6996. [Google Scholar] [CrossRef]
- Ohzuku, T.; Iwakoshi, Y.; Sawai, K. Formation of lithium-graphite intercalation compounds in nonaqueous electrolytes and their application as a negative electrode for a lithium ion (shuttlecock) cell. J. Electrochem. Soc. 1993, 140, 2490. [Google Scholar] [CrossRef]
- Avdeev, V.V.; Savchenkova, A.P.; Monyakina, L.A.; Nikol’skaya, I.V.; Khvostov, A.V. Intercalation reactions and carbide formation in graphite-lithium system. J. Phys. Chem. Solids 1996, 57, 947. [Google Scholar] [CrossRef]
- Langer, J.; Epp, V.; Heitjans, P.; Mautner, F.A.; Wilkening, M. Lithium motion in the anode material LiC6as seen via time-domain 7Li NMR. Phys. Rev. B 2013, 88, 094304. [Google Scholar] [CrossRef] [Green Version]
- Freiländer, P.; Heitjans, P.; Ackermann, H.; Bader, B.; Kiese, G.; Schirmer, A.; Stöckmann, H.J.; Van der Marel, C.; Magerl, A.; Zabel, H. Diffusion Processes in LiC6 Studied by β-NMR. Z. Phys. Chem. 1987, 151, 93. [Google Scholar] [CrossRef]
- Magerl, A.; Zabel, H.; Anderson, I. In-plane jump diffusion of Li in LiC6. Phys. Rev. Lett. 1985, 55, 222. [Google Scholar] [CrossRef] [PubMed]
- Toyoura, K.; Koyama, Y.; Kuwabara, A.; Tanaka, I. Effects of off-stoichiometry of LiC6 on the lithium diffusion mechanism and diffusivity by first principles calculations. J. Phys. Chem. C 2010, 114, 2375. [Google Scholar] [CrossRef]
- Persson, K.; Hinuma, Y.; Meng, Y.S.; Van der Ven, A.; Ceder, G. Thermodynamic and kinetic properties of the Li-graphite system from first-principles calculations. Phys. Rev. B 2010, 82, 125416. [Google Scholar] [CrossRef]
- Panosetti, C.; Anniés, S.B.; Grosu, C.; Seidlmayer, S.; Scheurer, C. DFTB Modeling of Lithium-Intercalated Graphite with Machine-Learned Repulsive Potential. J. Phys. Chem. C 2021, 125, 691. [Google Scholar] [CrossRef]
- Chou, C.P.; Nishimura, Y.; Fan, C.C.; Mazur, G.; Irle, S.; Witek, H.A. Automatized Parameterization of DFTB Using Particle Swarm Optimization. J. Chem. Theory Comput. 2016, 12, 53. [Google Scholar] [CrossRef]
- Panosetti, C.; Engelmann, A.; Nemec, L.; Reuter, K.; Margraf, J.T. Learning to Use the Force: Fitting Repulsive Potentials in Density-Functional Tight-Binding with Gaussian Process Regression. J. Chem. Theory Comput. 2020, 16, 2181. [Google Scholar] [CrossRef] [PubMed]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009, 180, 2175. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiStasio, R.A.; Gobre, V.V.; Tkatchenko, A. Many-body van der Waals interactions in molecules and condensed matter. J. Condens. Matter Phys. 2014, 26, 213202. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1. [Google Scholar] [CrossRef] [Green Version]
- Nichol, A.; Ackland, G.J. Property trends in simple metals: An empirical potential approach. Phys. Rev. B 2016, 93, 184101. [Google Scholar] [CrossRef] [Green Version]
- Bahn, S.R.; Jacobsen, K.W. An object-oriented scripting interface to a legacy electronic structure code. Comput. Sci. Eng. 2002, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Shanno, D.F. An example of numerical nonconvergence of a variable-metric method. J. Optim. Theory Appl. 1985, 46, 87. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901. [Google Scholar] [CrossRef] [Green Version]
- Bitzek, E.; Koskinen, P.; Gähler, F.; Moseler, M.; Gumbsch, P. Structural relaxation made simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.D. Modified Broyden’s method for accelerating convergence in self-consistent calculations. Phys. Rev. B 1988, 38, 12807. [Google Scholar] [CrossRef] [PubMed]
- Zhechkov, L.; Heine, T.; Patchkovskii, S.; Seifert, G.; Duarte, H.A. An efficient a posteriori treatment for dispersion interaction in density-functional-based tight binding. J. Chem. Theory Comput. 2005, 1, 841. [Google Scholar] [CrossRef] [PubMed]
- Chung, D. Review graphite. J. Mater. Sci. 2002, 37, 1475. [Google Scholar] [CrossRef]
- Wang, Z.; Selbach, S.M.; Grande, T. Van der Waals density functional study of the energetics of alkali metal intercalation in graphite. RSC Adv. 2014, 4, 4069. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Tkatchenko, A.; DiStasio, R.A., Jr.; Car, R.; Scheffler, M. Accurate and efficient method for many-body van der Waals interactions. Phys. Rev. Lett. 2012, 108, 236402. [Google Scholar] [CrossRef]
- Gould, T.; Lebègue, S.; Ángyán, J.G.; Bučko, T. A Fractionally Ionic Approach to Polarizability and van der Waals Many-Body Dispersion Calculations. J. Chem. Theory Comput. 2016, 12, 5920. [Google Scholar] [CrossRef] [Green Version]
- Dresselhaus, M.S.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 2002, 51, 1. [Google Scholar] [CrossRef]
- Andersen, M.; Panosetti, C.; Reuter, K. A Practical Guide to Surface Kinetic Monte Carlo Simulations. Front. Chem. 2019, 7, 202. [Google Scholar] [CrossRef]
- Mercer, M.P.; Peng, C.; Soares, C.; Hoster, H.E.; Kramer, D. Voltage hysteresis during lithiation/delithiation of graphite associated with meta-stable carbon stackings. J. Mater. Chem. A 2021, 9, 492. [Google Scholar] [CrossRef]
- Rana, K.; Kucukayan-Dogu, G.; Sen, H.S.; Boothroyd, C.; Gulseren, O.; Bengu, E. Analysis of charge transfer for in situ Li intercalated carbon nanotubes. J. Phys. Chem. C 2012, 116, 11364. [Google Scholar] [CrossRef]
- Axdal, S.A.; Chung, D. A theory for the kinetics of intercalation of graphite. Carbon 1987, 25, 377. [Google Scholar] [CrossRef]
- Kirczenow, G. Kinetics of stage ordering and stage transitions. Phys. Rev. Lett. 1985, 55, 2810. [Google Scholar] [CrossRef]
- Dimiev, A.M.; Ceriotti, G.; Behabtu, N.; Zakhidov, D.; Pasquali, M.; Saito, R.; Tour, J.M. Direct real-time monitoring of stage transitions in graphite intercalation compounds. ACS Nano 2013, 7, 2773. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Graetz, J.; Moreno, M.S.; Ma, C.; Wu, L.; Volkov, V.; Zhu, Y. Chemical distribution and bonding of lithium in intercalated graphite: Identification with optimized electron energy loss spectroscopy. ACS Nano 2011, 5, 1190. [Google Scholar] [CrossRef]
- Krishnan, S.; Brenet, G.; Machado-Charry, E.; Caliste, D.; Genovese, L.; Deutsch, T.; Pochet, P. Revisiting the domain model for lithium intercalated graphite. Appl. Phys. Lett. 2013, 103, 251904. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Wu, M.; Liu, G.; Ouyang, C. Understanding the effect of the layer-to-layer distance on Li-intercalated graphite. J. Appl. Phys. 2012, 111, 124325. [Google Scholar] [CrossRef]

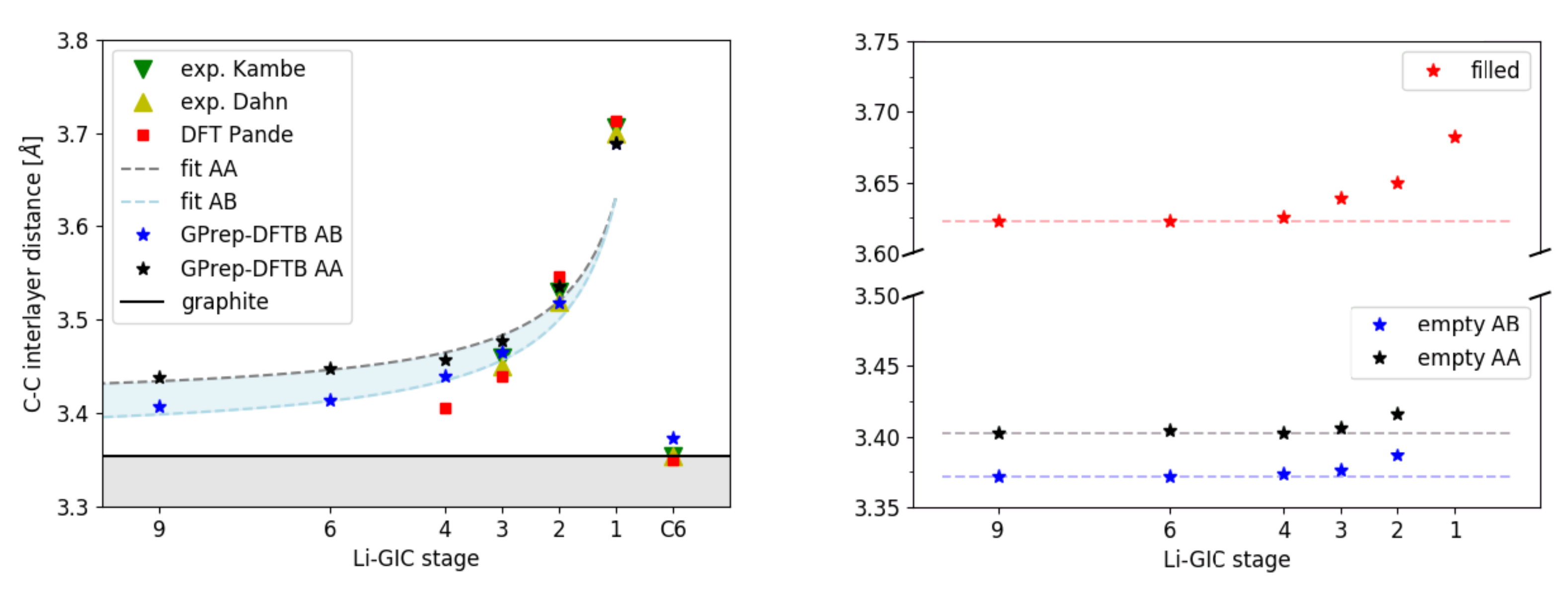

| DFTB | Experiment | DFT | ||||
|---|---|---|---|---|---|---|
| Filled | Empty / | Avg. / | Avg. Dahn [5] | Avg. Kambe [9] | Avg. Pande [13] | |
| stage 1 | 3.682 | - | 3.682/3.682 | 3.700 | 3.706 | 3.713 |
| stage 2 | 3.652 | 3.416/3.387 | 3.535/3.518 | 3.520 | 3.530 | 3.546 |
| stage 3 | 3.639 | 3.406/3.376 | 3.478/3.465 | 3.450 | 3.460 | 3.439 |
| stage 4 | 3.625 | 3.403/3.373 | 3.457/3.440 | - | - | 3.406 |
| stage 6 | 3.622 | 3.405/3.372 | 3.440/3.414 | - | - | - |
| stage 9 | 3.622 | 3.403/3.372 | 3.427/3.407 | - | - | - |
| graphite | - | 3.402/3.373 | 3.402/3.373 | 3.355 | 3.355 | 3.35 |
| Stage | Stacking | In-Plane % | Avg LS [Å] | Filled/Empty [Å] | Barrier [eV] | |
|---|---|---|---|---|---|---|
| LiC48 | III | 1/3 | 3.446 | 3.525/ | 0.493 | |
| Li2C48 | III | 2/3 | 3.478 | 3.616/ | 0.441 | |
| Li3C48 | III | 3/3 | 3.478 | 3.631/ | 0.424 | |
| LiC36 | II | 1/3 | 3.469 | 3.530/ | 0.504 | |
| Li2C36 | II | 2/3 | 3.512 | 3.614/ | 0.451 | |
| Li3C36 | II | 3/3 | 3.535 | 3.652/ | 0.426 | |
| Li2C36 | I | 1/3 | 3.539 | 0.492 | ||
| Li4C36 | I | 2/3 | 3.625 | 0.443 | ||
| Li4C36* | I | 2/3 | 3.658 | 0.412 | ||
| Li6C36 | I | 3/3 | 3.682 | 0.404 | ||
| Li6C36* | I | 3/3 | 3.758 | 0.396 |
| GPrep-DFTB | Experiment | DFT | ||||||
|---|---|---|---|---|---|---|---|---|
| Ref: | (a) | (b) | (c) | (d) | (e) | (f) | (g) | |
| LiC6 | * | |||||||
| LiC12 | * | |||||||
| LiC18 | * | − | − | − | − | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anniés, S.; Panosetti, C.; Voronenko, M.; Mauth, D.; Rahe, C.; Scheurer, C. Accessing Structural, Electronic, Transport and Mesoscale Properties of Li-GICs via a Complete DFTB Model with Machine-Learned Repulsion Potential. Materials 2021, 14, 6633. https://doi.org/10.3390/ma14216633
Anniés S, Panosetti C, Voronenko M, Mauth D, Rahe C, Scheurer C. Accessing Structural, Electronic, Transport and Mesoscale Properties of Li-GICs via a Complete DFTB Model with Machine-Learned Repulsion Potential. Materials. 2021; 14(21):6633. https://doi.org/10.3390/ma14216633
Chicago/Turabian StyleAnniés, Simon, Chiara Panosetti, Maria Voronenko, Dario Mauth, Christiane Rahe, and Christoph Scheurer. 2021. "Accessing Structural, Electronic, Transport and Mesoscale Properties of Li-GICs via a Complete DFTB Model with Machine-Learned Repulsion Potential" Materials 14, no. 21: 6633. https://doi.org/10.3390/ma14216633
APA StyleAnniés, S., Panosetti, C., Voronenko, M., Mauth, D., Rahe, C., & Scheurer, C. (2021). Accessing Structural, Electronic, Transport and Mesoscale Properties of Li-GICs via a Complete DFTB Model with Machine-Learned Repulsion Potential. Materials, 14(21), 6633. https://doi.org/10.3390/ma14216633