
Diptool—A Novel Numerical Tool for Membrane Interactions Analysis, Applying to Antimicrobial Detergents and Drug Delivery Aids


Abstract
:1. Introduction
2. Materials and Methods
2.1. Background
2.2. Theory and Calculation
2.3. Molecular Dynamics Validation
2.4. Data Visualization and Analysis
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Kramer, A. Hand Disinfection and Antiseptic of Skin, Mucous Membranes, and Wounds. In Dermatopharmacology of Topical Preparations; Springer: Berlin/Heidelberg, Germany, 2000; pp. 121–134. [Google Scholar]
- Russell, A.D. Antibiotic and biocide resistance in bacteria: Introduction. J. Appl. Microbiol. 2002, 92, 1S–3S. [Google Scholar] [CrossRef]
- Zhuang, W.; Wang, X.; Zhu, W.; Zhang, Y.; Sun, D.; Zhang, R.; Wu, C. Imidazoline Gemini Surfactants as Corrosion Inhibitors for Carbon Steel X70 in NaCl Solution. ACS Omega 2021, 6, 5653–5660. [Google Scholar] [CrossRef]
- Heakal, F.E.T.; Elkholy, A.E. Gemini surfactants as corrosion inhibitors for carbon steel. J. Mol. Liq. 2017, 230, 395–407. [Google Scholar] [CrossRef]
- Yang, W.; Cao, Y.; Ju, H.; Wang, Y.; Jiang, Y.; Geng, T. Amide Gemini surfactants linked by rigid spacer group 1,4-dibromo-2-butene: Surface properties, aggregate and application properties. J. Mol. Liq. 2021, 326, 115339. [Google Scholar] [CrossRef]
- Li, Y.; Wu, P.; Lei, C.; Liu, X.; Han, X. A novel cationic surfactant synthesized from carbon quantum dots and the versatility. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127088. [Google Scholar] [CrossRef]
- Zheng, L.C.; Tong, Q.X. Synthesis, surface adsorption, micellization behavior and antibacterial activity of novel gemini surfactants with morpholinium headgroup and benzene-based spacer. J. Mol. Liq. 2021, 331, 115781. [Google Scholar] [CrossRef]
- Sarıkaya, İ.; Bilgen, S.; Ünver, Y.; Bektaş, K.İ.; Akbaş, H. Synthesis, Characterization, Antibacterial Activity, and Interfacial and Micellar Features of Novel Cationic Gemini Surfactants with Different Spacers. J. Surfactants Deterg. 2021. [Google Scholar] [CrossRef]
- Fatma, N.; Panda, M.; Beg, M. Ester-bonded cationic gemini surfactants: Assessment of their cytotoxicity and antimicrobial activity. J. Mol. Liq. 2016, 222, 390–394. [Google Scholar] [CrossRef]
- Tawfik, S.M. Simple one step synthesis of gemini cationic surfactant-based ionic liquids: Physicochemical, surface properties and biological activity. J. Mol. Liq. 2015, 209, 320–326. [Google Scholar] [CrossRef]
- Hübner, N.-O.; Siebert, J.; Kramer, A. Octenidine Dihydrochloride, a Modern Antiseptic for Skin, Mucous Membranes and Wounds. Ski. Pharmacol. Physiol. 2010, 23, 244–258. [Google Scholar] [CrossRef]
- Brycki, B.E.; Kowalczyk, I.H.; Szulc, A.; Kaczerewska, O.; Pakiet, M. Multifunctional Gemini Surfactants: Structure, Synthesis, Properties and Applications. Appl. Charact. Surfactants 2017. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Qiao, W.; Li, Z. Vesicles from pH-regulated reversible gemini amino-acid surfactants as nanocapsules for delivery. Colloids Surf. B Biointerfaces 2016, 146, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Black, J.W.; Jennings, M.C.; Azarewicz, J.; Paniak, T.J.; Grenier, M.C.; Wuest, W.M.; Minbiole, K.P.C. TMEDA-derived biscationic amphiphiles: An economical preparation of potent antibacterial agents Dedicated to Professor Amos B. Smith, III, in celebration of his 40 years of mentoring scientists. Bioorg. Med. Chem. Lett. 2014, 24, 99–102. [Google Scholar] [CrossRef]
- Pernak, J.; Sobaszkiewicz, K.; Foksowicz-Flaczyk, J. Ionic liquids with symmetrical dialkoxymethyl-substituted imidazolium cations. Chem. A Eur. J. 2004, 10, 3479–3485. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, J.; Ma, J.; Li, M.; Li, X. Physicochemical and antimicrobial activities of cationic gemini surfactants with polyether siloxane linked group. J. Mol. Liq. 2017, 242, 8–15. [Google Scholar] [CrossRef]
- Muslim, A.A.; Ayyash, D.; Gujral, S.S.; Mekhail, G.M.; Rao, P.P.N.; Wettig, S.D. Synthesis and characterization of asymmetrical gemini surfactants. Phys. Chem. Chem. Phys. 2017, 19, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ding, S.; Yu, J.; Chen, X.; Lei, Q.; Fang, W. Antibacterial activity, in vitro cytotoxicity, and cell cycle arrest of gemini quaternary ammonium surfactants. Langmuir 2015, 31, 12161–12169. [Google Scholar] [CrossRef]
- Negm, N.A.; Abd-Elaal, A.A.; Mohamed, D.E.; El-Farargy, A.F.; Mohamed, S. Synthesis and evaluation of silver nanoparticles loaded with Gemini surfactants: Surface and antimicrobial activity. J. Ind. Eng. Chem. 2015, 24, 34–41. [Google Scholar] [CrossRef]
- Rzycki, M.; Drabik, D.; Szostak-Paluch, K.; Hanus-Lorenz, B.; Kraszewski, S. Unraveling the mechanism of octenidine and chlorhexidine on membranes: Does electrostatics matter? Biophys. J. 2021, 120, 3392–3408. [Google Scholar] [CrossRef]
- Van Oosten, B.; Marquardt, D.; Komljenović, I.; Bradshaw, J.P.; Sternin, E.; Harroun, T.A. Small molecule interaction with lipid bilayers: A molecular dynamics study of chlorhexidine. J. Mol. Graph. Model. 2014, 48, 96–104. [Google Scholar] [CrossRef]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular Docking in Modern Drug Discovery: Principles and Recent Applications. In Drug Discovery and Development—New Advances; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Paniak, T.J.; Jennings, M.C.; Shanahan, P.C.; Joyce, M.D.; Santiago, C.N.; Wuest, W.M.; Minbiole, K.P.C. The antimicrobial activity of mono-, bis-, tris-, and tetracationic amphiphiles derived from simple polyamine platforms. Bioorg. Med. Chem. Lett. 2014, 24, 5824–5828. [Google Scholar] [CrossRef] [PubMed]
- Ator, L.E.; Jennings, M.C.; McGettigan, A.R.; Paul, J.J.; Wuest, W.M.; Minbiole, K.P.C. Beyond paraquats: Dialkyl 3,3′- and 3,4′-bipyridinium amphiphiles as antibacterial agents. Bioorg. Med. Chem. Lett. 2014, 24, 3706–3709. [Google Scholar] [CrossRef]
- Mitchell, M.A.; Iannetta, A.A.; Jennings, M.C.; Fletcher, M.H.; Wuest, W.M.; Minbiole, K.P.C. Scaffold-Hopping of Multicationic Amphiphiles Yields Three New Classes of Antimicrobials. ChemBioChem 2015, 16, 2299–2303. [Google Scholar] [CrossRef]
- Forman, M.E.; Jennings, M.C.; Wuest, W.M.; Minbiole, K.P.C. Building a Better Quaternary Ammonium Compound (QAC): Branched Tetracationic Antiseptic Amphiphiles. ChemMedChem 2016, 11, 1401–1405. [Google Scholar] [CrossRef]
- Grenier, M.C.; Davis, R.W.; Wilson-Henjum, K.L.; Ladow, J.E.; Black, J.W.; Caran, K.L.; Seifert, K.; Minbiole, K.P.C. The antibacterial activity of 4,4′-bipyridinium amphiphiles with conventional, bicephalic and gemini architectures. Bioorg. Med. Chem. Lett. 2012, 22, 4055–4058. [Google Scholar] [CrossRef] [PubMed]
- Hemmateenejad, B.; Miri, R.; Akhond, M.; Shamsipur, M. QSAR study of the calcium channel antagonist activity of some recently synthesized dihydropyridine derivatives. An application of genetic algorithm for variable selection in MLR and PLS methods. Chemom. Intell. Lab. Syst. 2002, 64, 91–99. [Google Scholar] [CrossRef]
- Roy, K.; Ambure, P. The “double cross-validation” software tool for MLR QSAR model development. Chemom. Intell. Lab. Syst. 2016, 159, 108–126. [Google Scholar] [CrossRef]
- Alam, S.; Khan, F. QSAR and docking studies on xanthone derivatives for anticancer activity targeting DNA topoisomerase IIα. Drug Des. Devel. Ther. 2014, 8, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.K.; Gupta, S.; Rawal, R.K. Impact of Artificial Neural Networks in QSAR and Computational Modeling. In Artificial Neural Network for Drug Design, Delivery and Disposition; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 153–179. ISBN 9780128015599. [Google Scholar]
- Luo, J.; Hu, J.; Fu, L.; Liu, C.; Jin, X. Use of artificial neural network for a QSAR study on neurotrophic activities of N-p-tolyl/phenylsulfonyl L-amino acid thiolester derivatives. Proc. Proc. Eng. 2011, 15, 5158–5163. [Google Scholar] [CrossRef] [Green Version]
- Montañez-Godínez, N.; Martínez-Olguín, A.C.; Deeb, O.; Garduño-Juárez, R.; Ramírez-Galicia, G. Qsar/qspr as an application of artifi cial neural networks. Methods Mol. Biol. 2015, 1260, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Dahl, G.E.; Jaitly, N.; Salakhutdinov, R. Multi-task Neural Networks for QSAR Predictions. arXiv 2014, arXiv:1406.1231. [Google Scholar]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Duan, J.; Smith, E.; Von Bargen, C.D.; Sherman, W.; Repasky, M.P. AutoQSAR: An automated machine learning tool for best-practice quantitative structure-activity relationship modeling. Future Med. Chem. 2016, 8, 1825–1839. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Methods and Principles in Medicinal Chemistry; Wiley: Hoboken, NJ, USA, 2009; Volume 41, ISBN 9783527318520. [Google Scholar]
- Andrade, C.H.; Pasqualoto, K.F.M.; Ferreira, E.I.; Hopfinger, A.J. 4D-QSAR: Perspectives in Drug Design. Molecules 2010, 15, 3281–3294. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Lau, E. Preformulation Studies; Elsevier Inc.: Amsterdam, The Netherlands, 2001; Volume 3. [Google Scholar]
- Espinosa, J.R.; Wand, C.R.; Vega, C.; Sanz, E.; Frenkel, D. Calculation of the water-octanol partition coefficient of cholesterol for SPC, TIP3P, and TIP4P water. J. Chem. Phys. 2018, 149, 224501. [Google Scholar] [CrossRef]
- Stewart, J.P. MOPAC 2016; Stewart Computational Chemistry: Colorado Springs, CO, USA, 2016. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Stern, H.A.; Feller, S.E. Dielectric permittivity profiles of confined polar fluids. J. Chem. Phys. 2003, 118, 926. [Google Scholar] [CrossRef] [Green Version]
- Weaver, J.C.; Schoenbach, K.H. Biodielectrics. IEEE Trans. Dielectr. Electr. Insul. 2003, 10, 715–716. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Ben-tal, N.; Honig, B.; Peitzsch, R.M.; Denisov, G.; Mclaughlin, S. Binding of Small Basic Peptides to Membranes Containing Acidic Lipids: Theoretical Models and Experimental Results. Biophys. J. 1996, 71, 561–575. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Merz, K.M. Taking into account the ion-induced dipole interaction in the nonbonded model of ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1998, 103, 4613. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1998, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Bonhenry, D.; Tarek, M.; Dehez, F. Effects of Phospholipid Composition on the Transfer of a Small Cationic Peptide Across a Model Biological Membrane. J. Chem. Theory Comput. 2013, 9, 5675–5684. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.; Gumbart, J.C.; Hénin, J.; Lelièvre, T.; Pohorille, A.; Chipot, C. The Adaptive Biasing Force Method: Everything You Always Wanted To Know but Were Afraid To Ask. J. Phys. Chem. B 2014, 119, 1129–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hénin, J.; Fiorin, G.; Chipot, C.; Klein, M.L. Exploring Multidimensional Free Energy Landscapes Using Time-Dependent Biases on Collective Variables. J. Chem. Theory Comput. 2009, 6, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Petrache, H.I. 5.2 Lipid Bilayer Structure. Compr. Biophys. 2012, 5, 3–15. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Ön, A.; Pabst, G.; Zellner, A.; Lohner, K. Octenidine: Novel insights into the detailed killing mechanism of Gram-negative bacteria at a cellular and molecular level. Int. J. Antimicrob. Agents 2020, 56, 106146. [Google Scholar] [CrossRef]
- Van Oosten, B.; Marquardt, D.; Harroun, T.A. Testing High Concentrations of Membrane Active Antibiotic Chlorhexidine Via Computational Titration and Calorimetry. J. Phys. Chem. B 2017, 121, 4657–4668. [Google Scholar] [CrossRef]
- Wilson, M.A.; Pohorille, A. Mechanism of Unassisted Ion Transport across Membrane Bilayers. J. Am. Chem. Soc. 1996, 118, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, K.; Shinoda, W. Comparative molecular dynamics study of ether-and ester-linked phospholipid bilayers. J. Chem. Phys. 2004, 121, 9648. [Google Scholar] [CrossRef] [PubMed]
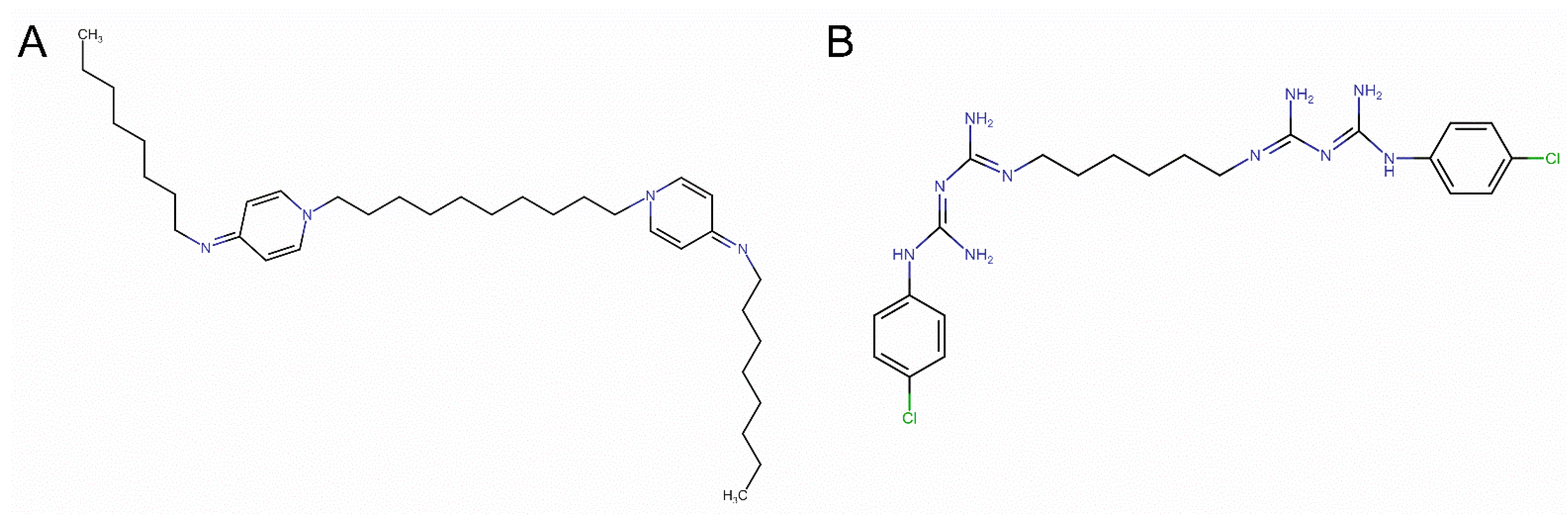


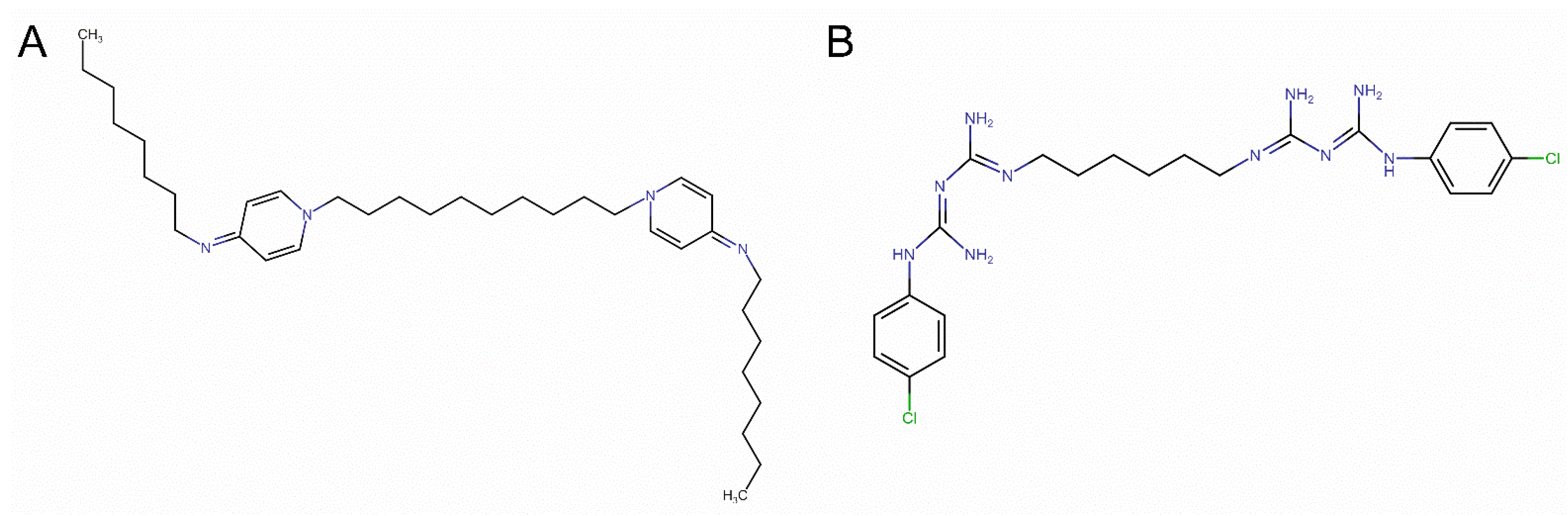{kind=link}
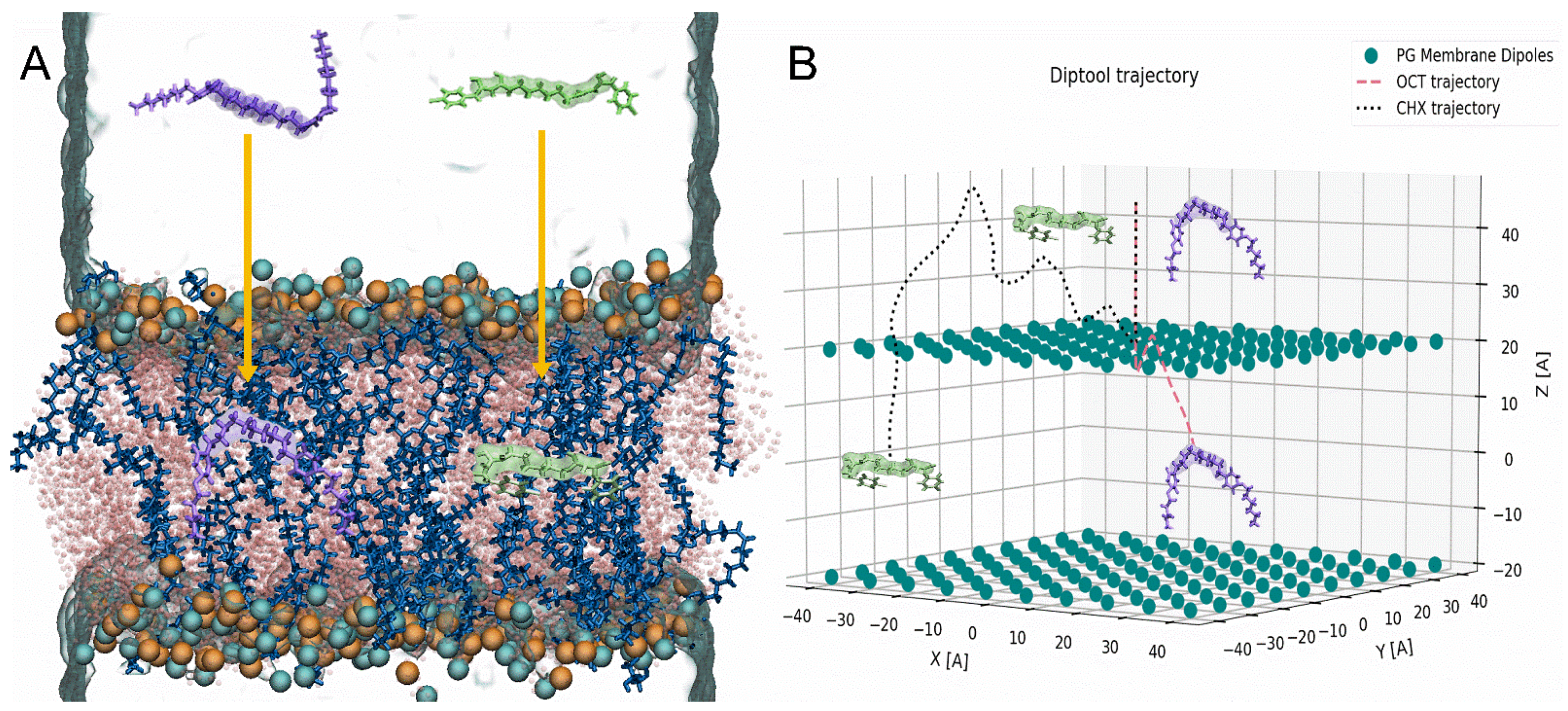
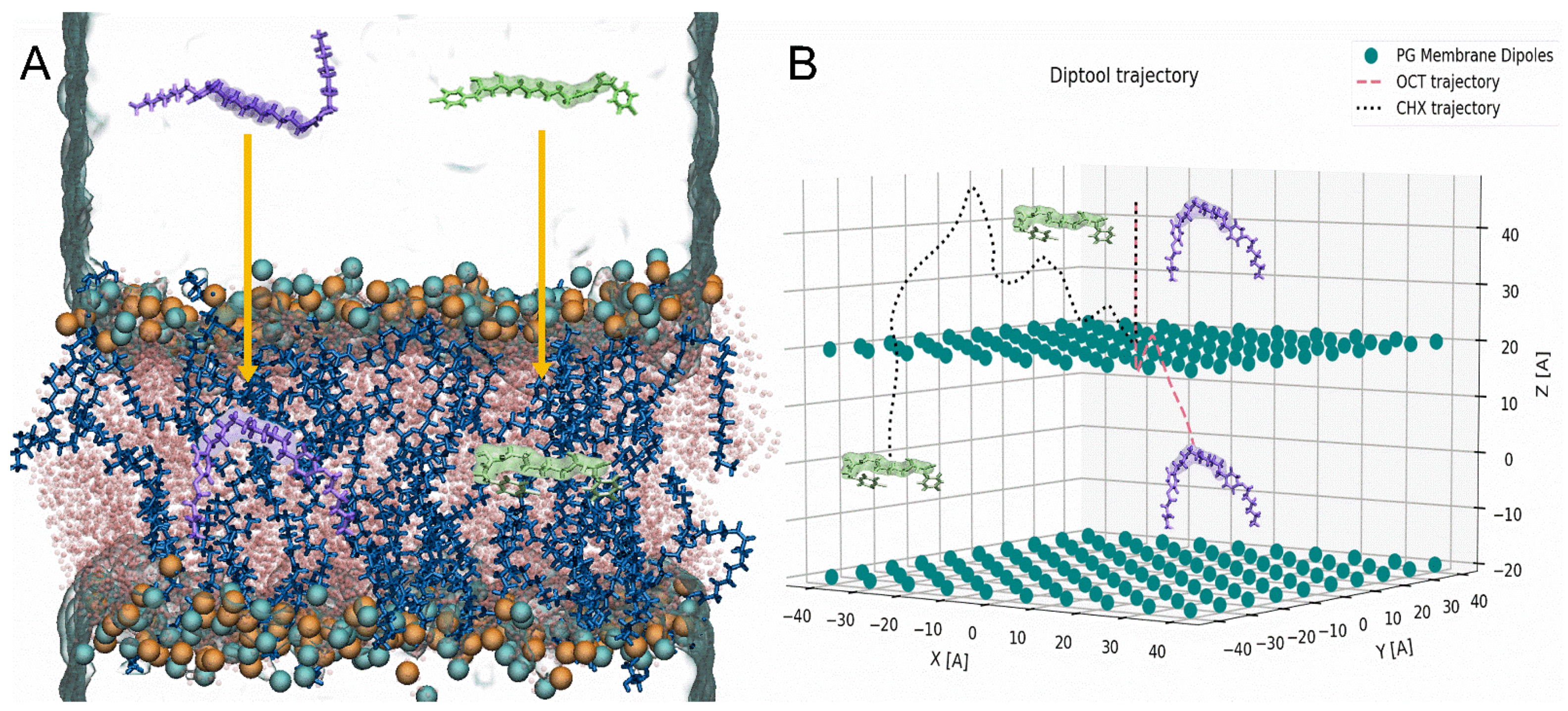{kind=link}
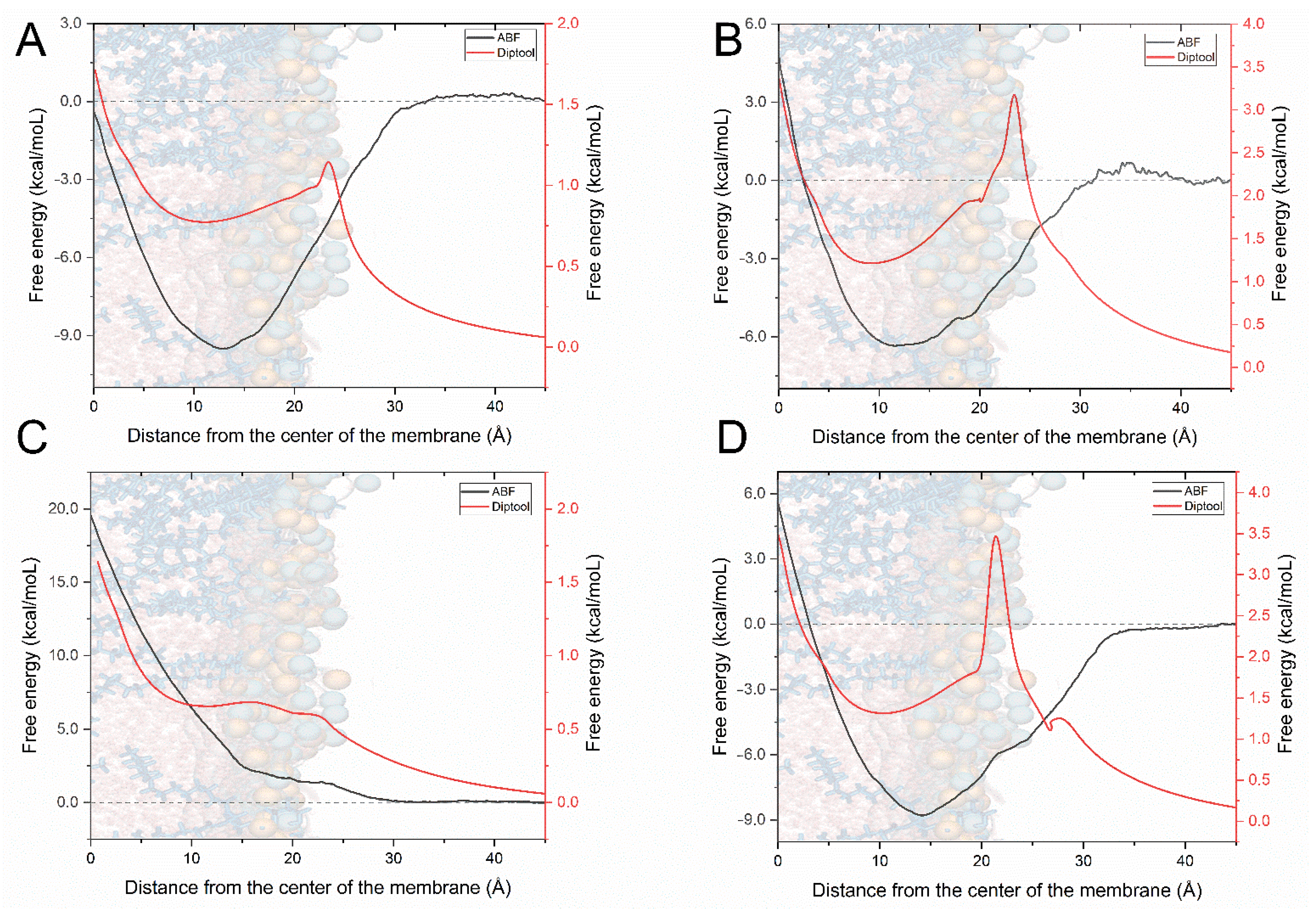{kind=link}
| E. faecalis | E. coli | ||
| Descriptor | Relative Importance | Descriptor | Relative Importance |
| length/width | 0.1920 | length/width | 0.2316 |
| logP | 0.8700 | logP | 0.8331 |
| hydrophobic dipole moment | 0.3655 | hydrophobic dipole moment | 0.3816 |
| Hydrogen count | −0.1418 | double bond count | −0.2713 |
| 1.0/Csp3 bonded to 2 C | 1.0000 | 1.0/Csp3 bonded to 2 C | 1.0000 |
| S. aureus | P. aeruginosa | ||
|---|---|---|---|
| Descriptor | Relative Importance | Descriptor | Relative Importance |
| length/width | 0.3422 | length/width | 0.3644 |
| logP | 0.8358 | logP | 0.8829 |
| hydrophobic dipole moment | 0.4667 | hydrophobic dipole moment | 0.4782 |
| atomic charge weighted positive area-atomic charge weighted negative area | 0.1409 | charge weighted nonpolar area | −0.2206 |
| 1.0/Csp3 bonded to 2 C | 1.0000 | 1.0/Csp3 bonded to 2 C | 1.0000 |
| Dipole Moment in Particular Axis | X (D) | Y (D) | Z (D) | TOTAL (D) | |
|---|---|---|---|---|---|
| Particle type | PC | 0.34 ± 11.29 | −0.37 ± 11.39 | 1.65 ± 8.44 | 18.27 ± 2.32 |
| PG | 0.14 ± 10.17 | 0.29 ± 10.09 | −35.08 ± 8.29 | 39.19 ± 4.69 | |
| OCT | 0.69 ± 7.08 | 2.12 ± 8.14 | −0.51 ± 12.78 | 16.12 ± 4.55 | |
| CHX | 2.74 ± 15.46 | 2.21 ± 10.37 | −0.55 ± 10.25 | 24.73 ± 8.72 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzycki, M.; Kraszewski, S.; Gładysiewicz-Kudrawiec, M. Diptool—A Novel Numerical Tool for Membrane Interactions Analysis, Applying to Antimicrobial Detergents and Drug Delivery Aids. Materials 2021, 14, 6455. https://doi.org/10.3390/ma14216455
Rzycki M, Kraszewski S, Gładysiewicz-Kudrawiec M. Diptool—A Novel Numerical Tool for Membrane Interactions Analysis, Applying to Antimicrobial Detergents and Drug Delivery Aids. Materials. 2021; 14(21):6455. https://doi.org/10.3390/ma14216455
Chicago/Turabian StyleRzycki, Mateusz, Sebastian Kraszewski, and Marta Gładysiewicz-Kudrawiec. 2021. "Diptool—A Novel Numerical Tool for Membrane Interactions Analysis, Applying to Antimicrobial Detergents and Drug Delivery Aids" Materials 14, no. 21: 6455. https://doi.org/10.3390/ma14216455
APA StyleRzycki, M., Kraszewski, S., & Gładysiewicz-Kudrawiec, M. (2021). Diptool—A Novel Numerical Tool for Membrane Interactions Analysis, Applying to Antimicrobial Detergents and Drug Delivery Aids. Materials, 14(21), 6455. https://doi.org/10.3390/ma14216455