
LCAO Electronic Structure of Nucleic Acid Bases and Other Heterocycles and Transfer Integrals in B-DNA, Including Structural Variability



Abstract
1. Introduction
2. Theory
2.1. LCAO with All Valence Orbitals for Nucleic Acid Bases or Similar Molecules
2.2. LCAO with All Valence Orbitals for B-DNA Base Pairs
2.3. Coherent Charge Transfer and Transport Parameters for a TB Wire Model
2.3.1. Eigenstates
2.3.2. Coherent Charge Transfer
2.3.3. Coherent Charge Transport
2.3.4. TB Parameters for a Wire Model Description
2.4. DNA Fragments Generated by MD
3. Results and Discussion
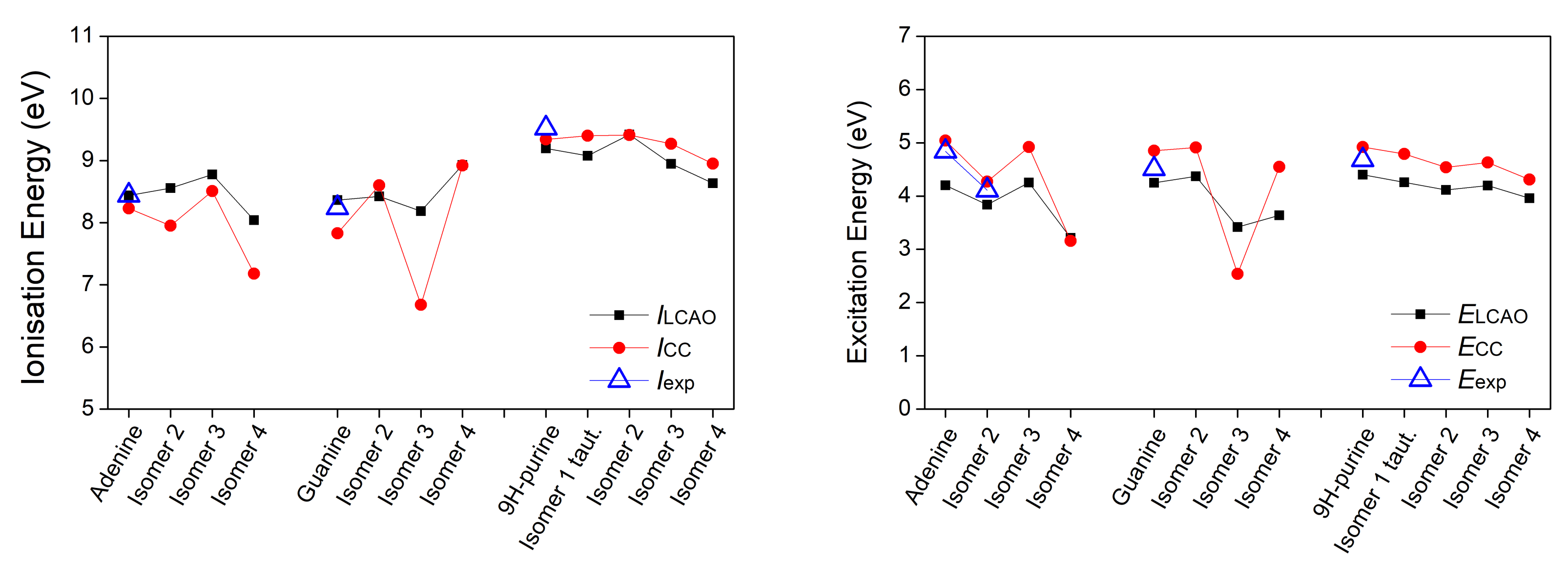
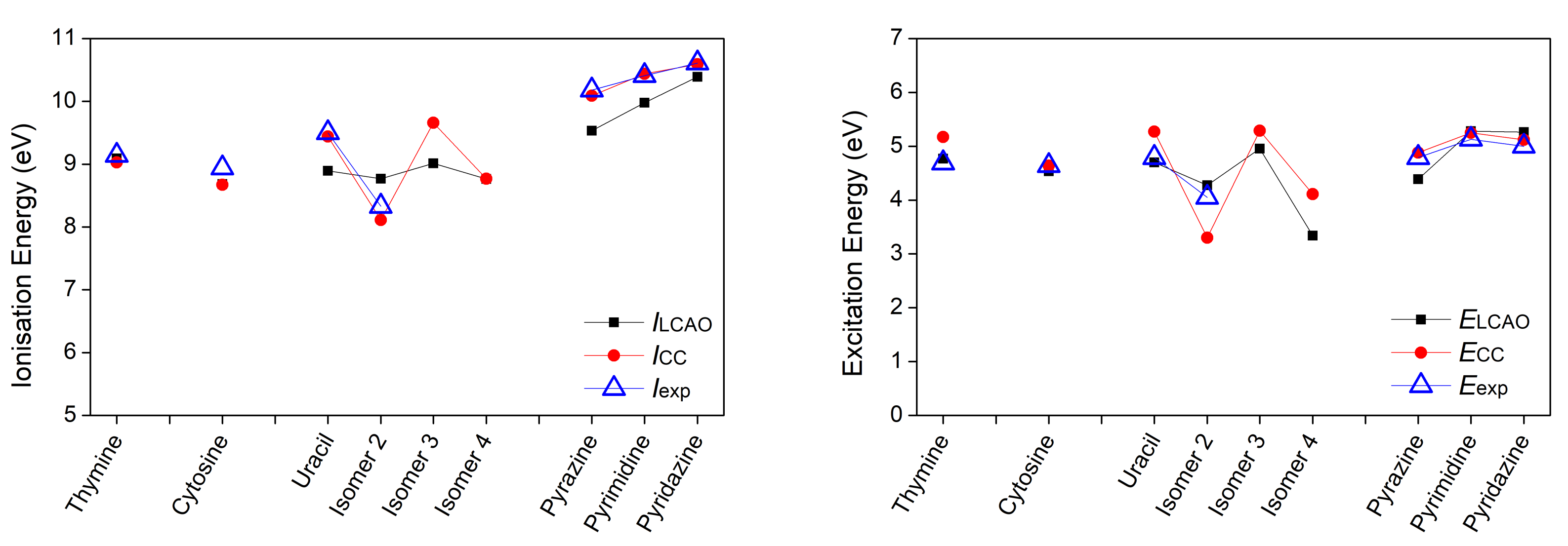
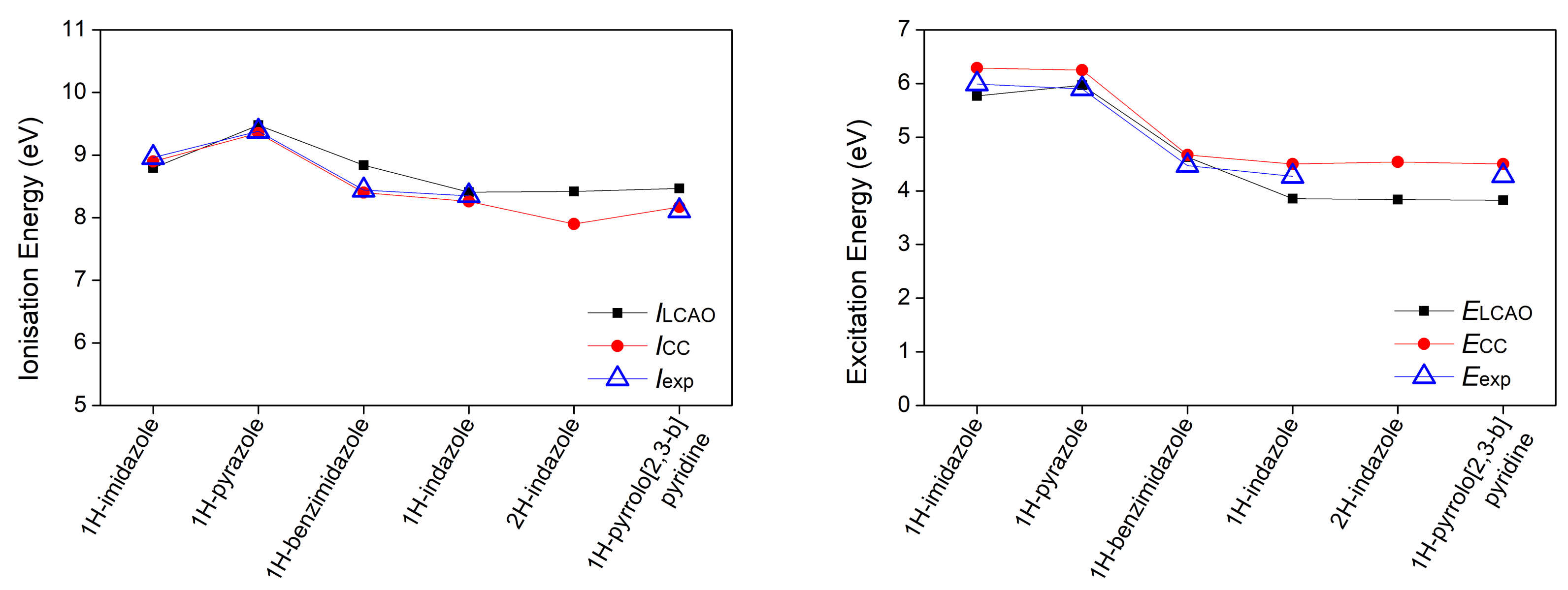
3.1. Heterocyclic Planar Molecules including Nucleic Acid Bases
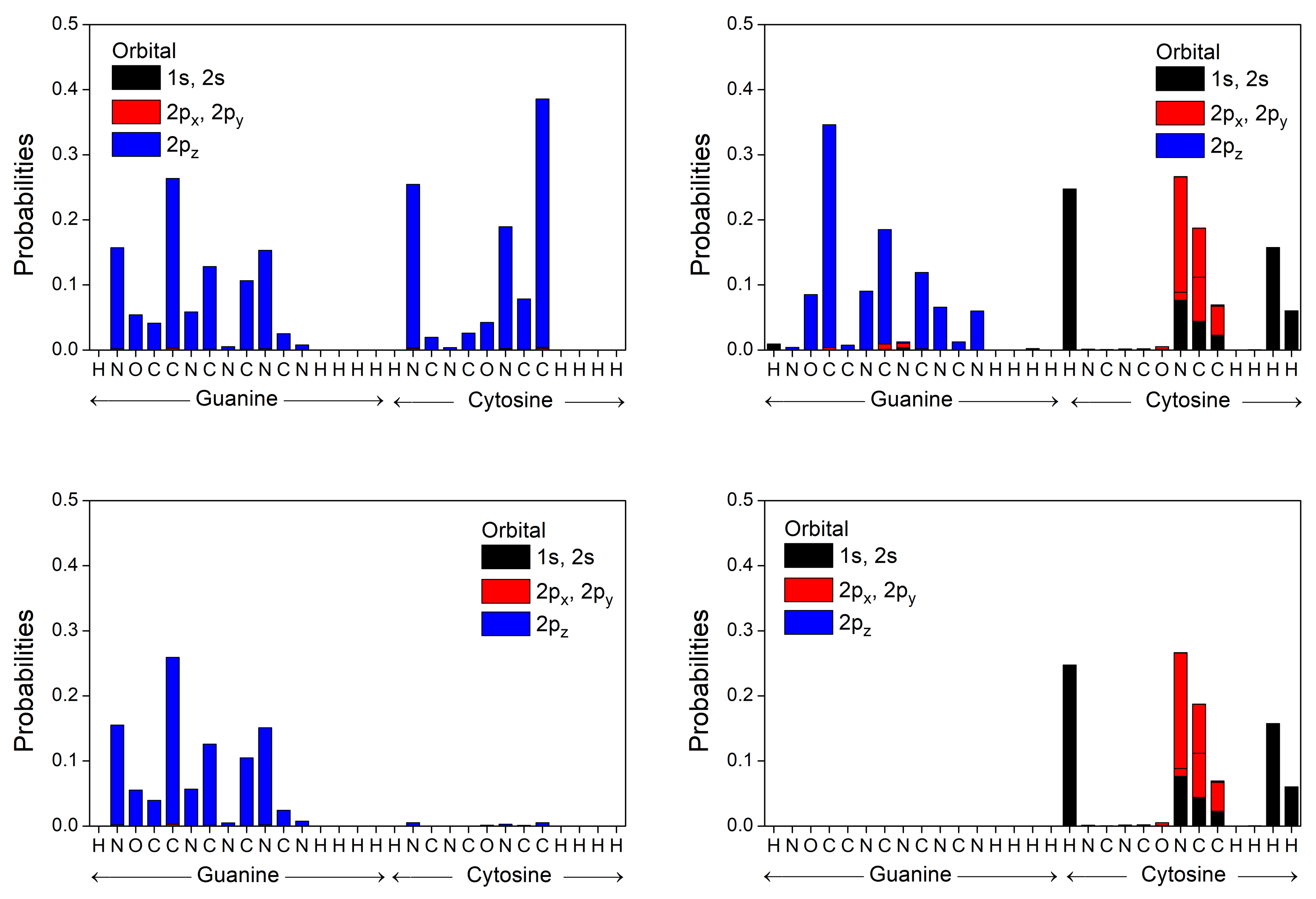
3.2. B-DNA Base Pairs
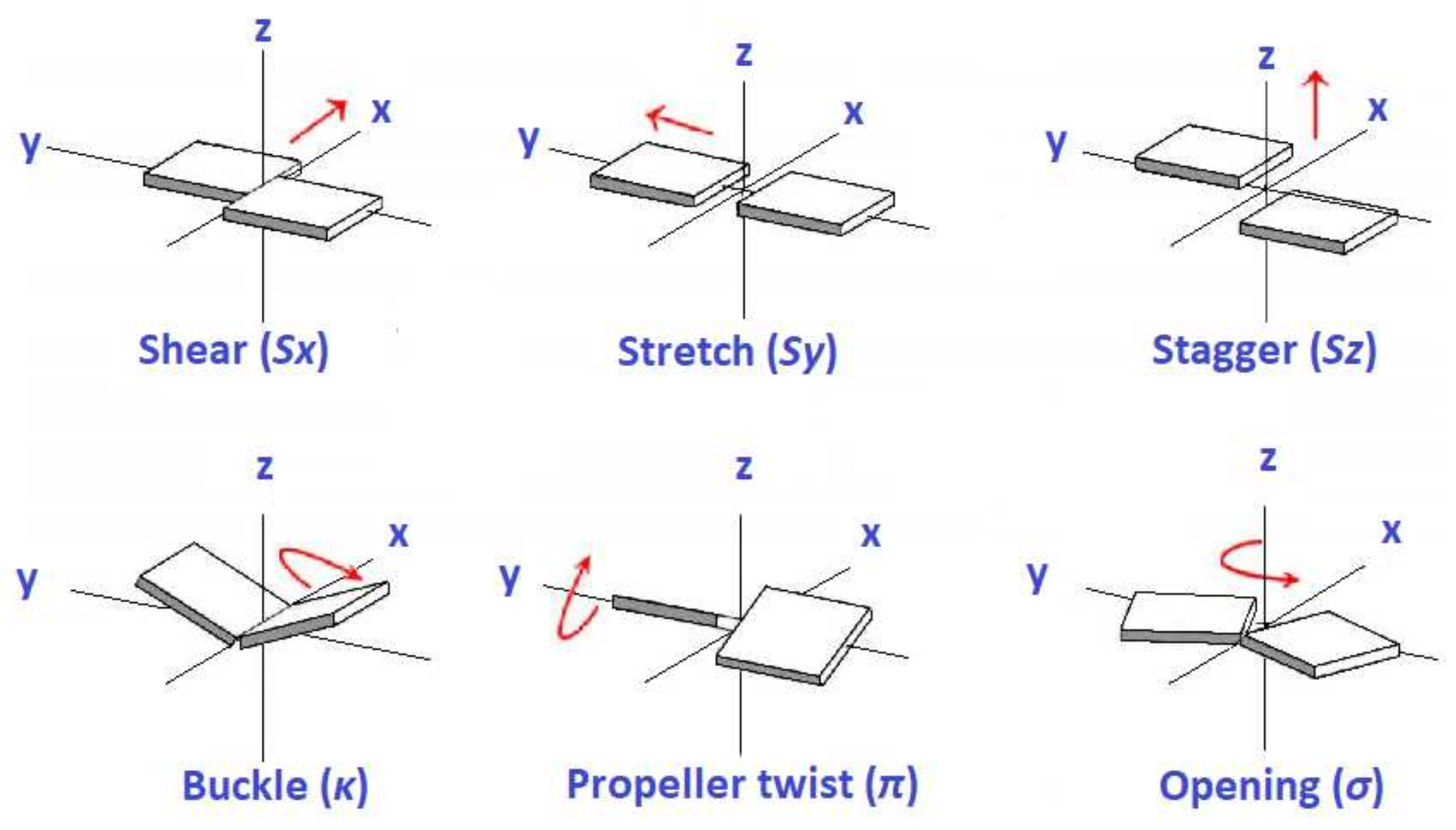
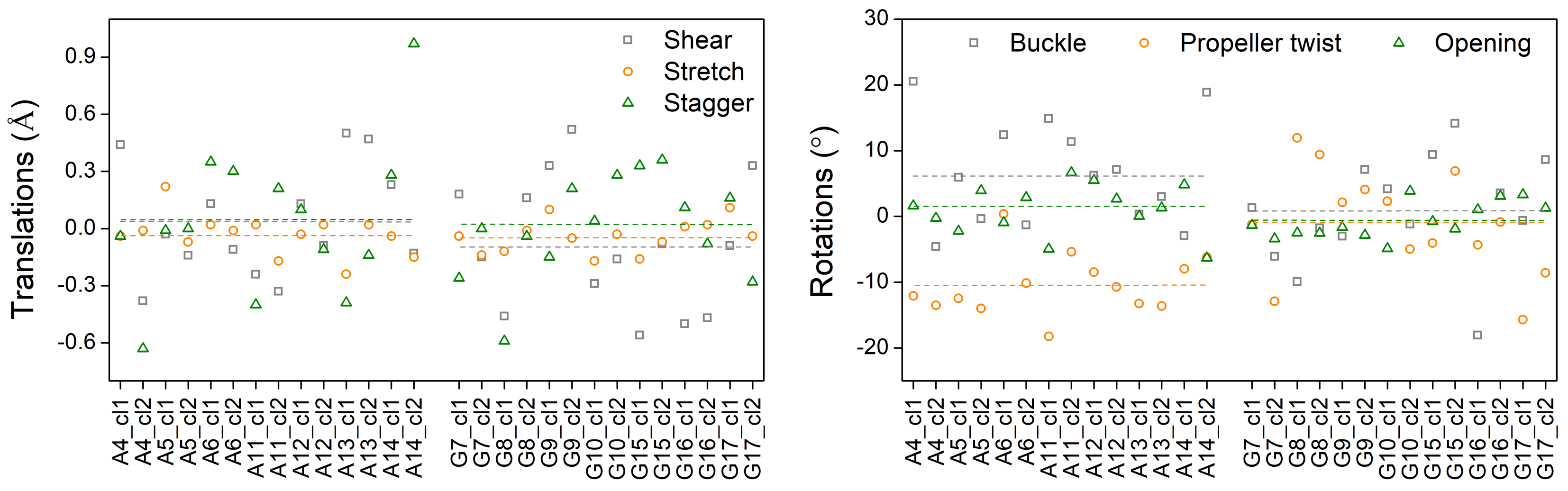
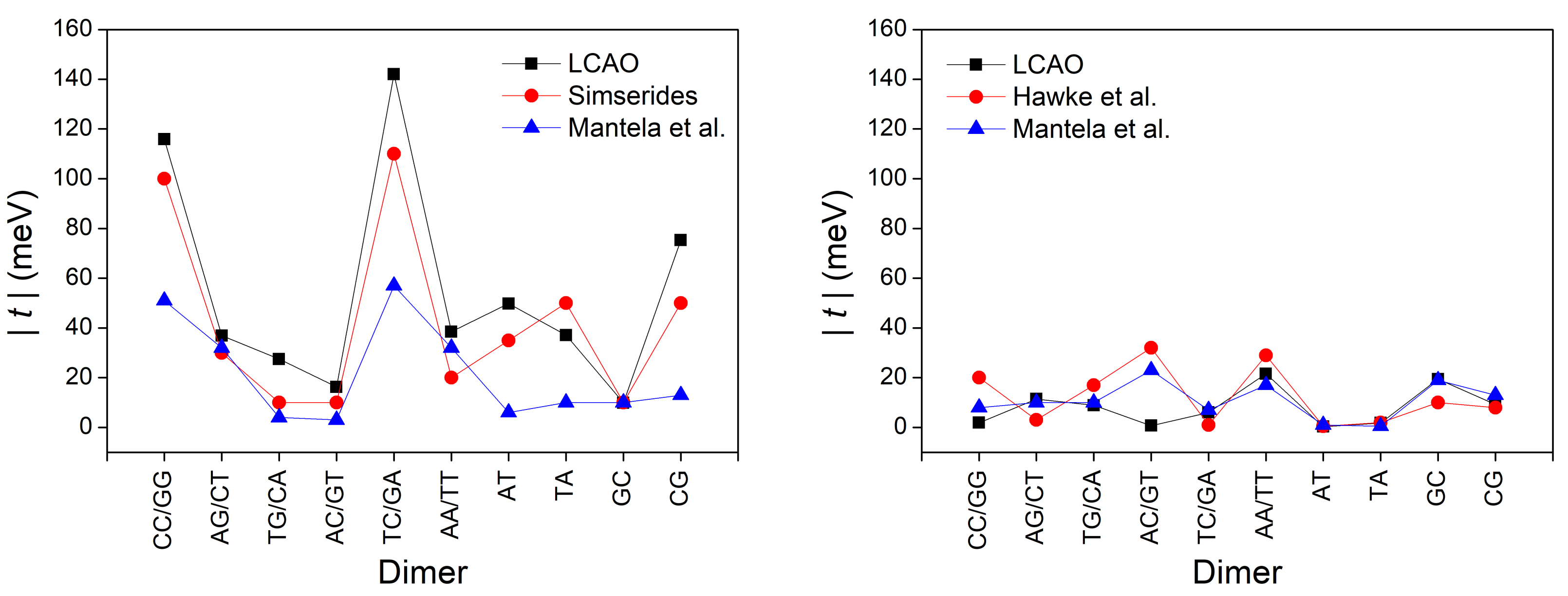
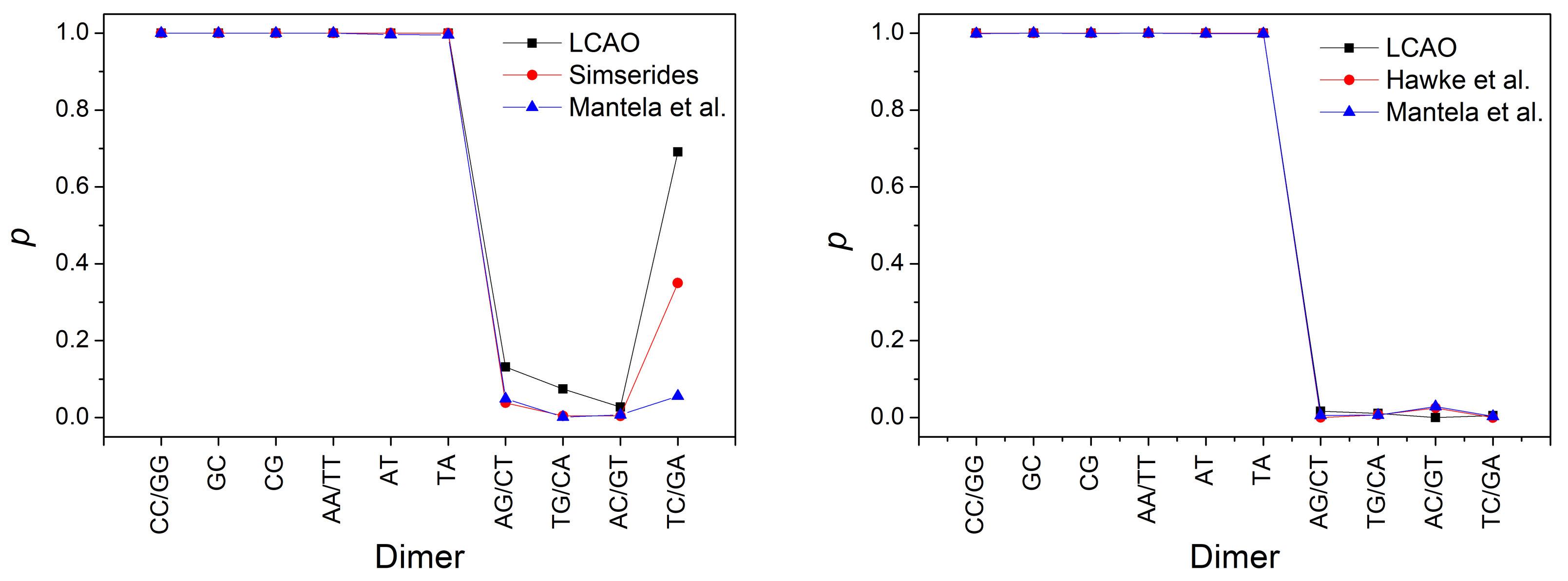
3.3. Effects of Structural Variability
4. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LCAO | Linear Combination of Atomic Orbitals |
| MD | Molecular Dynamics |
| DNA | Deoxyribonucleic Acid |
| TB | Tight Binding |
| IP-EOMCCSD | Ionization Potential Equation of Motion |
| Coupled Cluster with Singles and Doubles | |
| CR-EOMCCSD(T) | Completely Renormalized Equation Of Motion |
| Coupled Cluster with Singles, Doubles, and non-Iterative Triples | |
| RMSPE | Root Mean Square Percentage Error |
| HOMO | Highest Occupied Molecular Orbital |
| LUMO | Lowest Unoccupied Molecular Orbital |
References
- Kawai, K.; Majima, T. Hole Transfer Kinetics of DNA. Acc. Chem. Res. 2013, 46, 2616–2625. [Google Scholar] [CrossRef]
- Dandliker, P.J.; Holmlin, R.E.; Barton, J.K. Oxidative Thymine Dimer Repair in the DNA Helix. Science 1997, 275, 1465–1468. [Google Scholar] [CrossRef]
- Rajski, S.R.; Jackson, B.A.; Barton, J.K. DNA repair: Models for damage and mismatch recognition. Mutat. Res. 2000, 447, 49–72. [Google Scholar] [CrossRef]
- Giese, B. Electron transfer through DNA and peptides. Bioorgan. Med. Chem. 2006, 14, 6139–6143. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.T.; Cheng, Y.Y.; Wells, S.A.; Hsu, C.L.; Römer, R.A. Charge transport in cancer-related genes and early carcinogenesis. Comput. Phys. Commun. 2011, 182, 36–38. [Google Scholar] [CrossRef]
- Páez, C.J.; Schulz, P.A.; Wilson, N.R.; Römer, R.A. Robust signatures in the current–voltage characteristics of DNA molecules oriented between two graphene nanoribbon electrodes. New J. Phys. 2012, 14, 093049. [Google Scholar] [CrossRef]
- Shih, C.T.; Roche, S.; Römer, R.A. Point-Mutation Effects on Charge-Transport Properties of the Tumor-Suppressor Gene p53. Phys. Rev. Lett. 2008, 100, 018105. [Google Scholar] [CrossRef]
- Oliveira, J.I.N.; Albuquerque, E.L.; Fulco, U.L.; Mauriz, P.W.; Sarmento, R.G.; Caetano, E.W.S.; Freire, V.N. Conductance of single microRNAs chains related to the autism spectrum disorder. Europhys. Lett. 2014, 107, 68006. [Google Scholar] [CrossRef]
- Wohlgamuth, C.H.; McWilliams, M.A.; Slinker, J.D. DNA as a Molecular Wire: Distance and Sequence Dependence. Anal. Chem. 2013, 85, 8634–8640. [Google Scholar] [CrossRef]
- Lewis, F.D.; Wasielewski, M.R. Dynamics and efficiency of photoinduced charge transport in DNA: Toward the elusive molecular wire. Pure Appl. Chem. 2013, 85, 1379–1387. [Google Scholar] [CrossRef]
- Seeman, N.C. DNA in a material world. Nat. Nanotechnol. 2003, 421, 427–431. [Google Scholar] [CrossRef]
- Shapir, E.; Cohen, H.; Calzolari, A.; Cavazzoni, C.; Ryndik, D.; Cuniberti, G.; Kotlyar, A.; Di Felice, R.; Porath, D. Electronic structure of single DNA molecules resolved by transverse scanning tunnelling spectroscopy. Nat. Mater. 2008, 7, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Stern, A.; Rotem, D.; Borovok, N.; Eidelshtein, G.; Migliore, A.; Penzo, E.; Wind, S.; Di Felice, R.; Skourtis, S.; et al. Long-range charge transport in single G-quadruplex DNA molecules. Nat. Nanotechnol. 2014, 9, 1040–1046. [Google Scholar] [CrossRef]
- Wang, K. DNA-Based Single-Molecule Electronics: From Concept to Function. J. Funct. Biomater. 2018, 9, 8. [Google Scholar] [CrossRef]
- Mantela, M.; Morphis, A.; Lambropoulos, K.; Simserides, C.; Di Felice, R. Effects of Structural Dynamics on Charge Carrier Transfer in B-DNA: A Combined MD and RT-TDDFT Study. J. Phys. Chem. B 2021, 125, 3986–4003. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C.; Koster, G.F. Simplified LCAO Method for the Periodic Potential Problem. Phys. Rev. 1954, 94, 1498–1524. [Google Scholar] [CrossRef]
- Harrison, W.A. Electronic Structure and the Properties of Solids: The Physics of the Chemical Bond, 2nd ed.; Dover: New York, NY, USA, 1989. [Google Scholar]
- Harrison, W.A. Elementary Electronic Structure; World Scientific: River Edge, NJ, USA, 1999. [Google Scholar]
- Menon, M.; Allen, R.E. Simulations of atomic processes at semiconductor surfaces: General method and chemisorption on GaAs(110). Phys. Rev. B 1988, 38, 6196–6205. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Subbaswamy, K.R. Nonorthogonal tight-binding molecular-dynamics study of silicon clusters. Phys. Rev. B 1993, 47, 12754–12759. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.; Connolly, J.; Lathiotakis, N.; Andriotis, A. Tight-binding molecular-dynamics study of transition-metal clusters. Phys. Rev. B 1994, 50, 8903–8906. [Google Scholar] [CrossRef]
- Lambropoulos, K.; Simserides, C. Periodic, quasiperiodic, fractal, Kolakoski, and random binary polymers: Energy structure and carrier transport. Phys. Rev. E 2019, 99, 032415. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, R. Definitions and nomenclature of nucleic acid structure components. Nucleic Acids Res. 1989, 17, 1797–1803. [Google Scholar] [CrossRef]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.J.; Neidle, S.; Shakked, Z.; et al. A Standard Reference Frame for the Description of Nucleic Acid Base-pair Geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar] [CrossRef]
- Mazur, J.; Jernigan, R.L. Comparison of Rotation Models for Describing DNA Conformations: Application to Static and Polymorphic Forms. Biophys. J. 1995, 68, 1472–1489. [Google Scholar] [CrossRef][Green Version]
- Ussery, D. DNA Structure: A-, B- and Z-DNA Helix Families. Encycl. Life Sci. 2002, 1–7. [Google Scholar] [CrossRef]
- Mantela, M.; Morphis, A.; Tassi, M.; Simserides, C. Lowest ionisation and excitation energies of biologically important heterocyclic planar molecules. Mol. Phys. 2016, 114, 709–718. [Google Scholar] [CrossRef]
- Hilborn, R.C. Einstein coefficients, cross sections, f values, dipole moments, and all that. Am. J. Phys. 1982, 50, 982–986. [Google Scholar] [CrossRef]
- Hush, N.; Cheung, A.S. Ionization potentials and donor properties of nucleic acid bases and related compounds. Chem. Phys. Lett. 1975, 34, 11–13. [Google Scholar] [CrossRef]
- Clark, L.B.; Peschel, G.G.; Tinoco, I. Vapor Spectra and Heats of Vaporization of Some Purine and Pyrimidine Bases. J. Phys. Chem. 1965, 69, 3615–3618. [Google Scholar] [CrossRef]
- Voet, D.; Gratzer, W.B.; Cox, R.A.; Doty, P. Absorption spectra of nucleotides, polynucleotides, and nucleic acids in the far ultraviolet. Biopolymers 1963, 1, 193–208. [Google Scholar] [CrossRef]
- Santhosh, C.; Mishra, P. Electronic spectra of 2-aminopurine and 2,6-diaminopurine: Phototautomerism and fluorescence reabsorption. Spectrochim. Acta Part A Mol. Spectrosc. 1991, 47, 1685–1693. [Google Scholar] [CrossRef]
- Clark, L.B.; Tinoco, I. Correlations in the Ultraviolet Spectra of the Purine and Pyrimidine Bases1. J. Am. Chem. Soc. 1965, 87, 11–15. [Google Scholar] [CrossRef]
- Maier, J.P.; Muller, J.F.; Kubota, T. Ionisation Energies and the Electronic Structures of the N-oxides of diazabenzenes. Helv. Chim. Acta 1975, 58, 1634–1640. [Google Scholar] [CrossRef]
- Kubota, T. Electronic Spectra and Electronic Structures of Some Basic Heterocyclic N-Oxides. Bull. Chem. Soc. Jpn. 1962, 35, 946–955. [Google Scholar] [CrossRef]
- Gleiter, R.; Heilbronner, E.; Hornung, V. Photoelectron Spectra of Azabenzenes and Azanaphthalenes: I. Pyridine, diazines, s-triazine and s-tetrazine. Helv. Chim. Acta 1972, 55, 255–274. [Google Scholar] [CrossRef]
- Pisanias, M.N.; Christophorou, L.G.; Carter, J.G.; McCorkle, D.L. Compound-negative-ion resonance states and threshold-electron excitation spectra of N-heterocyclic molecules: Pyridine, pyridazine, pyrimidine, pyrazine, and sym-triazine. J. Chem. Phys. 1973, 58, 2110–2124. [Google Scholar] [CrossRef]
- Bolovinos, A.; Tsekeris, P.; Philis, J.; Pantos, E.; Andritsopoulos, G. Absolute vacuum ultraviolet absorption spectra of some gaseous azabenzenes. J. Mol. Spectrosc. 1984, 103, 240–256. [Google Scholar] [CrossRef]
- Halverson, F.; Hirt, R.C. Near Ultraviolet Solution Spectra of the Diazines. J. Chem. Phys. 1951, 19, 711–718. [Google Scholar] [CrossRef]
- Ramsey, B.G. Substituent effects on imidazole basicity and photoelectron spectroscopy determined ionization energies. J. Org. Chem. 1979, 44, 2093–2097. [Google Scholar] [CrossRef]
- Caswell, D.S.; Spiro, T.G. Ultraviolet resonance Raman spectroscopy of imidazole, histidine, and Cu(imidazole)42+: Implications for protein studies. J. Am. Chem. Soc. 1986, 108, 6470–6477. [Google Scholar] [CrossRef]
- Lichtenberger, D.L.; Copenhaver, A.S. Ionization band profile analysis in valence photoelectron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 1990, 50, 335–352. [Google Scholar] [CrossRef]
- Noyce, D.S.; Ryder, E.; Walker, B.H. The ultraviolet absorption spectra of substituted pyrazoles. J. Org. Chem. 1955, 20, 1681–1686. [Google Scholar] [CrossRef]
- Gordon, R.D.; Yang, R.F. Vapor absorption spectra of benzoxazole, benzimidazole, and benzothiazole near 2850 Å. Can. J. Chem. 1970, 48, 1722–1729. [Google Scholar] [CrossRef]
- Kovać, B.; Klasinc, L.; Stanovnik, B.; Tišler, M. Photoelectron spectroscopy of heterocycles. Azaindenes and azaindolizines. J. Heterocycl. Chem. 1980, 17, 689–694. [Google Scholar] [CrossRef]
- Cané, E.; Trombetti, A.; Velino, B.; Caminati, W. Assignment of the 290-nm electronic band system of indazole [1,2-benzodiazole] as π*-π by rotational band contour analysis. J. Mol. Spectrosc. 1992, 155, 307–314. [Google Scholar] [CrossRef]
- Fuke, K.; Yoshiuchi, H.; Kaya, K.; Achiba, Y.; Sato, K.; Kimura, K. Multiphoton ionization photoelectron spectroscopy and two-color multiphoton ionization threshold spectroscopy on the hydrogen bonded phenol and 7-azaindole in a supersonic jet. Chem. Phys. Lett. 1984, 108, 179–184. [Google Scholar] [CrossRef]
- Ilich, P. 7-Azaindole: The low-temperature near-UV/vis spectra and electronic structure. J. Mol. Struct. 1995, 354, 37–47. [Google Scholar] [CrossRef]
- Pluhařová, E.; Slavíček, P.; Jungwirth, P. Modeling Photoionization of Aqueous DNA and Its Components. Acc. Chem. Res. 2015, 48, 1209–1217. [Google Scholar] [CrossRef]
- Hawke, L.G.D.; Kalosakas, G.; Simserides, C. Electronic parameters for charge transfer along DNA. Eur. Phys. J. E 2010, 32, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Varsano, D.; Di Felice, R.; Marques, M.; Rubio, A. A TDDFT Study of the Excited States of DNA Bases and Their Assemblies. J. Phys. Chem. B 2006, 110, 7129–7138. [Google Scholar] [CrossRef]
- Mallajosyula, S.S.; Datta, A.; Pati, S.K. Structure and electronic properties of the Watson–Crick base pairs: Role of hydrogen bonding. Synth. Met. 2005, 155, 398–401. [Google Scholar] [CrossRef]
- Simserides, C. A systematic study of electron or hole transfer along DNA dimers, trimers and polymers. Chem. Phys. 2014, 440, 31–41. [Google Scholar] [CrossRef]
- Peluso, A.; Caruso, T.; Landi, A.; Capobianco, A. The Dynamics of Hole Transfer in DNA. Molecules 2019, 24, 4044. [Google Scholar] [CrossRef]
- Endres, R.G.; Cox, D.; Singh, R.R.P. Colloquium: The quest for high-conductance DNA. Rev. Mod. Phys. 2004, 76, 195–214. [Google Scholar] [CrossRef]
- Voityuk, A.A.; Jortner, J.; Bixon, M.; Rösch, N. Electronic coupling between Watson–Crick pairs for hole transfer and transport in desoxyribonucleic acid. J. Chem. Phys. 2001, 114, 5614–5620. [Google Scholar] [CrossRef]
- Migliore, A.; Corni, S.; Varsano, D.; Klein, M.L.; Di Felice, R. First Principles Effective Electronic Couplings for Hole Transfer in Natural and Size-Expanded DNA. J. Phys. Chem. B 2009, 113, 9402–9415. [Google Scholar] [CrossRef]
- Kubař, T.; Woiczikowski, P.B.; Cuniberti, G.; Elstner, M. Efficient Calculation of Charge-Transfer Matrix Elements for Hole Transfer in DNA. J. Phys. Chem. B 2008, 112, 7937–7947. [Google Scholar] [CrossRef]
- Ivanova, A.; Shushkov, P.; Rösch, N. Systematic Study of the Influence of Base-Step Parameters on the Electronic Coupling between Base-Pair Dimers: Comparison of A-DNA and B-DNA Forms. J. Phys. Chem. A 2008, 112, 7106–7114. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| b | ||||
|---|---|---|---|---|
| Parameter | A-T | G-C | [25] | [26] | [27] | [28] |
|---|---|---|---|---|---|---|
| shear (Å) | ||||||
| stretch (Å) | ||||||
| stagger (Å) | ||||||
| buckle () | ||||||
| propeller twist () | ||||||
| opening () |
| Name Formula | |||||||
|---|---|---|---|---|---|---|---|
| Adenine | |||||||
| 8.44 | 4.20 | 0.330 | 8.23 | 5.04 | 8.44 [31] | 4.84 [32,33] | |
| (Isomer 1) | |||||||
| 2-Aminopurine | |||||||
| 8.56 | 3.84 | 0.239 | 7.95 | 4.27 | 4.11 [34] | ||
| (Isomer 2) | |||||||
| 1H-pyrazolo[3,4-d] | |||||||
| pyrimidin-4-amine | |||||||
| 8.78 | 4.25 | 0.328 | 8.51 | 4.92 | |||
| (Isomer 3) | |||||||
| Pyrimido [5,4-e]-as- | |||||||
| triazine, 1,2-dihydro- | |||||||
| 8.04 | 3.21 | 0.282 | 7.18 | 3.16 | |||
| (Isomer 4) | |||||||
| Guanine | 4.77 () | ||||||
| 8.36 | 4.25 | 0.288 | 7.83 | 4.85 | 8.24 [31] | 4.51 [33] | |
| (Isomer 1) | |||||||
| 7-Amino-S-triazolo(1,5-a) | |||||||
| pyrimidin-5(4H)-one | |||||||
| 8.42 | 4.37 | 0.285 | 8.60 | 4.91 | |||
| (Isomer 2) | |||||||
| Pyrimido[5,4-e]-as-triazin- | |||||||
| 5[6h]-one, 1,2-dihydro- | |||||||
| 8.19 | 3.42 | 0.198 | 6.68 | 2.54 | |||
| (Isomer 3) | |||||||
| 7H-imidazo[4,5-d]-v | |||||||
| triazin-4-one, 6-methyl- | 4.47 () | ||||||
| 8.93 | 3.64 | 0.302 | 8.92 | 4.55 | |||
| (Isomer 4) | |||||||
| 9H-purine | 4.49 () | 4.28 [35] () | |||||
| 9.20 | 4.40 | 0.313 | 9.34 | 4.92 | 9.52 [31] | 4.68 [35] | |
| (Isomer 1) | |||||||
| 7H-purine | 9.34 (n) | 4.36 () | |||||
| 9.08 | 4.26 | 0.295 | 9.40 | 4.79 | |||
| (Isomer 1 taut.) | |||||||
| 1H-1,2,3-triazolo | |||||||
| [4,5-b]pyridine | 4.49 | ||||||
| 9.42 | 4.12 | 0.340 | 9.41 | 4.54 | |||
| (Isomer 2) | |||||||
| [1,2,4]Triazolo | |||||||
| [1,5-a]pyrazine | |||||||
| 8.95 | 4.20 | 0.230 | 9.27 | 4.63 | |||
| (Isomer 3) | |||||||
| [1,2,3]Triazolo | |||||||
| [1,5-a]pyrazine | |||||||
| 8.64 | 3.96 | 0.172 | 8.95 | 4.31 | |||
| (Isomer 4) | |||||||
| Formula | |||||||
| Thymine | 5.07 () | ||||||
| 9.09 | 4.77 | 0.316 | 9.03 | 5.17 | 9.14 [31] | 4.69 [33] | |
| Cytosine | |||||||
| 8.68 | 4.54 | 0.306 | 8.67 | 4.64 | 8.94 [31] | 4.64 [33] | |
| Uracil | 5.03 () | ||||||
| 8.89 | 4.70 | 0.286 | 9.44 | 5.27 | 9.50 [31] | 4.79 [33,35] | |
| (Isomer 1) | |||||||
| Pyrazine, 1,4-dioxide | |||||||
| 8.77 | 4.28 | 0.403 | 8.11 | 3.30 | 8.33 [36] | 4.05 [37] | |
| (Isomer 2) | |||||||
| 4(1H)-pyrimidinone, | |||||||
| 6-hydroxy- | |||||||
| 9.01 | 4.95 | 0.103 | 9.66 | 5.29 | |||
| (Isomer 3) | |||||||
| Maleic hydrazide | |||||||
| 8.77 | 3.34 | 0.113 | 8.77 | 4.11 | |||
| (Isomer 4) | |||||||
| Pyrazine | 9.49 (n) | 4.07 () | 9.63 [38] | 4.20 [39] | |||
| 9.53 | 4.39 | 0.258 | 10.09 | 4.88 | 10.18 [38] | 4.79 [40,41] | |
| (Isomer 1) | |||||||
| Pyrimidine | 4.41 () | 4.35 [39] | |||||
| 9.56 (n) | 4.84 () | 9.73 [38] | 4.62 [40] | ||||
| (Isomer 2) | 9.98 | 5.28 | 0.249 | 10.44 | 5.25 | 10.41 [38] | 5.13 [33,35,40,41] |
| Pyridazine | 3.76 () | 3.70 [39] | |||||
| 9.41 (n) | 4.28 | 0.000 () | 9.07 (n) | 4.47 () | 9.31 [38] | ||
| (Isomer 3) | 10.39 | 5.26 | 0.253 | 10.59 | 5.12 | 10.61 [38] | 5.00 [41] |
| 1H-imidazole | 4.97 | 0.000 () | 5.50 () | ||||
| 8.80 | 5.77 | 0.171 | 8.90 | 6.29 | 8.96 [42] | 5.99 [43] | |
| (Isomer 1) | |||||||
| 1H-pyrazole | 5.69 | 0.000 () | |||||
| 9.69 | 5.90 | 0.000 () | 6.11 () | ||||
| (Isomer 2) | 9.48 | 5.97 | 0.196 | 9.35 | 6.25 | 9.38 [44] | 5.90 [45] |
| 1H-benzimidazole | |||||||
| 8.84 | 4.63 | 0.245 | 8.40 | 4.67 | 8.44 [42] | 4.47 [46] | |
| (Isomer 1) | |||||||
| 1H-indazole | |||||||
| 8.41 | 3.85 | 0.217 | 8.26 | 4.50 | 8.35 [47] | 4.27 [48] | |
| (Isomer 2) | |||||||
| 2H-indazole | |||||||
| 8.42 | 3.84 | 0.229 | 7.90 | 4.54 | |||
| (Isomer 2 taut.) | |||||||
| 1H-pyrrolo[2,3-b] | |||||||
| pyridine | |||||||
| 8.47 | 3.82 | 0.184 | 8.17 | 4.50 | 8.11 [49] | 4.28 [50] | |
| (Isomer 3) |
| Base or Base Pair | [52] | [52] | [52] | |||
|---|---|---|---|---|---|---|
| A | ||||||
| T | ||||||
| G | ||||||
| () | () | |||||
| C | ||||||
| A-T | ||||||
| () | () | |||||
| G-C |
| XY | [55] | [29] | [55] | [29] | ||
|---|---|---|---|---|---|---|
| 92 () | ||||||
| GG, CC | 116 | 100 | 51 | 2 | 20 | 8 |
| 11 () | ||||||
| AG, CT | 37 | 30 | 32 | 11 | 3 | 10 |
| 2 () | ||||||
| TG, CA | 28 | 10 | 4 | 9 | 17 | 10 |
| 1 () | ||||||
| AC, GT | 16 | 10 | 3 | 1 | 32 | 23 |
| 3 () | ||||||
| TC, GA | 142 | 110 | 57 | 6 | 1 | 7 |
| AA, TT | 38 | 20 | 32 | 22 | 29 | 17 |
| AT | 50 | 35 | 6 | 1 | 1 | 1 |
| TA | 37 | 50 | 10 | 2 | 2 | 1 |
| 2 () | ||||||
| GC | 10 | 10 | 10 | 19 | 10 | 19 |
| 1 () | ||||||
| CG | 75 | 50 | 13 | 9 | 8 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mantela, M.; Simserides, C.; Di Felice, R. LCAO Electronic Structure of Nucleic Acid Bases and Other Heterocycles and Transfer Integrals in B-DNA, Including Structural Variability. Materials 2021, 14, 4930. https://doi.org/10.3390/ma14174930
Mantela M, Simserides C, Di Felice R. LCAO Electronic Structure of Nucleic Acid Bases and Other Heterocycles and Transfer Integrals in B-DNA, Including Structural Variability. Materials. 2021; 14(17):4930. https://doi.org/10.3390/ma14174930
Chicago/Turabian StyleMantela, Marilena, Constantinos Simserides, and Rosa Di Felice. 2021. "LCAO Electronic Structure of Nucleic Acid Bases and Other Heterocycles and Transfer Integrals in B-DNA, Including Structural Variability" Materials 14, no. 17: 4930. https://doi.org/10.3390/ma14174930
APA StyleMantela, M., Simserides, C., & Di Felice, R. (2021). LCAO Electronic Structure of Nucleic Acid Bases and Other Heterocycles and Transfer Integrals in B-DNA, Including Structural Variability. Materials, 14(17), 4930. https://doi.org/10.3390/ma14174930