
Anisotropic to Isotropic Transition in Monolayer Group-IV Tellurides


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
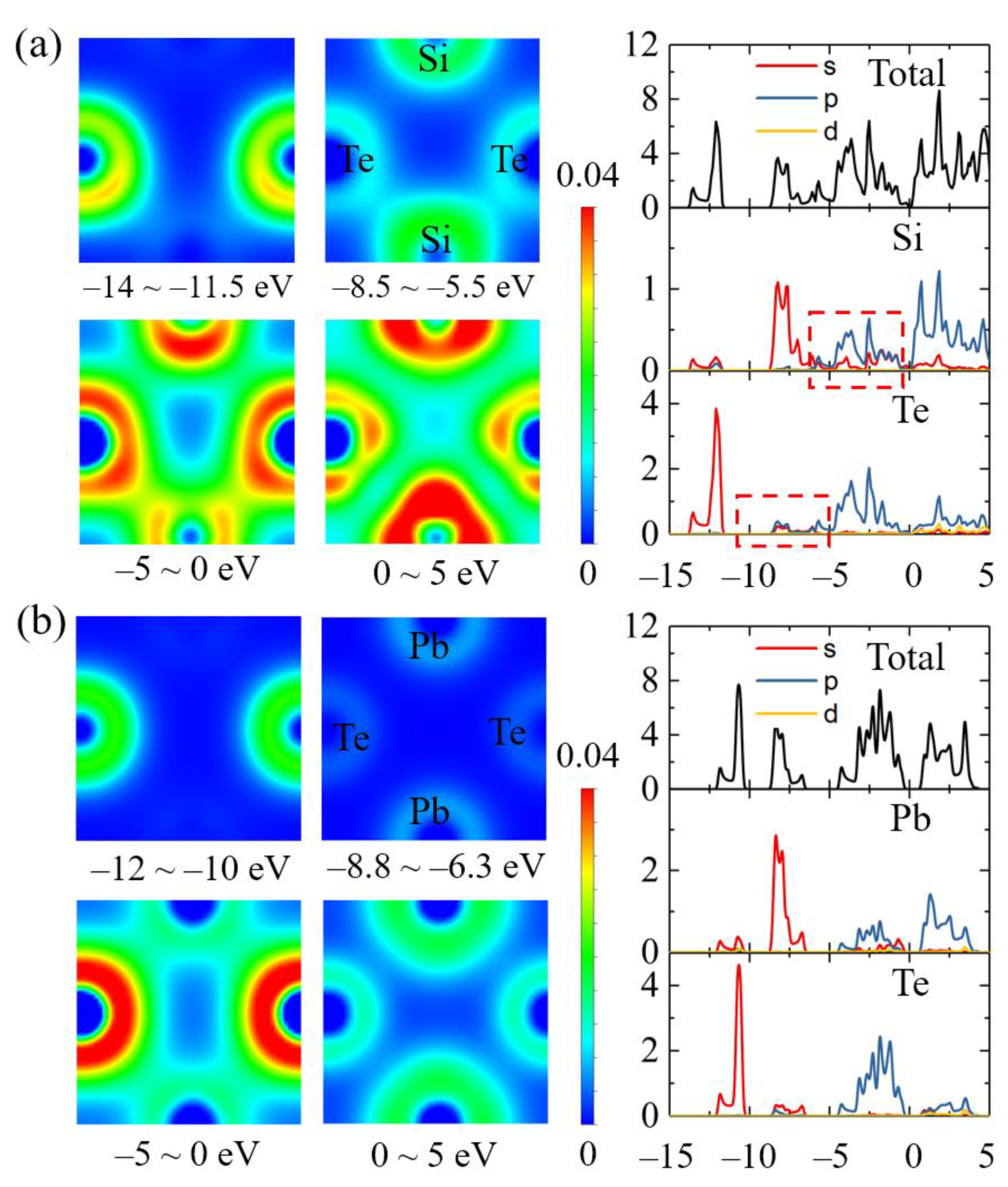
3.1. Geometry Structures
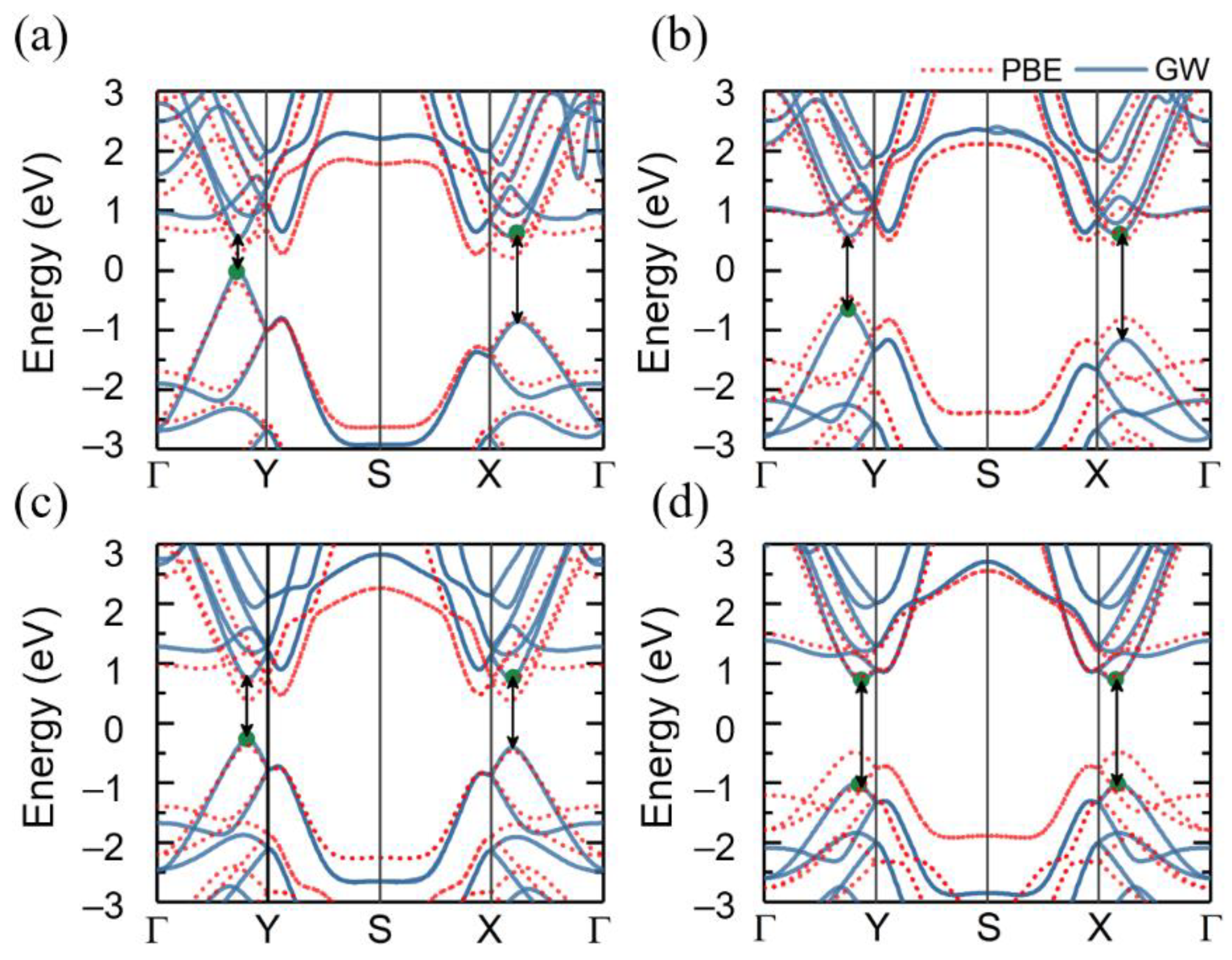
3.2. Quasiparticle Band Structures
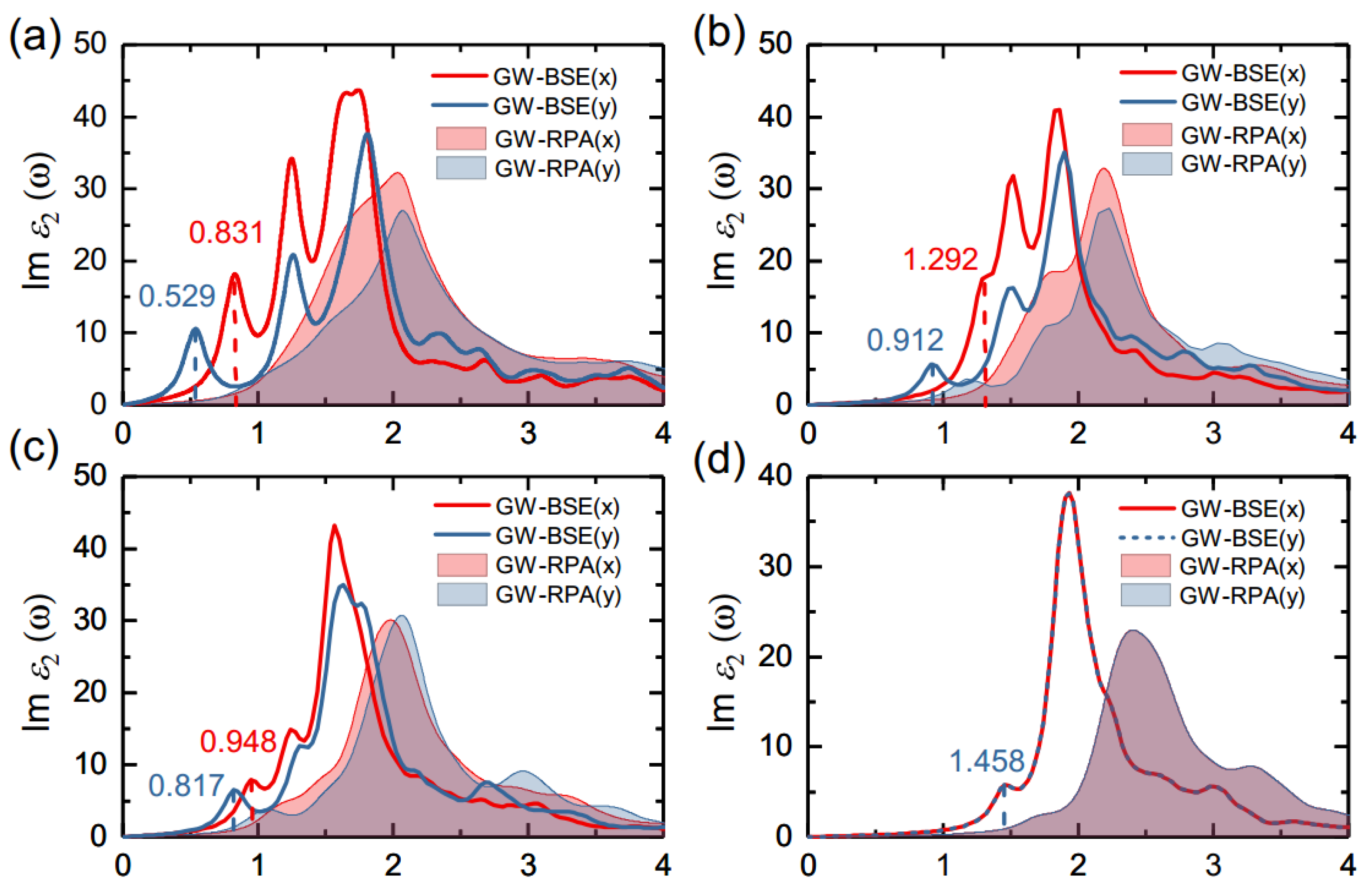
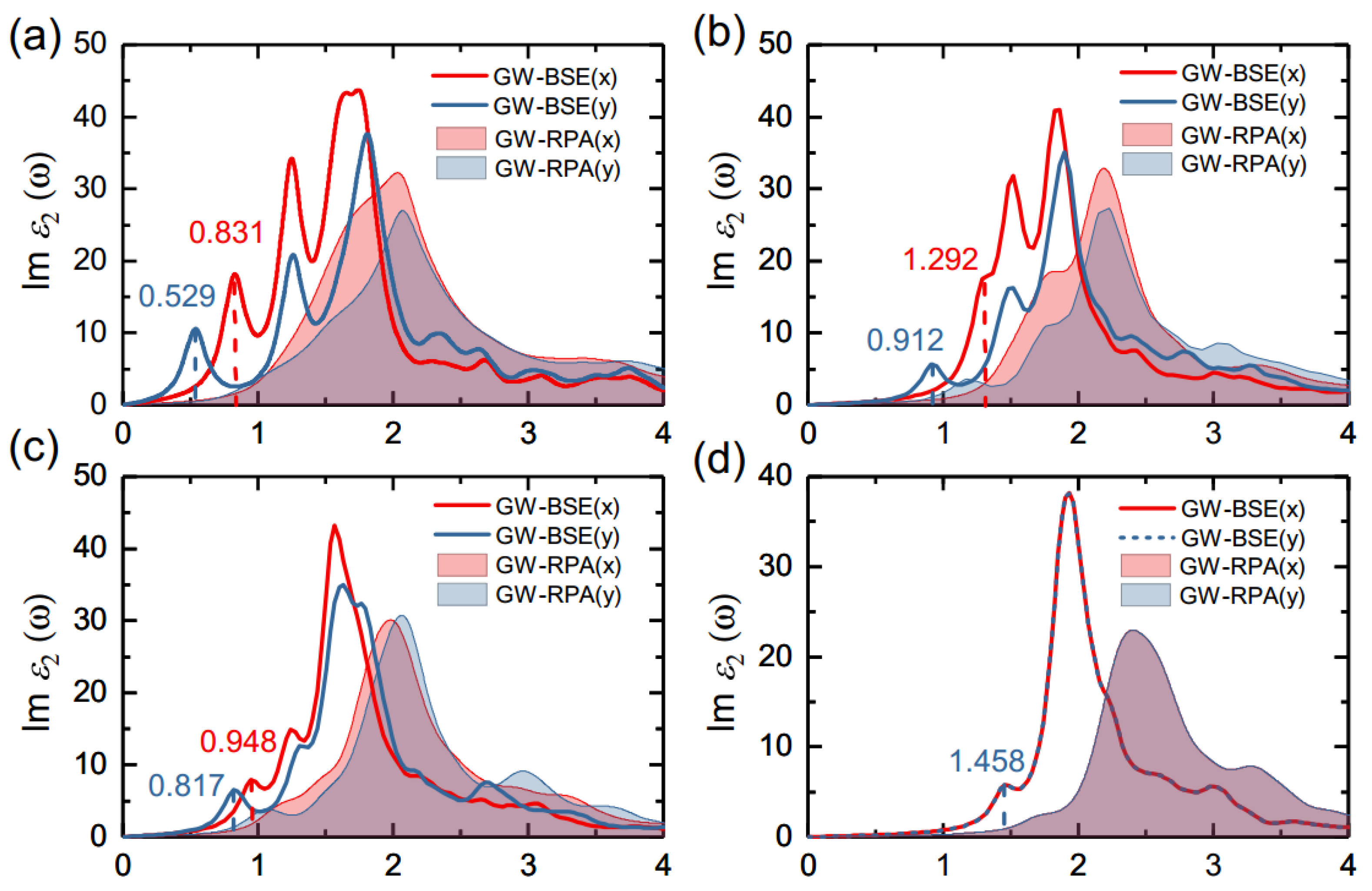
3.3. Optical Properties of Monolayer Group-IV Tellurides
3.4. Anisotropic to Isotropic Transition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pei, Y.; LaLonde, A.; Iwanaga, S.; Snyder, G.J. High Thermoelectric Figure of Merit in Heavy Hole Dominated PbTe. Energy Environ. Sci. 2011, 4, 2085. [Google Scholar] [CrossRef] [Green Version]
- Li, S.P.; Li, J.Q.; Wang, Q.B.; Wang, L.; Liu, F.S.; Ao, W.Q. Synthesis and Thermoelectric Properties of the (GeTe)1-x(PbTe)x Alloys. Solid State Sci. 2011, 13, 399–403. [Google Scholar] [CrossRef]
- Edwards, A.; Pineda, A.; Schultz, P.; Martin, M.; Thompson, A.; Hjalmarson, H.; Umrigar, C. Electronic Structure of Intrinsic Defects in Crystalline Germanium Telluride. Phys. Rev. B 2006, 73, 045210. [Google Scholar] [CrossRef]
- Yin, Z.; Zhu, J.; He, Q.; Cao, X.; Tan, C.; Chen, H.; Yan, Q.; Zhang, H. Graphene-Based Materials for Solar Cell Applications. Adv. Energy Mater. 2014, 4, 1300574. [Google Scholar] [CrossRef]
- Das, S.; Pandey, D.; Thomas, J.; Roy, T. The Role of Graphene and Other 2D Materials in Solar Photovoltaics. Adv. Mater. 2019, 31, 1802722. [Google Scholar] [CrossRef] [Green Version]
- Buscema, M.; Groenendijk, D.J.; Blanter, S.I.; Steele, G.A.; van der Zant, H.S.; Castellanos-Gomez, A. Fast and Broadband Photoresponse of Few-Layer Black Phosphorus Field-Effect Transistors. Nano Lett. 2014, 14, 3347–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voiry, D.; Yang, J.; Chhowalla, M. Recent Strategies for Improving the Catalytic Activity of 2D TMD Nanosheets toward the Hydrogen Evolution Reaction. Adv. Mater. 2016, 28, 6197–6206. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; O’Mullane, A.P.; Du, A. 2D MXenes: A New Family of Promising Catalysts for the Hydrogen Evolution Reaction. ACS Catal. 2017, 7, 494–500. [Google Scholar] [CrossRef]
- Liang, D.; Zhang, Y.-W.; Lu, P.; Yu, Z.G. Strain and Defect Engineered Monolayer Ni-MoS2 for PH-Universal Hydrogen Evolution Catalysis. Nanoscale 2019, 11, 18329–18337. [Google Scholar] [CrossRef]
- Lu, P. Recent Development of Two-dimensional Stanene. J. Sichuan Norm. Univ. Nat. Sci. 2020, 43, 1–20. [Google Scholar] [CrossRef]
- Mohan Kumar, G.; Fu, X.; Ilanchezhiyan, P.; Yuldashev, S.U.; Lee, D.J.; Cho, H.D.; Kang, T.W. Highly Sensitive Flexible Photodetectors Based on Self-Assembled Tin Monosulfide Nanoflakes with Graphene Electrodes. ACS Appl. Mater. Interfaces 2017, 9, 32142–32150. [Google Scholar] [CrossRef]
- Xue, D.-J.; Liu, S.-C.; Dai, C.-M.; Chen, S.; He, C.; Zhao, L.; Hu, J.-S.; Wan, L.-J. GeSe Thin-Film Solar Cells Fabricated by Self-Regulated Rapid Thermal Sublimation. J. Am. Chem. Soc. 2017, 139, 958–965. [Google Scholar] [CrossRef]
- Li, X.-B.; Guo, P.; Zhang, Y.-N.; Peng, R.-F.; Zhang, H.; Liu, L.-M. High Carrier Mobility of Few-Layer PbX (X = S, Se, Te). J. Mater. Chem. C 2015, 3, 6284–6290. [Google Scholar] [CrossRef]
- Xue, D.-J.; Tan, J.; Hu, J.-S.; Hu, W.; Guo, Y.-G.; Wan, L.-J. Anisotropic Photoresponse Properties of Single Micrometer-Sized GeSe Nanosheet. Adv. Mater. 2012, 24, 4528–4533. [Google Scholar] [CrossRef] [PubMed]
- Antunez, P.D.; Buckley, J.J.; Brutchey, R.L. Tin and Germanium Monochalcogenide IV–VI Semiconductor Nanocrystals for Use in Solar Cells. Nanoscale 2011, 3, 2399. [Google Scholar] [CrossRef] [PubMed]
- Higashitarumizu, N.; Kawamoto, H.; Ueno, K.; Nagashio, K. Fabrication and Surface Engineering of Two-Dimensional SnS Toward Piezoelectric Nanogenerator Application. MRS Adv. 2018, 3, 2809–2814. [Google Scholar] [CrossRef] [Green Version]
- Mounet, N.; Gibertini, M.; Schwaller, P.; Campi, D.; Merkys, A.; Marrazzo, A.; Sohier, T.; Castelli, I.E.; Cepellotti, A.; Pizzi, G.; et al. Two-Dimensional Materials from High-Throughput Computational Exfoliation of Experimentally Known Compounds. Nat. Nanotechnol. 2018, 13, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, T.J.; Chen, X.; Meng, X.Y.; Zhao, X.B.; He, J. Anisotropic Growth of Cubic PbTe Nanoparticles to Nanosheets: Controlled Synthesis and Growth Mechanisms. Cryst. Growth Des. 2010, 10, 3727–3731. [Google Scholar] [CrossRef]
- Li, L.; Chen, Z.; Hu, Y.; Wang, X.; Zhang, T.; Chen, W.; Wang, Q. Single-Layer Single-Crystalline SnSe Nanosheets. J. Am. Chem. Soc. 2013, 135, 1213–1216. [Google Scholar] [CrossRef]
- Ramasamy, P.; Kwak, D.; Lim, D.-H.; Ra, H.-S.; Lee, J.-S. Solution Synthesis of GeS and GeSe Nanosheets for High-Sensitivity Photodetectors. J. Mater. Chem. C 2016, 4, 479–485. [Google Scholar] [CrossRef]
- Chang, K.; Liu, J.; Lin, H.; Wang, N.; Zhao, K.; Zhang, A.; Jin, F.; Zhong, Y.; Hu, X.; Duan, W.; et al. Discovery of Robust In-Plane Ferroelectricity in Atomic-Thick SnTe. Science 2016, 353, 274–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Zhang, R.; Jia, B.; Wu, L.; Zhou, B.; Fan, L.; Liu, G.; Wang, Y.; Lu, P.; Peng, G.-D. Fluorine Passivation of ODC Defects in Amorphous Germanium Dioxide. J. Non-Cryst. Solids 2020, 550, 120388. [Google Scholar] [CrossRef]
- Yu, Y.; Ji, Y.; Zhang, Z.; Qiao, H.; Huang, Z.; Qi, X.; Liu, Y.; Zhong, J. Photo-Response of Solution-Processed Hybrid Germanium Selenide Nanosheets Based Photoelectrochemical Devices. Ceram. Int. 2021, 47, 17411–17416. [Google Scholar] [CrossRef]
- Li, F.; Chen, H.; Xu, L.; Zhang, F.; Yin, P.; Yang, T.; Shen, T.; Qi, J.; Zhang, Y.; Li, D.; et al. Defect Engineering in Ultrathin SnSe Nanosheets for High-Performance Optoelectronic Applications. ACS Appl. Mater. Interfaces 2021, 13, 33226–33236. [Google Scholar] [CrossRef]
- Boland, J.B.; Tian, R.; Harvey, A.; Vega-Mayoral, V.; Griffin, A.; Horvath, D.V.; Gabbett, C.; Breshears, M.; Pepper, J.; Li, Y.; et al. Liquid Phase Exfoliation of GeS Nanosheets in Ambient Conditions for Lithium Ion Battery Applications. 2D Mater. 2020, 7, 035015. [Google Scholar] [CrossRef]
- Samal, S.; Molnárová, O.; Průša, F.; Kopeček, J.; Heller, L.; Šittner, P.; Škodová, M.; Abate, L.; Blanco, I. Net-Shape NiTi Shape Memory Alloy by Spark Plasma Sintering Method. Appl. Sci. 2021, 11, 1802. [Google Scholar] [CrossRef]
- Samal, S.; Tyc, O.; Cizek, J.; Klecka, J.; Lukáč, F.; Molnárová, O.; de Prado, E.; Weiss, Z.; Kopeček, J.; Heller, L.; et al. Fabrication of Thermal Plasma Sprayed NiTi Coatings Possessing Functional Properties. Coatings 2021, 11, 610. [Google Scholar] [CrossRef]
- Singh, A.K.; Hennig, R.G. Computational Prediction of Two-Dimensional Group-IV Mono-Chalcogenides. Appl. Phys. Lett. 2014, 105, 042103. [Google Scholar] [CrossRef]
- Wu, M.; Wei, S.-H.; Huang, L. Origin of Polymorphism of the Two-Dimensional Group-IV Monochalcogenides. Phys. Rev. B 2017, 96, 205411. [Google Scholar] [CrossRef]
- Jain, A.; McGaughey, A.J.H. Strongly Anisotropic In-Plane Thermal Transport in Single-Layer Black Phosphorene. Sci. Rep. 2015, 5, 8501. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Yang, J.; Zhu, Y.; Yan, H.; Pei, J.; Myint, Y.W.; Zhang, S.; Lu, Y. Layer-Dependent Surface Potential of Phosphorene and Anisotropic/Layer-Dependent Charge Transfer in Phosphorene–Gold Hybrid Systems. Nanoscale 2015, 8, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Ke, Q.; Zhang, G.; Feng, Y.P.; Shenoy, V.B.; Zhang, Y.-W. Giant Phononic Anisotropy and Unusual Anharmonicity of Phosphorene: Interlayer Coupling Strain Engineering. Adv. Funct. Mater. 2015, 25, 2230–2236. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.Y.; Lu, W.J.; Shao, D.F.; Sun, Y.P. Enhanced Thermoelectric Performance of Phosphorene by Strain-Induced Band Convergence. Phys. Rev. B 2014, 90, 085433. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, M.N.; Berman, O.L.; Kezerashvili, R.Y. Optical Properties of Anisotropic Excitons in Phosphorene. Phys. Rev. B 2019, 100, 155433. [Google Scholar] [CrossRef] [Green Version]
- Shafique, A.; Shin, Y.-H. Thermoelectric and Phonon Transport Properties of Two-Dimensional IV–VI Compounds. Sci. Rep. 2017, 7, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medrano Sandonas, L.; Teich, D.; Gutierrez, R.; Lorenz, T.; Pecchia, A.; Seifert, G.; Cuniberti, G. Anisotropic Thermoelectric Response in Two-Dimensional Puckered Structures. J. Phys. Chem. C 2016, 120, 18841–18849. [Google Scholar] [CrossRef]
- Huang, L.; Wu, F.; Li, J. Structural Anisotropy Results in Strain-Tunable Electronic and Optical Properties in Monolayer GeX and SnX (X = S, Se, Te). J. Chem. Phys. 2016, 144, 114708. [Google Scholar] [CrossRef]
- Tian, Z.; Guo, C.; Zhao, M.; Li, R.; Xue, J. Two-Dimensional SnS: A Phosphorene Analogue with Strong In-Plane Electronic Anisotropy. ACS Nano 2017, 11, 2219–2226. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Y.; Wu, M.; Zhao, L.-D.; Lin, Z.; Cheng, H.; Wang, Y.; Jiang, C.; Wei, S.-H.; Huang, L.; et al. Highly-Anisotropic Optical and Electrical Properties in Layered SnSe. Nano Res. 2018, 11, 554–564. [Google Scholar] [CrossRef]
- He, W.; Chen, H.; Ouyang, H.; Zhou, J.; Sui, Y.; Zhang, C.; Zheng, X.; Zhang, R.; Yuan, X.; Xu, Z.; et al. Tunable Anisotropic Plasmon Response of Monolayer GeSe Nanoribbon Arrays. Nanoscale 2020, 12, 16762–16769. [Google Scholar] [CrossRef]
- Fei, R.; Kang, W.; Yang, L. Ferroelectricity and Phase Transitions in Monolayer Group-IV Monochalcogenides. Phys. Rev. Lett. 2016, 117, 097601. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zeng, X.C. Intrinsic Ferroelasticity and/or Multiferroicity in Two-Dimensional Phosphorene and Phosphorene Analogues. Nano Lett. 2016, 16, 3236–3241. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qian, X. Two-Dimensional Multiferroics in Monolayer Group IV Monochalcogenides. 2D Mater. 2017, 4, 015042. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Chen, Y. Novel Two-Dimensional Ferroelectric PbTe under Tension: A First-Principles Prediction. J. Appl. Phys. 2017, 122, 064101. [Google Scholar] [CrossRef]
- Wan, W.; Liu, C.; Xiao, W.; Yao, Y. Promising Ferroelectricity in 2D Group IV Tellurides: A First-Principles Study. Appl. Phys. Lett. 2017, 111, 132904. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, M.N.; Ding, T.; Ma, K.; Liu, F.S.; Ao, W.Q.; Li, J.Q. Promising Thermoelectric Properties and Anisotropic Electrical and Thermal Transport of Monolayer SnTe. Appl. Phys. Lett. 2019, 114, 083901. [Google Scholar] [CrossRef]
- Kobayashi, K. Electronic States of SnTe and PbTe (001) Monolayers with Supports. Surf. Sci. 2015, 639, 54–65. [Google Scholar] [CrossRef]
- Zhang, P.; Zhao, F.; Long, P.; Wang, Y.; Yue, Y.; Liu, X.; Feng, Y.; Li, R.; Hu, W.; Li, Y.; et al. Sonication-Assisted Liquid-Phase Exfoliated α-GeTe: A Two-Dimensional Material with High Fe3+ Sensitivity. Nanoscale 2018, 10, 15989–15997. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Rohlfing, M.; Louie, S.G. Electron-Hole Excitations and Optical Spectra from First Principles. Phys. Rev. B 2000, 62, 4927–4944. [Google Scholar] [CrossRef]
- Onida, G.; Reining, L.; Rubio, A. Electronic Excitations: Density-Functional versus Many-Body Green’s-Function Approaches. Rev. Mod. Phys. 2002, 74, 601–659. [Google Scholar] [CrossRef] [Green Version]
- Marzari, N.; Mostofi, A.A.; Yates, J.R.; Souza, I.; Vanderbilt, D. Maximally Localized Wannier Functions: Theory and Applications. Rev. Mod. Phys. 2012, 84, 1419–1475. [Google Scholar] [CrossRef] [Green Version]
- Marzari, N.; Vanderbilt, D. Maximally Localized Generalized Wannier Functions for Composite Energy Bands. Phys. Rev. B 1997, 56, 12847–12865. [Google Scholar] [CrossRef] [Green Version]
- Mostofi, A.A.; Yates, J.R.; Pizzi, G.; Lee, Y.-S.; Souza, I.; Vanderbilt, D.; Marzari, N. An Updated Version of Wannier90: A Tool for Obtaining Maximally-Localised Wannier Functions. Comput. Phys. Commun. 2014, 185, 2309–2310. [Google Scholar] [CrossRef] [Green Version]
- Bohm, D.; Pines, D. A Collective Description of Electron Interactions: III. Coulomb Interactions in a Degenerate Electron Gas. Phys. Rev. 1953, 92, 609–625. [Google Scholar] [CrossRef] [Green Version]
- Ehrenreich, H.; Cohen, M.H. Self-Consistent Field Approach to the Many-Electron Problem. Phys. Rev. 1959, 115, 786–790. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, Q.; Jena, P. SiTe Monolayers: Si-Based Analogues of Phosphorene. J. Mater. Chem. C 2016, 4, 6353–6361. [Google Scholar] [CrossRef]
- Wang, Q.; Quhe, R.; Guan, Z.; Wu, L.; Bi, J.; Guan, P.; Lei, M.; Lu, P. High N-Type and p-Type Thermoelectric Performance of Two-Dimensional SiTe at High Temperature. RSC Adv. 2018, 8, 21280–21287. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Yang, M.; Wang, S.J.; Feng, Y.P. Electronic and Optical Properties of the Monolayer Group-IV Monochalcogenides M X (M = Ge, Sn; X = S, Se, Te). Phys. Rev. B 2017, 95, 235434. [Google Scholar] [CrossRef]
- Kamal, C.; Chakrabarti, A.; Ezawa, M. Direct Band Gaps in Group IV-VI Monolayer Materials: Binary Counterparts of Phosphorene. Phys. Rev. B 2016, 93, 125428. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Singh, A.K. Semiconductor-Metal Transition in Semiconducting Bilayer Sheets of Transition-Metal Dichalcogenides. Phys. Rev. B 2012, 86, 075454. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Tongay, S.; Zhou, J.; Li, J.; Wu, J. Band Offsets and Heterostructures of Two-Dimensional Semiconductors. Appl. Phys. Lett. 2013, 102, 012111. [Google Scholar] [CrossRef] [Green Version]
- Bafekry, A.; Stampfl, C.; Peeters, F.M. The Electronic, Optical, and Thermoelectric Properties of Monolayer PbTe and the Tunability of the Electronic Structure by External Fields and Defects. Phys. Status Solidi B 2020, 257, 2000182. [Google Scholar] [CrossRef]
- Cudazzo, P.; Tokatly, I.V.; Rubio, A. Dielectric Screening in Two-Dimensional Insulators: Implications for Excitonic and Impurity States in Graphane. Phys. Rev. B 2011, 84, 085406. [Google Scholar] [CrossRef] [Green Version]
- Berkelbach, T.C.; Hybertsen, M.S.; Reichman, D.R. Theory of Neutral and Charged Excitons in Monolayer Transition Metal Dichalcogenides. Phys. Rev. B 2013, 88, 045318. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D.; Edgecombe, K.E. A Simple Measure of Electron Localization in Atomic and Molecular Systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a (Å) | b (Å) | b/a | Δd (Å) | |
|---|---|---|---|---|
| Phosphorene | 3.30 | 4.63 | 1.403 | 0.83 |
| SiTe | 4.11 | 4.29 | 1.044 | 0.31 |
| GeTe | 4.23 | 4.39 | 1.038 | 0.28 |
| SnTe | 4.55 | 4.59 | 1.009 | 0.15 |
| PbTe | 4.64 | 4.64 | 1 | 0 |
| me (m0) | mh (m0) | EgPBE (eV) | EgGW (eV) | Egopt (eV) | Eb (eV) | Δρ (e−) | Ref | |
|---|---|---|---|---|---|---|---|---|
| Direction | x/y | x/y | x/y | x/y | x/y | x/y | - | |
| SiTe | 0.24/0.09 | 0.21/0.09 | 0.99/0.47 | 1.43/0.56 | 0.83/0.53 | 0.60/0.03 | 0.37 | |
| GeTe | 0.24/0.14 | 0.28/0.13 | 1.24/0.92 | 1.76/1.24 | 1.29/0.91 | 0.47/0.31 | 0.39 | |
| - | - | - | 1.68/1.23 | 1.34/1.00 | 0.34/0.23 | - | ref [67] | |
| SnTe | 0.11/0.10 | 0.13/0.11 | 0.84/0.75 | 1.16/1.04 | 0.95/0.82 | 0.22/0.21 | 0.63 | |
| - | - | - | 1.04/1.02 | 0.85/0.83 | 0.19/0.19 | - | ref [67] | |
| PbTe | 0.20 | 0.18 | 1.26 | 1.74 | 1.46 | 0.28 | 0.66 | |
| - | - | 1.3 | - | - | - | - | ref [71] | |
| 0.169 | 0.190 | 1.26 | - | - | - | - | ref [13] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Wu, L.; Urban, A.; Cao, H.; Lu, P. Anisotropic to Isotropic Transition in Monolayer Group-IV Tellurides. Materials 2021, 14, 4495. https://doi.org/10.3390/ma14164495
Wang Q, Wu L, Urban A, Cao H, Lu P. Anisotropic to Isotropic Transition in Monolayer Group-IV Tellurides. Materials. 2021; 14(16):4495. https://doi.org/10.3390/ma14164495
Chicago/Turabian StyleWang, Qian, Liyuan Wu, Alexander Urban, Huawei Cao, and Pengfei Lu. 2021. "Anisotropic to Isotropic Transition in Monolayer Group-IV Tellurides" Materials 14, no. 16: 4495. https://doi.org/10.3390/ma14164495
APA StyleWang, Q., Wu, L., Urban, A., Cao, H., & Lu, P. (2021). Anisotropic to Isotropic Transition in Monolayer Group-IV Tellurides. Materials, 14(16), 4495. https://doi.org/10.3390/ma14164495