
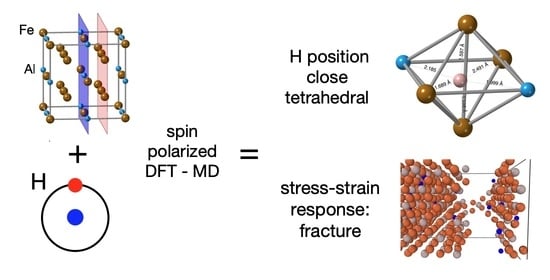
The Effect of Hydrogen on the Stress-Strain Response in Fe3Al: An ab initio Molecular-Dynamics Study




Abstract
:
1. Introduction
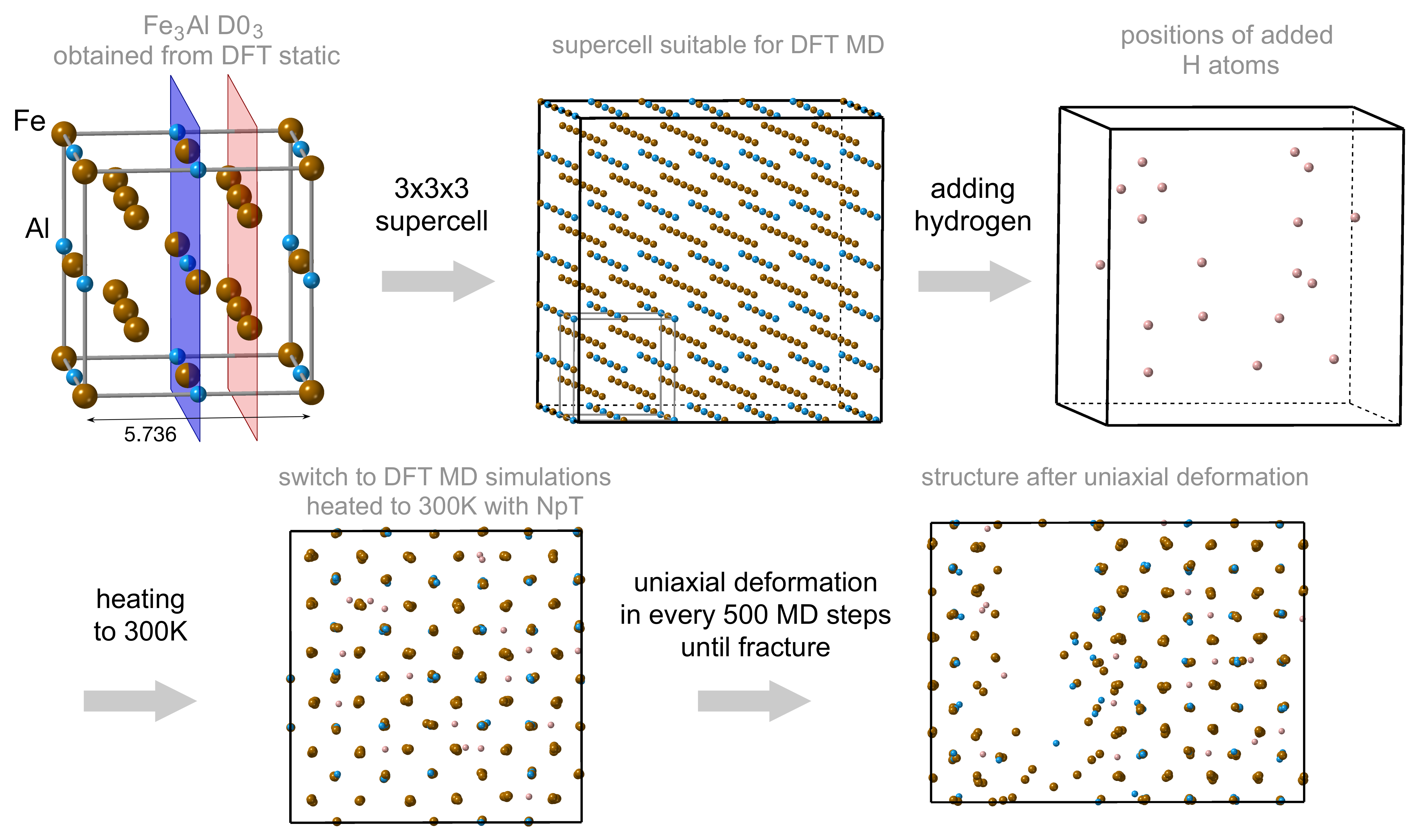
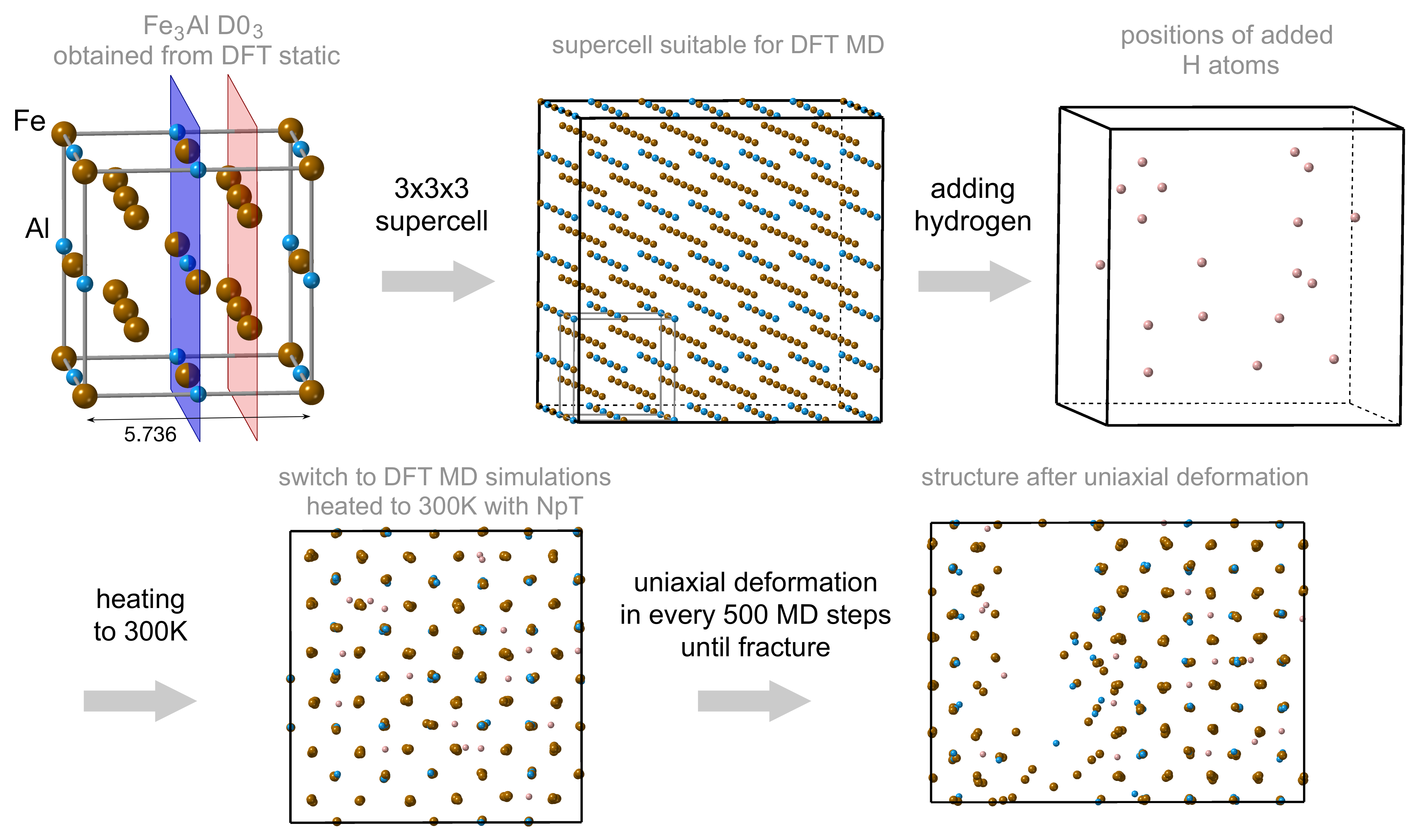
2. Methods
3. Results
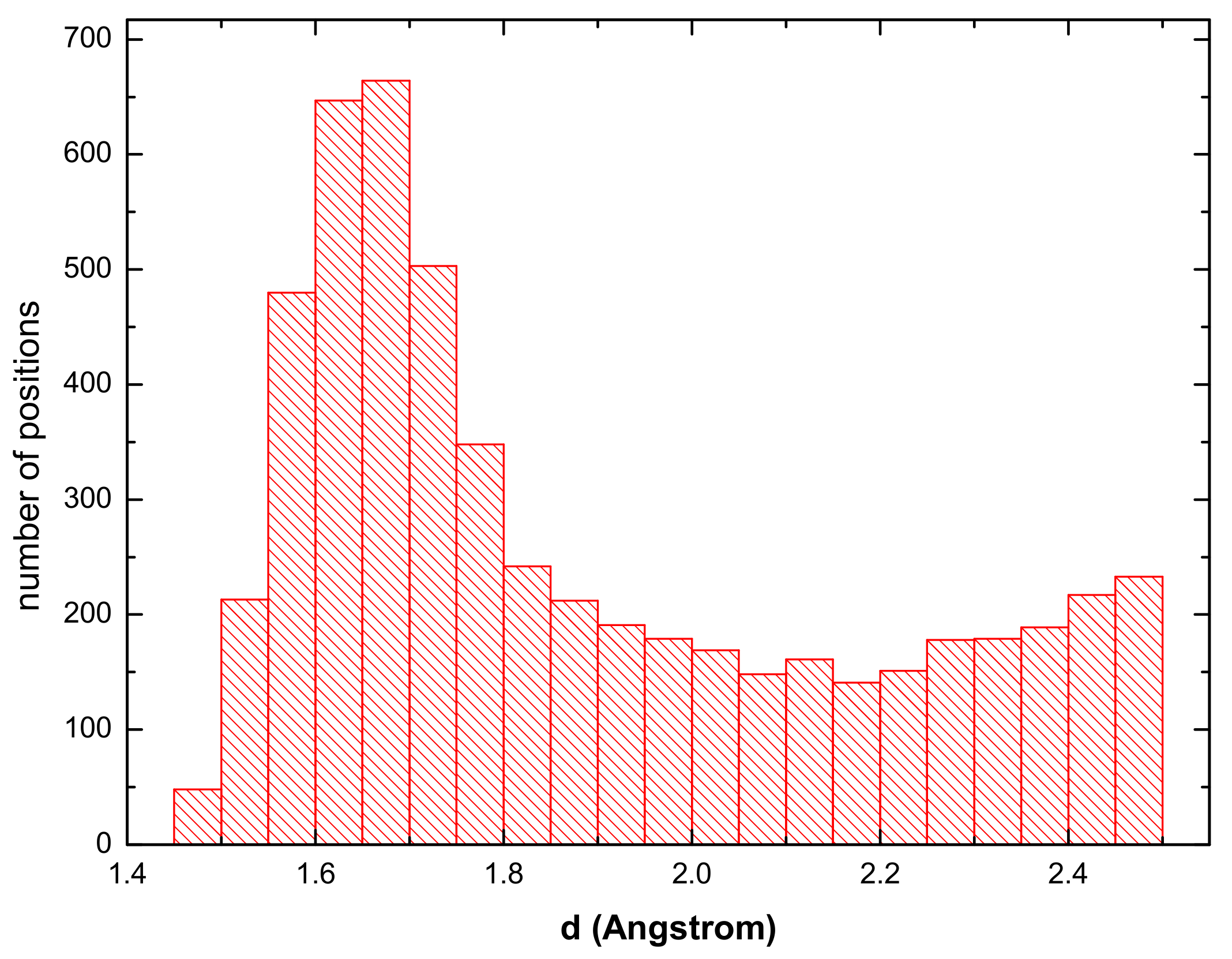
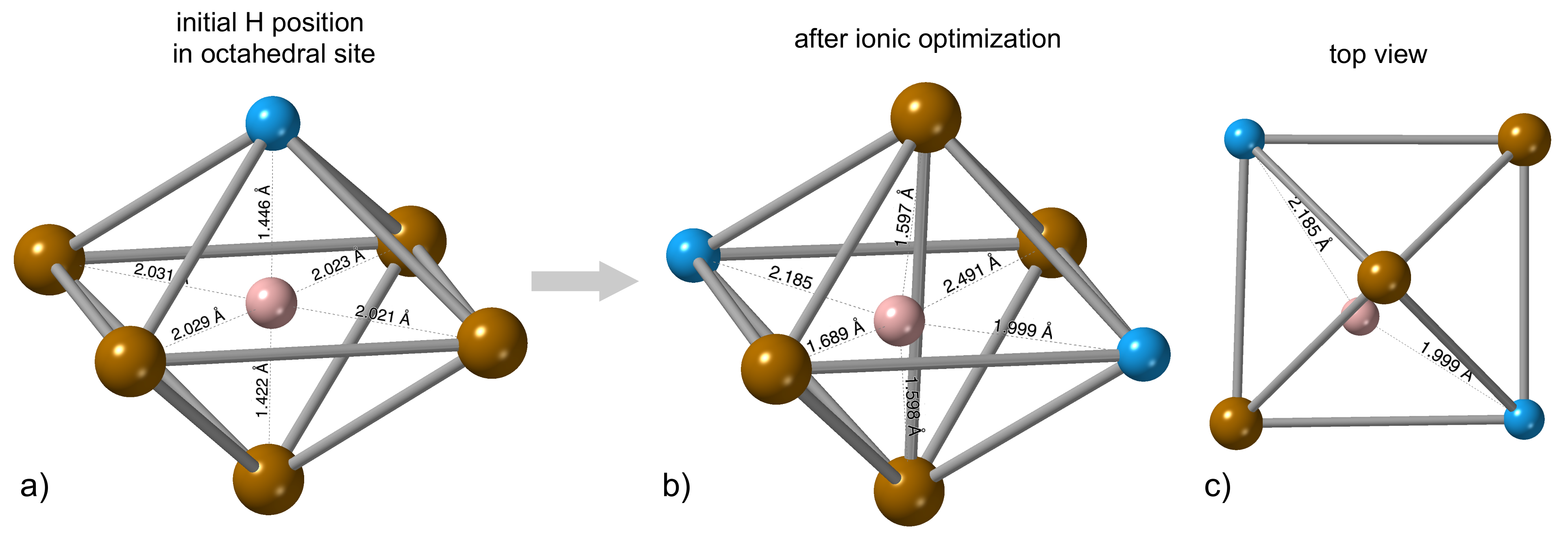
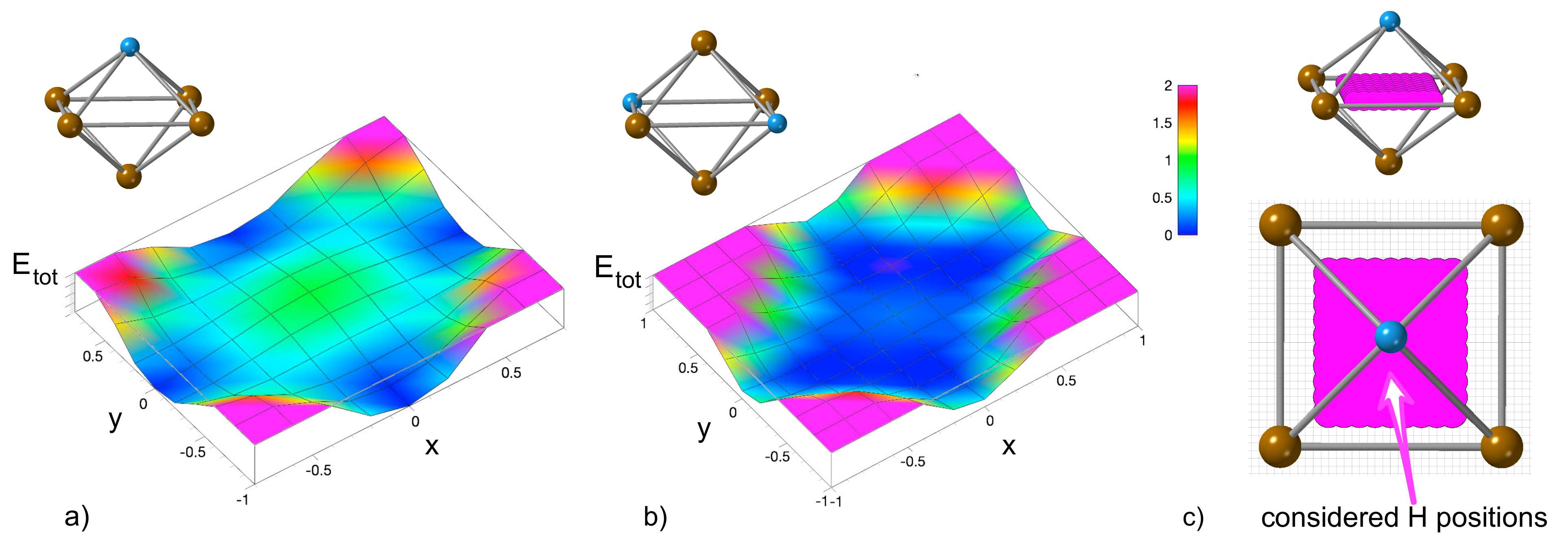
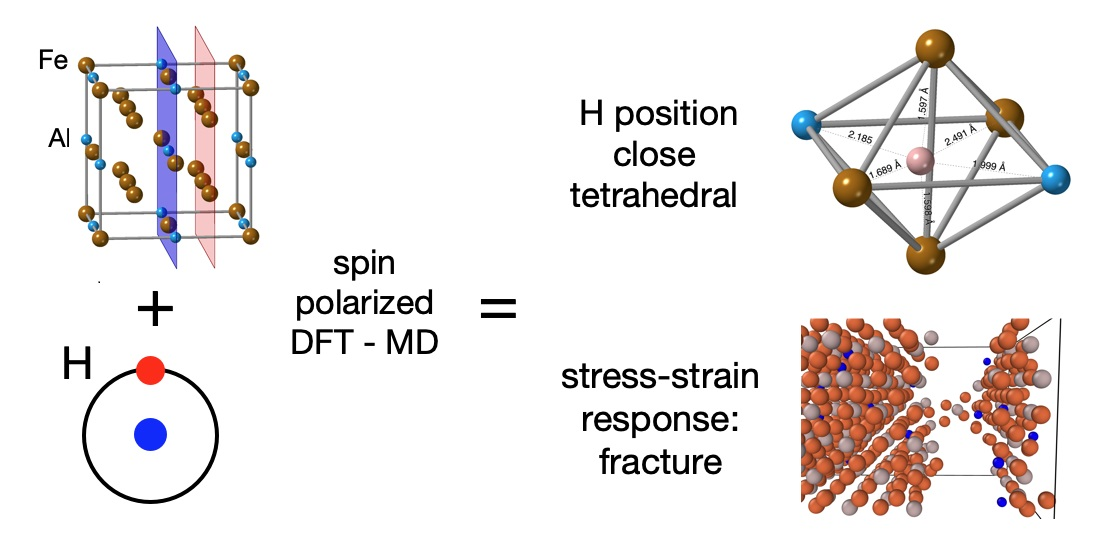
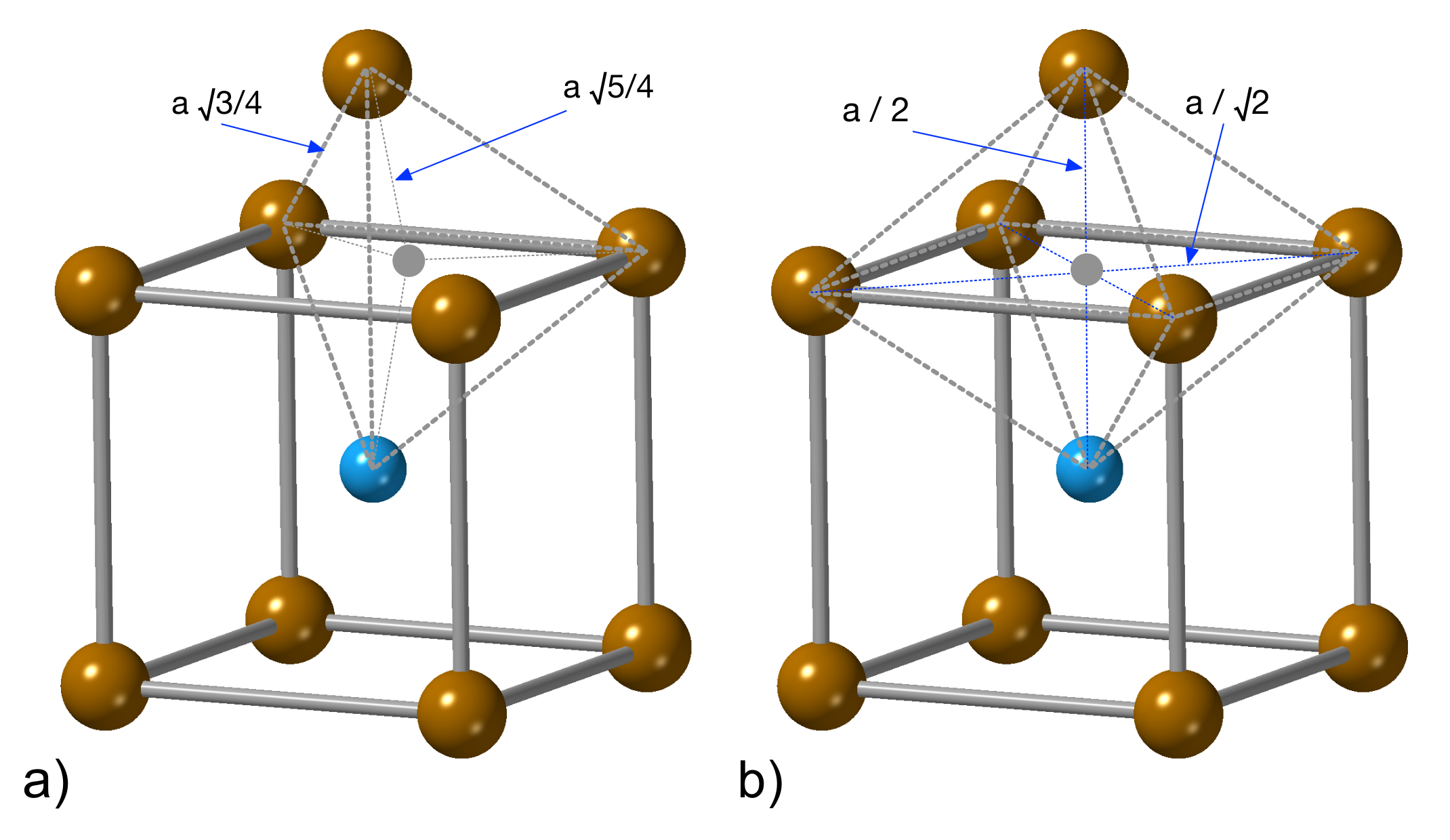
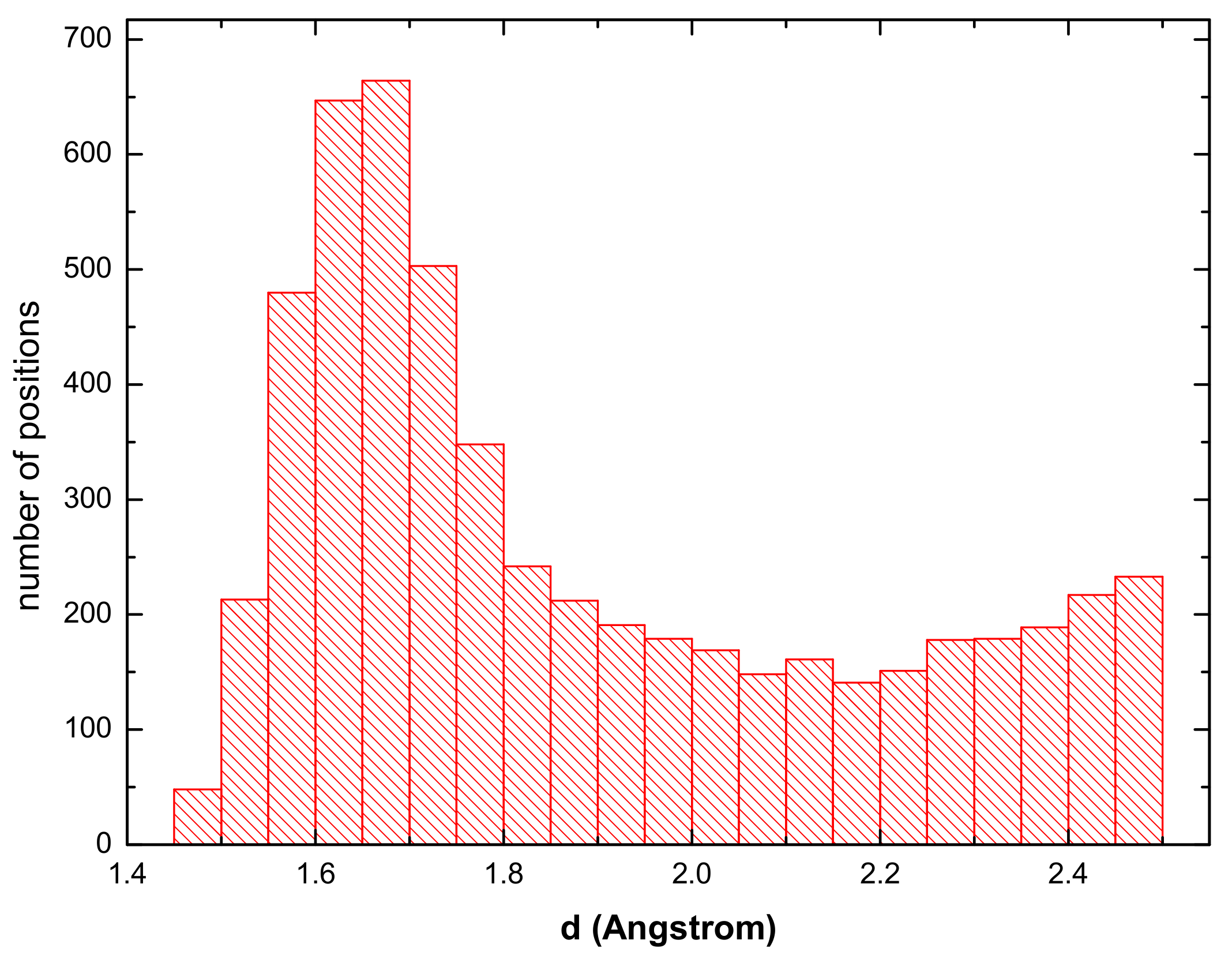
3.1. Hydrogen Position in the FeAl with the D0 Structure
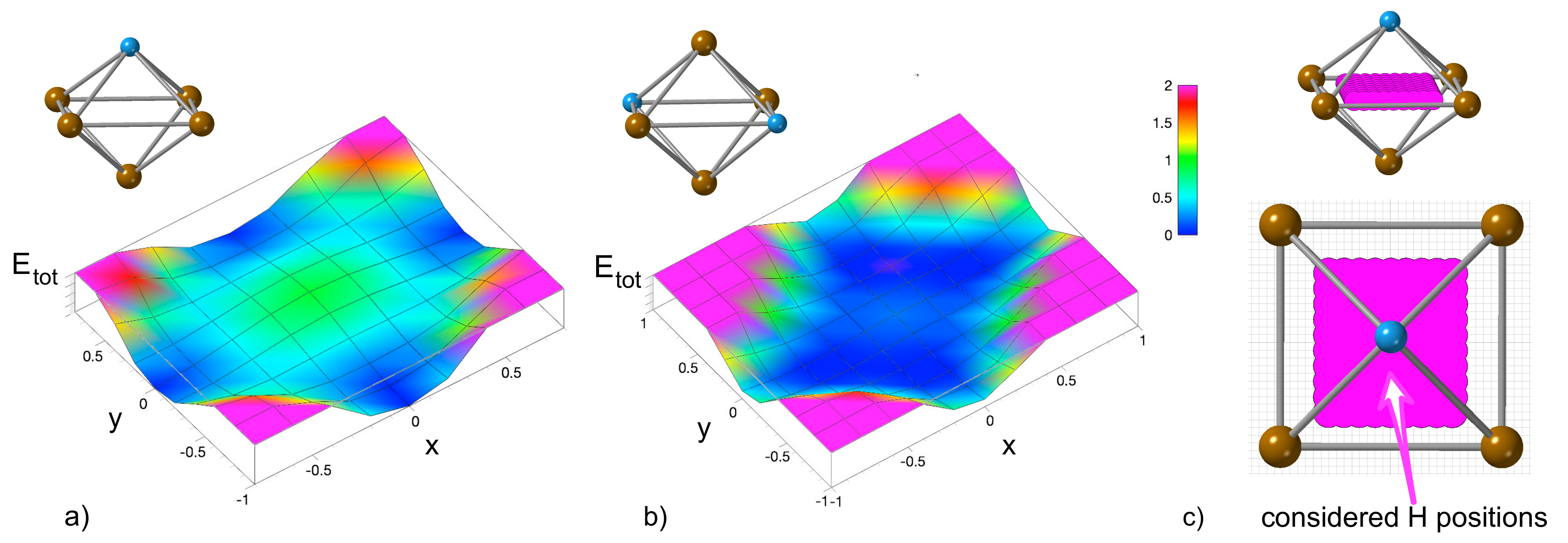
3.2. Effect of Hydrogen on the Stress-Strain Behavior
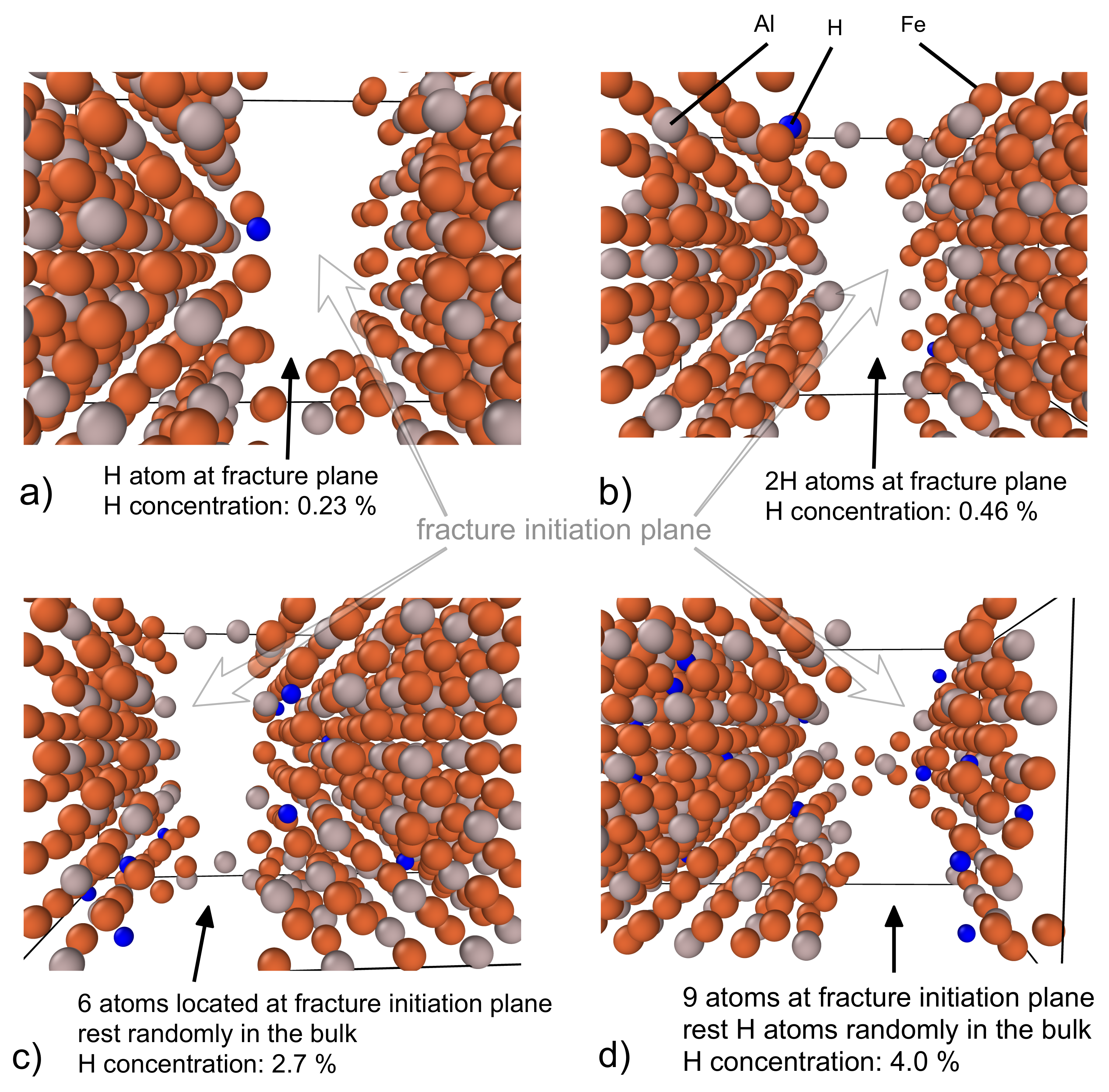
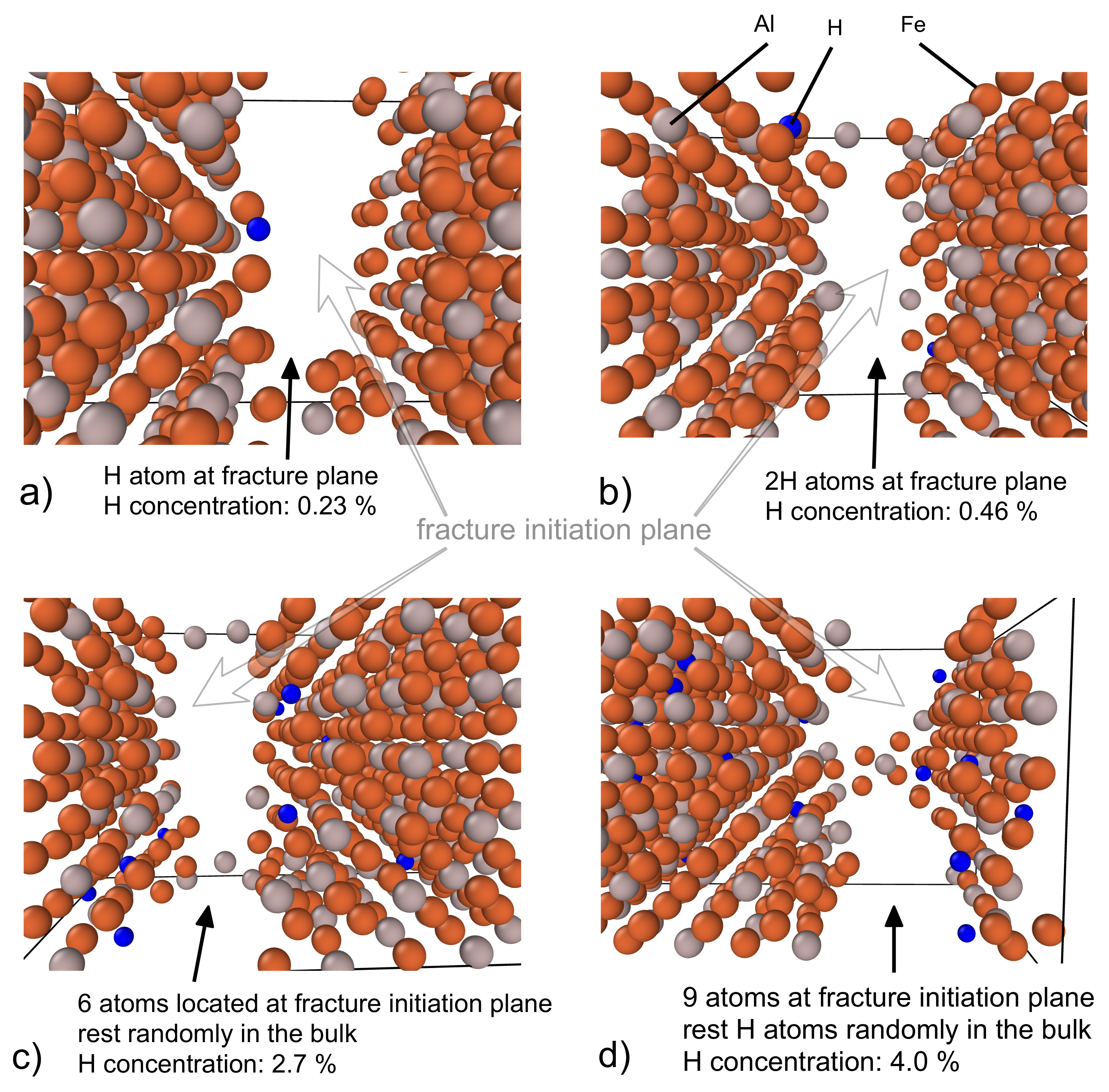
3.3. Fracture Initiation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sauthoff, G. Intermetallics; VCH Verlagsgesellschaft: Weinheim, Germany, 1995. [Google Scholar]
- Liu, C.T.; Stringer, J.; Mundy, J.N.; Horton, L.L.; Angelini, P. Ordered intermetallic alloys: An assessment. Intermetallics 1997, 5, 579–596. [Google Scholar] [CrossRef]
- Xu, Z.; McLellan, R. The solubility of hydrogen in Fe3Al-intermetallics. J. Phys. Chem. Solids 1997, 58, 2127–2129. [Google Scholar] [CrossRef]
- Stoloff, N.S. Iron aluminides: Present status and future prospects. Mater. Sci. Eng. A 1998, 258, 1–14. [Google Scholar] [CrossRef]
- Cheng, X.; Wan, X. Hydrogen Diffusivity in a Fe3Al-Based Alloy. Scr. Mater. 1998, 38, 1505–1509. [Google Scholar] [CrossRef]
- Gu, B.; Chu, W.Y.; Qiao, L.J. TEM observation on stress corrosion cracking of Fe3Al alloy in acetone and water. J. Mater. Sci. Lett. 1999, 18, 1291–1293. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C. Moisture induced environmental embrittlement of intermetallics. Int. Mater. Rev. 2001, 46, 253–270. [Google Scholar] [CrossRef]
- Kattner, U.; Burton, B. Al-Fe (Aluminium-Iron). Phase Diagrams of Binary Iron Alloys; Okamoto, H., Ed.; ASM International: Materials Park, OH, USA, 1993; pp. 12–28. [Google Scholar]
- Palm, M.; Inden, G.; Thomas, N. The Fe-Al-Ti system. J. Phase Equilibria 1995, 16, 209–222. [Google Scholar] [CrossRef]
- Palm, M.; Lacaze, J. Assessment of the Al-Fe-Ti system. Intermetallics 2006, 14, 1291–1303. [Google Scholar] [CrossRef] [Green Version]
- Palm, M.; Sauthoff, G. Deformation behaviour and oxidation resistance of single-phase and two-phase L21-ordered Fe-Al-Ti alloys. Intermetallics 2004, 12, 1345–1359. [Google Scholar] [CrossRef]
- Sundman, B.; Ohnuma, I.; Dupin, N.; Kattner, U.R.; Fries, S.G. An assessment of the entire Al-Fe system including D03 ordering. Acta Mater. 2009, 57, 2896–2908. [Google Scholar] [CrossRef]
- Zamanzade, M.; Barnoush, A.; Motz, C. A Review on the Properties of Iron Aluminide Intermetallics. Crystals 2016, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Castagna, A.; Stoloff, N. Hydrogen embrittlement of Fe3Al alloys. Mater. Sci. Eng. A 1995, 192–193, 399–406. [Google Scholar] [CrossRef]
- Jirásková, Y.; Pizúrová, N.; Titov, A.; Janičkovič, D.; Friák, M. Phase separation in Fe-Ti-Al alloy—Structural, magnetic, and Mössbauer study. J. Magn. Magn. Mater. 2018, 468, 91–99. [Google Scholar] [CrossRef]
- Dobeš, F.; Dymáček, P.; Friák, M. Force-to-Stress Conversion Methods in Small Punch Testing Exemplified by Creep Results of Fe-Al Alloy with Chromium and Cerium Additions. IOP Conf. Ser. Mater. Sci. Eng. 2018, 461, 012017. [Google Scholar] [CrossRef]
- Dobeš, F.; Dymáček, P.; Friák, M. Small punch creep of Fe-Al-Cr alloy with Ce addition and its relation to uniaxial creep tests. Kov. Mater. Met. Mater. 2018, 56, 205. [Google Scholar] [CrossRef] [Green Version]
- Dymáček, P.; Dobeš, F.; Jirásková, Y.; Pizúrová, N.; Friák, M. Tensile, creep and fracture testing of prospective Fe-Al-based alloys using miniature specimens. Theor. Appl. Fract. Mech. 2019, 99, 18–26. [Google Scholar] [CrossRef]
- Dobeš, F.; Dymáček, P.; Friák, M. The Influence of Niobium Additions on Creep Resistance of Fe-27 at. % Al Alloys. Metals 2019, 9, 739. [Google Scholar] [CrossRef] [Green Version]
- Grigorchik, A.N.; Astrashab, V.E.; Kukareko, V.A.; Belotserkovsky, M.A.; Sosnovsky, V.A. High-temperature heat treatment of hypersonic metallization coatings from pseudoalloy “Fe-Al”. Lett. Mater. 2021, 11, 198–203. [Google Scholar] [CrossRef]
- Deevi, S.C. Advanced intermetallic iron aluminide coatings for high temperature applications. Prog. Mater. Sci. 2021, 118, 100769. [Google Scholar] [CrossRef]
- Tolochyn, O.I.; Baglyuk, G.A.; Tolochyna, O.V.; Evych, Y.I.; Podrezov, Y.M.; Molchanovska, H.M. Structure and Physicomechanical Properties of the Fe3Al Intermetallic Compound Obtained by Impact Hot Compaction. Mater. Sci. 2021, 56, 499–508. [Google Scholar] [CrossRef]
- Komarov, O.N.; Zhilin, S.G.; Predein, V.V.; Popov, A.V. Mechanisms for Forming Iron-Containing Intermetallics Prepared by Aluminothermy and the Effect of Special Treatment Methods on their Properties. Metallurgist 2020, 64, 810–821. [Google Scholar] [CrossRef]
- Vodičková, V.; Švec, M.; Hanus, P.; Novák, P.; Záděra, A.; Keller, V.; Prokopčaková, P.P. The Effect of Simultaneous Si and Ti/Mo Alloying on High-Temperature Strength of Fe3Al-Based Iron Aluminides. Molecules 2020, 25, 4268. [Google Scholar] [CrossRef]
- Luo, X.; Cao, J.; Meng, G.; Chuan, Y.; Yao, Z.; Xie, H. Systematical investigation on the microstructures and tribological properties of Fe-Al laser cladding coatings. Appl. Surf. Sci. 2020, 516, 146121. [Google Scholar] [CrossRef]
- Luo, X.; Cao, J.; Meng, G.; Yu, F.; Jiang, Q.; Zhang, P.; Xie, H. Double Glow Plasma Surface Metallurgy Technology Fabricated Fe-Al-Cr Coatings with Excellent Corrosion Resistance. Coatings 2020, 10, 575. [Google Scholar] [CrossRef]
- Teker, T.; Yilmaz, S.O. Synthesis and structural characterization of Fe based Ti+Ni3Al+Al2O3 reinforcement composite produced by mechanical alloying. Rev. Metal. 2020, 56, e178. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Niu, M.; Shao, M.; Geng, X. Microstructure and mechanical behavior of in situ TiC reinforced Fe3Al (Fe-23Al-3Cr) matrix composites by mechanical alloying and vacuum hot-pressing sintering technology. Vacuum 2020, 180, 109544. [Google Scholar] [CrossRef]
- Ghazanfari, H.; Blais, C.; Gariepy, M.; Savoie, S.; Schulz, R.; Alamdari, H. Improving wear resistance of metal matrix composites using reinforcing particles in two length-scales: Fe3Al/TiC composites. Surf. Coat. Technol. 2020, 386, 125502. [Google Scholar] [CrossRef]
- Khodaei, M. Characterization of Al2O3 in Fe3Al-30 vol.% Al2O3 Nanocomposite Powder Synthesized by Mechanochemical Process. J. Nanostruct. 2020, 10, 456–462. [Google Scholar] [CrossRef]
- Altunin, R.R.; Moiseenko, E.T.; Zharkov, S.M. Structural Phase Transformations during a Solid-State Reaction in a Bilayer Al/Fe Thin-Film Nanosystem. Phys. Solid State 2020, 62, 200–205. [Google Scholar] [CrossRef]
- Tolochyn, O.I.; Tolochyna, O.V.; Bagliuk, H.A.; Yevych, Y.I.; Podrezov, Y.M.; Mamonova, A.A. Influence of Sintering Temperature on the Structure and Properties of Powder Iron Aluminide Fe3Al. Powder Metall. Met. Ceram. 2020, 59, 150–159. [Google Scholar] [CrossRef]
- Adler, L.; Fu, Z.; Koerner, C. Electron beam based additive manufacturing of Fe3Al based iron aluminides—Processing window, microstructure and properties. Mater. Sci. Eng. A Struct. Mater. Prop. Microstruct. Process. 2020, 785, 139369. [Google Scholar] [CrossRef]
- Michalcova, A.; Ozkan, M.; Mikula, P.; Marek, I.; Knaislova, A.; Kopecek, J.; Vojtech, D. The Influence of Powder Milling on Properties of SPS Compacted FeAl. Molecules 2020, 25, 2263. [Google Scholar] [CrossRef] [PubMed]
- Peska, M.; Karczewski, K.; Rzeszotarska, M.; Polanski, M. Direct Synthesis of Fe-Al Alloys from Elemental Powders Using Laser Engineered Net Shaping. Materials 2020, 13, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Cao, J.; Meng, G.; Zhou, Y.; Xie, H. Long-range-ordered Fe3Al with excellent electromagnetic wave absorption. J. Mater. Sci. Mater. Electron. 2020, 31, 15608–15615. [Google Scholar] [CrossRef]
- Ismail, A.; Bahanan, W.; Bin Hussain, P.; Saat, A.M.; Shaik, N.B. Diffusion Bonding of Al-Fe Enhanced by Gallium. Processes 2020, 8, 824. [Google Scholar] [CrossRef]
- Watson, R.E.; Weinert, M. Transition-metal aluminide formation: Ti, V, Fe, and Ni aluminides. Phys. Rev. B 1998, 58, 5981–5988. [Google Scholar] [CrossRef]
- Gonzales-Ormeno, P.; Petrilli, H.; Schön, C. Ab-initio calculations of the formation energies of BCC-based superlattices in the Fe-Al system. Calphad 2002, 26, 573–582. [Google Scholar] [CrossRef]
- Connetable, D.; Maugis, P. First principle calculations of the kappa-Fe3AlC perovskite and iron-aluminium intermetallics. Intermetallics 2008, 16, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Kellou, A.; Grosdidier, T.; Raulot, J.M.; Aourag, H. Atomistic study of magnetism effect on structural stability in Fe3Al and Fe3AlX (X = H, B, C, N, O) alloys. Phys. Status Solidi B Basic Solid State Phys. 2008, 245, 750–755. [Google Scholar] [CrossRef]
- Šesták, P.; Friák, M.; Holec, D.; Všianská, M.; Šob, M. Strength and brittleness of interfaces in Fe-Al superalloy nanocomposites under multiaxial loading: An ab initio and atomistic study. Nanomaterials 2018, 8, 873. [Google Scholar] [CrossRef] [Green Version]
- Lechermann, F.; Fähnle, M.; Meyer, B.; Elsässer, C. Electronic correlations, magnetism, and structure of Fe-Al subsystems: An LDA+U study. Phys. Rev. B 2004, 69, 165116. [Google Scholar] [CrossRef]
- Airiskallio, E.; Nurmi, E.; Heinonen, M.H.; Vayrynen, I.J.; Kokko, K.; Ropo, M.; Punkkinen, M.P.J.; Pitkanen, H.; Alatalo, M.; Kollar, J.; et al. High temperature oxidation of Fe-Al and Fe-Cr-Al alloys: The role of Cr as a chemically active element. Corros. Sci. 2010, 52, 3394–3404. [Google Scholar] [CrossRef]
- Lechermann, F.; Welsch, F.; Elsässer, C.; Ederer, C.; Fähnle, M.; Sanchez, J.; Meyer, B. Density-functional study of Fe3Al: LSDA versus GGA. Phys. Rev. B 2002, 65, 132104. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Slávik, A.; Miháliková, I.; Holec, D.; Všianská, M.; Šob, M.; Palm, M.; Neugebauer, J. Origin of the low magnetic moment in Fe2AlTi: An Ab initio study. Materials 2018, 11, 1732. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.; Kang, M.; Zhou, Y.; Yang, C.; Wang, K.; Li, J.; Wang, R.; Fu, H.; Wang, J. First-principles investigations of the stability, electronic structures, mechanical properties and thermodynamic properties of FexAlyCz compounds in Fe-Cr-B-Al-C alloy. J. Phys. Chem. Solids 2020, 143. [Google Scholar] [CrossRef]
- Miháliková, I.; Friák, M.; Jirásková, Y.; Holec, D.; Koutná, N.; Šob, M. Impact of Nano-Scale Distribution of Atoms on Electronic and Magnetic Properties of Phases in Fe-Al Nanocomposites: An Ab Initio Study. Nanomaterials 2018, 8, 1059. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Holec, D.; Šob, M. Quantum-Mechanical Study of Nanocomposites with Low and Ultra-Low Interface Energies. Nanomaterials 2018, 8, 1057. [Google Scholar] [CrossRef] [Green Version]
- Kulikov, N.I.; Postnikov, A.V.; Borstel, G.; Braun, J. Onset of magnetism in B2 transition-metal aluminides. Phys. Rev. B 1999, 59, 6824–6833. [Google Scholar] [CrossRef] [Green Version]
- Friák, M.; Deges, J.; Krein, R.; Frommeyer, G.; Neugebauer, J. Combined ab initio and experimental study of structural and elastic properties of Fe3Al-based ternaries. Intermetallics 2010, 18, 1310. [Google Scholar] [CrossRef]
- Friák, M.; Neugebauer, J. Ab initio study of the anomalous volume-composition dependence in Fe-Al alloys. Intermetallics 2010, 18, 1316–1321. [Google Scholar] [CrossRef]
- Ipser, H.; Semenova, O.; Krachler, R. Intermetallic phases with D0(3)-structure: A statistical-thermodynamic model. J. Alloy. Compd. 2002, 338, 20–25. [Google Scholar] [CrossRef]
- Friák, M.; Všianská, M.; Šob, M. A Quantum–Mechanical Study of Clean and Cr—Segregated Antiphase Boundaries in Fe3Al. Materials 2019, 12, 3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fähnle, M.; Drautz, R.; Lechermann, F.; Singer, R.; Diaz-Ortiz, A.; Dosch, H. Thermodynamic properties from ab-initio calculations: New theoretical developments, and applications to various materials systems. Phys. Status Solidi B Basic Solid State Phys. 2005, 242, 1159–1173. [Google Scholar] [CrossRef]
- Kirklin, S.; Saal, J.E.; Hegde, V.I.; Wolverton, C. High-throughput computational search for strengthening precipitates in alloys. Acta Mater. 2016, 102, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Duan, S.; Ma, B. First-principles calculation of vibrational entropy for Fe-Al compounds. Phys. Rev. B 1998, 58, 9705–9709. [Google Scholar]
- Čížek, J.; Lukáč, F.; Procházka, I.; Kužel, R.; Jirásková, Y.; Janičkovič, D.; Anwand, W.; Brauer, G. Characterization of quenched-in vacancies in Fe-Al alloys. Phys. B Condens. Matter 2012, 407, 2659–2664. [Google Scholar] [CrossRef]
- Miháliková, I.; Friák, M.; Koutná, N.; Holec, D.; Šob, M. An Ab Initio Study of Vacancies in Disordered Magnetic Systems: A Case Study of Fe-Rich Fe-Al Phases. Materials 2019, 12, 1430. [Google Scholar] [CrossRef] [Green Version]
- Amara, H.; Fu, C.C.; Soisson, F.; Maugis, P. Aluminum and vacancies in α-iron: Dissolution, diffusion, and clustering. Phys. Rev. B 2010, 81, 174101. [Google Scholar] [CrossRef]
- Friák, M.; Černý, M.; Všianská, M.; Šob, M. Impact of Antiphase Boundaries on Structural, Magnetic and Vibrational Properties of Fe3Al. Materials 2020, 13, 4884. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Yang, J. First principle calculations and mechanical properties of the intermetallic compounds in a laser welded steel/aluminum joint. Opt. Laser Technol. 2020, 122, 105875. [Google Scholar] [CrossRef]
- Friák, M.; Černý, M.; Šob, M. The impact of vibrational entropy on the segregation of Cu to antiphase boundaries in Fe3Al. Magnetochemistry 2021, 7. submitted for publication. [Google Scholar]
- Liu, C.T.; Lee, E.H.; McKamey, C.G. An environmental-effect as the major cause for room-temperature embrittlement in FeAl. Scr. Metall. Mater. 1989, 23, 875–880. [Google Scholar] [CrossRef]
- Lynch, R.J.; Heldt, L.A.; Milligan, W.W. Effects of alloy composition on environmental embrittlement of B2 ordered iron aluminides. Scr. Metall. Mater. 1991, 25, 2147–2151. [Google Scholar] [CrossRef]
- Lin, J.; Chu, W.; Hsiao, C. Hydrogen induced cracking in Fe3Al + Cr. Scr. Metall. Mater. 1994, 30, 583–586. [Google Scholar] [CrossRef]
- Tu, J.; Meng, L.; Liu, M. Evaluation of Hydrogen Embrittlement Characteristics of Fe3Al Intermetallic Compounds. Scr. Mater. 1998, 38, 833–838. [Google Scholar] [CrossRef]
- Liu, C.T.; McKamey, C.G.; Lee, E.H. Environmental-effects on room-temperature ductility and fracture in Fe3Al. Scr. Metall. Mater 1990, 24, 385–389. [Google Scholar] [CrossRef]
- Lynch, R.J.; Gee, K.A.; Heldt, L.A. Environmental embrittlement of single-crystal and thermomechanically processed B2-ordered iron aluminides. Scr. Metall. Mater. 1994, 30, 945–950. [Google Scholar] [CrossRef]
- Balasubramaniam, R. Hydrogen in iron aluminides. J. Alloy. Compd. 2002, 330–332, 506–510. [Google Scholar] [CrossRef]
- Alven, D.; Stoloff, N. The influence of composition on the environmental embrittlement of Fe3Al alloys. Mater. Sci. Eng. A 1997, 239–240, 362–368. [Google Scholar] [CrossRef]
- Zamanzade, M.; Barnoush, A. An Overview of the Hydrogen Embrittlement of Iron Aluminides. Procedia Mater. Sci. 2014, 3, 2016–2023. [Google Scholar] [CrossRef] [Green Version]
- Zamanzade, M.; Vehoff, H.; Barnoush, A. Cr effect on hydrogen embrittlement of Fe3Al-based iron aluminide intermetallics: Surface or bulk effect. Acta Mater. 2014, 69, 210–223. [Google Scholar] [CrossRef]
- Mubarak, A.A. The elastic, electronic and magnetism structure of the MAl and M3Al (M = Fe and Ni) alloy with and without hydrogen atoms. J. Magn. Magn. Mater. 2016, 401, 816–822. [Google Scholar] [CrossRef]
- Kellou, A.; Raulot, J.; Grosdidier, T. Structural and thermal properties of Fe3Al, Fe3AlC and hypothetical Fe3AlX (X = H, B, N, O) compounds: Ab initio and quasi-harmonic Debye modelling. Intermetallics 2010, 18, 1293–1296. [Google Scholar] [CrossRef]
- Rao, V.S. Fe3Al-Fe3AlC intermetallics for high temperature applications: An assessment. J. Mater. Sci. 2004, 39, 4193–4198. [Google Scholar] [CrossRef]
- Prakash, U.; Parvathavarthini, N.; Dayal, R. Effect of composition on hydrogen permeation in Fe–Al alloys. Intermetallics 2007, 15, 17–19. [Google Scholar] [CrossRef]
- Deng, Y.; Rogne, B.R.S.; Barnoush, A. In-situ microscale examination of hydrogen effect on fracture toughness: A case study on B2 and D03 ordered iron aluminides intermetallic alloys. Eng. Fract. Mech. 2019, 217, 106551. [Google Scholar] [CrossRef]
- Barnoush, A.; Dake, J.; Kheradmand, N.; Vehoff, H. Examination of hydrogen embrittlement in FeAl by means of in situ electrochemical micropillar compression and nanoindentation techniques. Intermetallics 2010, 18, 1385–1389. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Všianská, M.; Friák, M.; Šob, M. An ab initio study of Fe3Al: A critical review of generalized gradient approximation. (to be published).
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshi, A.; Balasubramaniam, R. On critical hydrogen concentration for hydrogen embrittlement of Fe3Al. Bull. Mater. Sci. 2001, 24, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Černý, M.; Šesták, P.; Řehák, P.; Všianská, M.; Šob, M. Atomistic approaches to cleavage of interfaces. Modell. Simul. Mater. Sci. Eng. 2019, 27, 035007. [Google Scholar] [CrossRef]
- Batyrev, I.; Alavi, A.; Finnis, M. Equilibrium and adhesion of Nb/sapphire: The effect of oxygen partial pressure. Phys. Rev. B Condens. Matter Mater. Phys. 2000, 62, 4698–4706. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Qi, Y.; Li, X. Adhesion at diamond/metal interfaces: A density functional theory study. J. Appl. Phys. 2010, 107, 033722. [Google Scholar] [CrossRef] [Green Version]
- Janisch, R.; Ahmed, N.; Hartmaier, A. Ab initio tensile tests of Al bulk crystals and grain boundaries: Universality of mechanical behavior. Phys. Rev. B 2010, 81, 184108. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Løvvik, O.M.; Marthinsen, K.; Li, Y. Segregation of Mg, Cu and their effects on the strength of Al Σ5 (210)[001] symmetrical tilt grain boundary. Acta Mater. 2018, 145, 235–246. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, G.H.; Deng, S.; Wang, T.; Xu, H.; Kohyama, M.; Yamamoto, R. Weakening of an aluminum grain boundary induced by sulfur segregation: A first-principles computational tensile test. Phys. Rev. B 2007, 75, 174101. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, G.H.; Hu, X.; Wang, T.; Kohyama, M.; Yamamoto, R. First-principles computational tensile test on a Na-segregated Al grain boundary with an Si additive and an intergranular embrittlement suppression mechanism. J. Phys. Condens. Matter 2007, 19, 456225. [Google Scholar] [CrossRef]
- Elsner, B.A.M.; Müller, S. Size effects and strain localization in atomic-scale cleavage modeling. J. Phys. Condens. Matter 2015, 27, 345002. [Google Scholar] [CrossRef] [PubMed]
- Tahir, A.; Janisch, R.; Hartmaier, A. Hydrogen embrittlement of a carbon segregated Σ5(310)[001] symmetrical tilt grain boundary in α-Fe. Mater. Sci. Eng. A 2014, 612, 462–467. [Google Scholar] [CrossRef]
- Khalid, M.Z.; Friis, J.; Ninive, P.H.; Marthinsen, K.; Strandlie, A. Ab-initio study of atomic structure and mechanical behaviour of Al/Fe intermetallic interfaces. Comput. Mater. Sci. 2020, 174, 109481. [Google Scholar] [CrossRef]
- Černý, M.; Šesták, P.; Řehák, P.; Všianská, M.; Šob, M. Ab initio tensile tests of grain boundaries in the fcc crystals of Ni and Co with segregated sp-impurities. Mater. Sci. Eng. A 2016, 669, 218–225. [Google Scholar] [CrossRef]
- Lazar, P.; Podloucky, R. Cleavage fracture of a crystal: Density functional theory calculations based on a model which includes structural relaxations. Phys. Rev. B 2008, 78, 104114. [Google Scholar] [CrossRef] [Green Version]
- Geng, W.T.; Freeman, A.J.; Wu, R.; Olson, G.B. Effect of Mo and Pd on the grain-boundary cohesion of Fe. Phys. Rev. B 2000, 62, 6208. [Google Scholar] [CrossRef]
- Fu, C.; Painter, G. First principles investigation of hydrogen embrittlement in FeAl. J. Mater. Res. 1991, 6, 719–723. [Google Scholar] [CrossRef]
- Jiang, D.E.; Carter, E.A. Diffusion of interstitial hydrogen into and through bcc Fe from first principles. Phys. Rev. B 2004, 70, 064102. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.F.; Carter, E.A. First-principles assessment of hydrogen absorption into FeAl and Fe3Si: Towards prevention of steel embrittlement. Acta Mater. 2010, 58, 638–648. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H (at.%) | 0.0000 | 0.2309 | 0.4608 | 1.3699 | 2.7027 | 4.0000 |
|---|---|---|---|---|---|---|
| (Å) | 5190.91 | 5193.05 | 5195.04 | 5203.40 | 5215.15 | 5228.55 |
| (GPa) | 21.2 | 21.9 | 20.8 | 21.1 | 20.6 | 20.9 |
| 0.336 | 0.336 | 0.322 | 0.328 | 0.306 | 0.285 | |
| () | 683.7 | 683.0 | 681.7 | 678.5 | 673.2 | 668.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šesták, P.; Friák, M.; Šob, M. The Effect of Hydrogen on the Stress-Strain Response in Fe3Al: An ab initio Molecular-Dynamics Study. Materials 2021, 14, 4155. https://doi.org/10.3390/ma14154155
Šesták P, Friák M, Šob M. The Effect of Hydrogen on the Stress-Strain Response in Fe3Al: An ab initio Molecular-Dynamics Study. Materials. 2021; 14(15):4155. https://doi.org/10.3390/ma14154155
Chicago/Turabian StyleŠesták, Petr, Martin Friák, and Mojmír Šob. 2021. "The Effect of Hydrogen on the Stress-Strain Response in Fe3Al: An ab initio Molecular-Dynamics Study" Materials 14, no. 15: 4155. https://doi.org/10.3390/ma14154155
APA StyleŠesták, P., Friák, M., & Šob, M. (2021). The Effect of Hydrogen on the Stress-Strain Response in Fe3Al: An ab initio Molecular-Dynamics Study. Materials, 14(15), 4155. https://doi.org/10.3390/ma14154155