
Strain Effects on the Electronic and Thermoelectric Properties of n(PbTe)-m(Bi2Te3) System Compounds


Abstract
1. Introduction
2. Methods and Computational Details
3. Results and Discussion
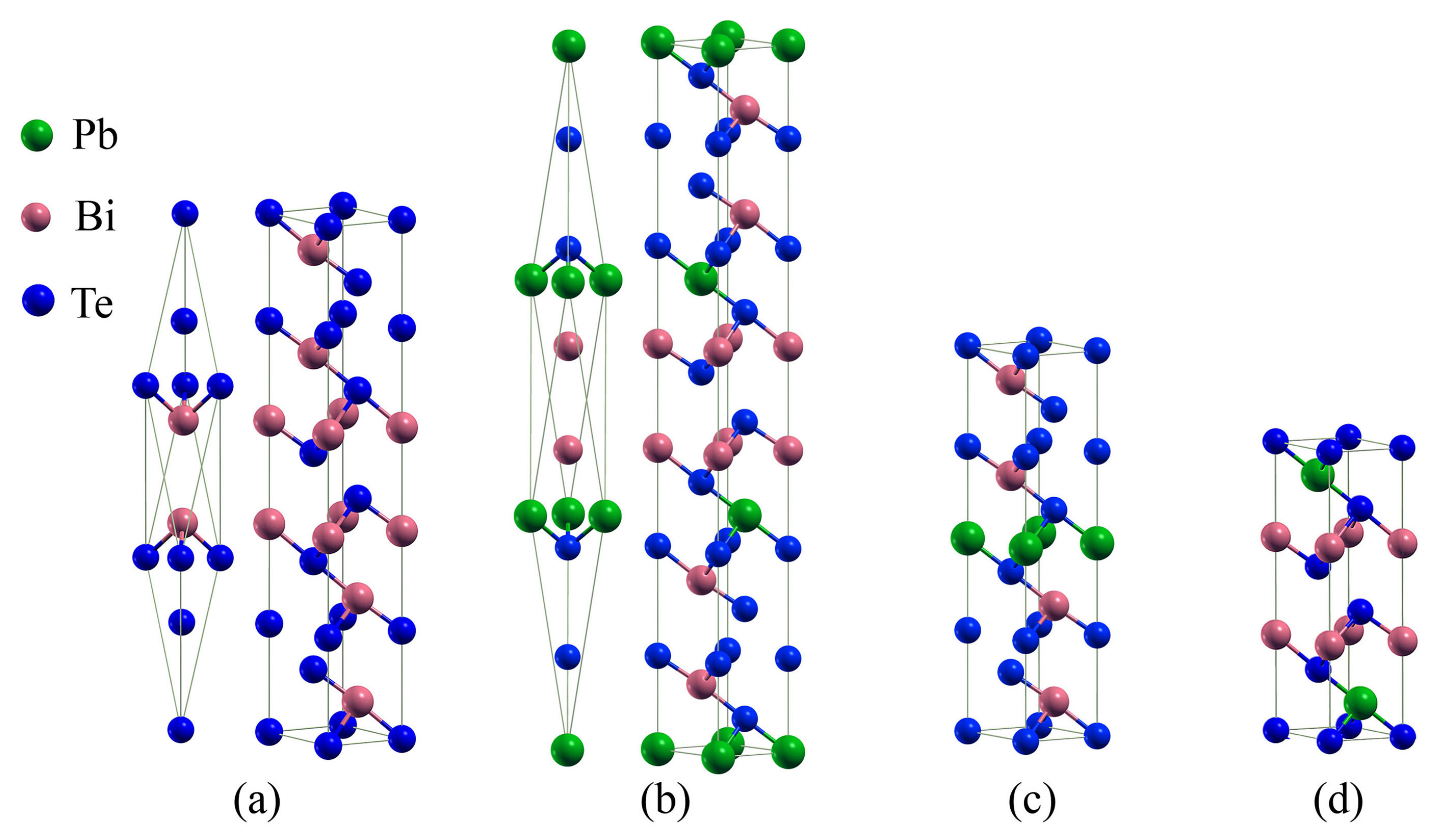
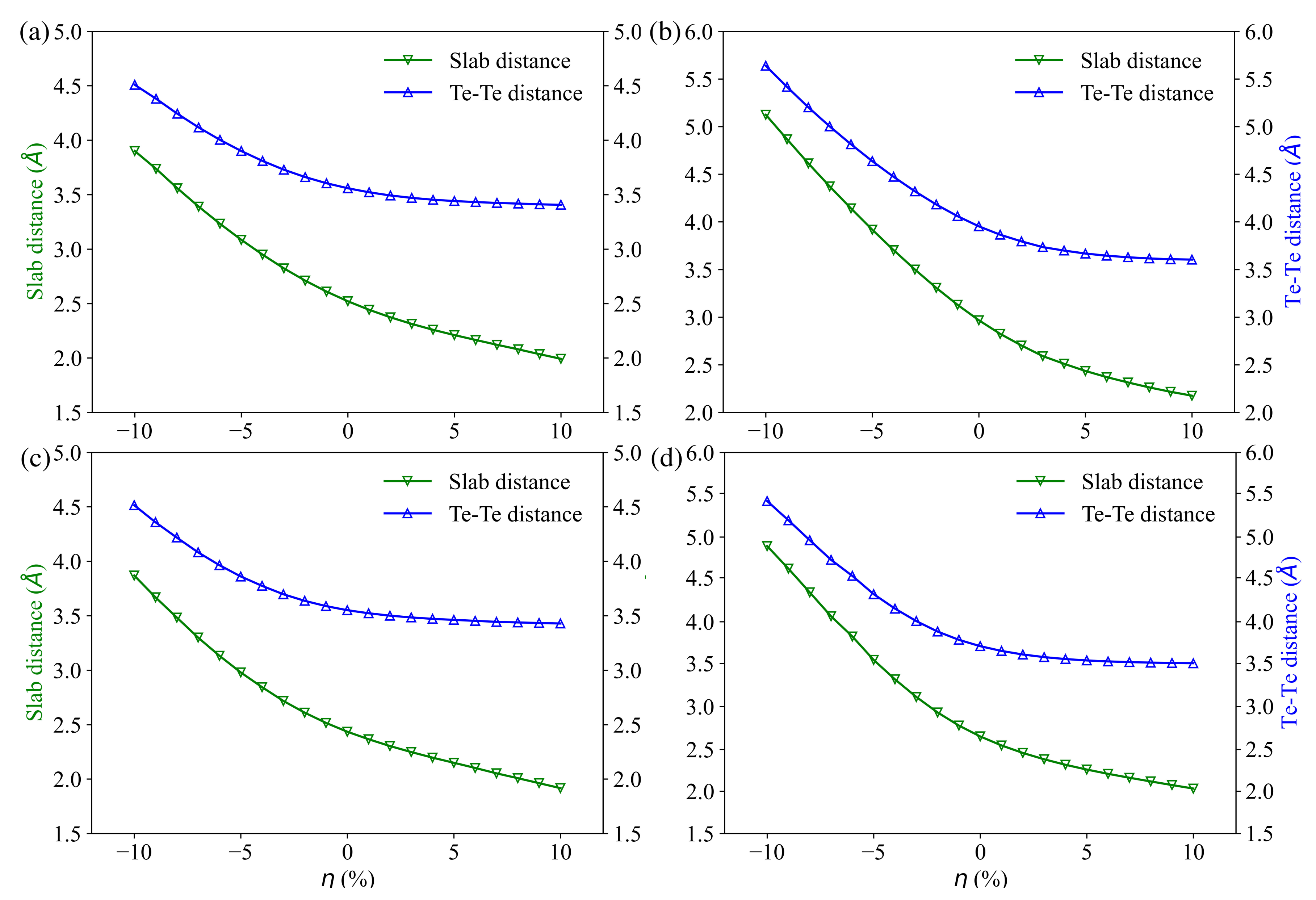
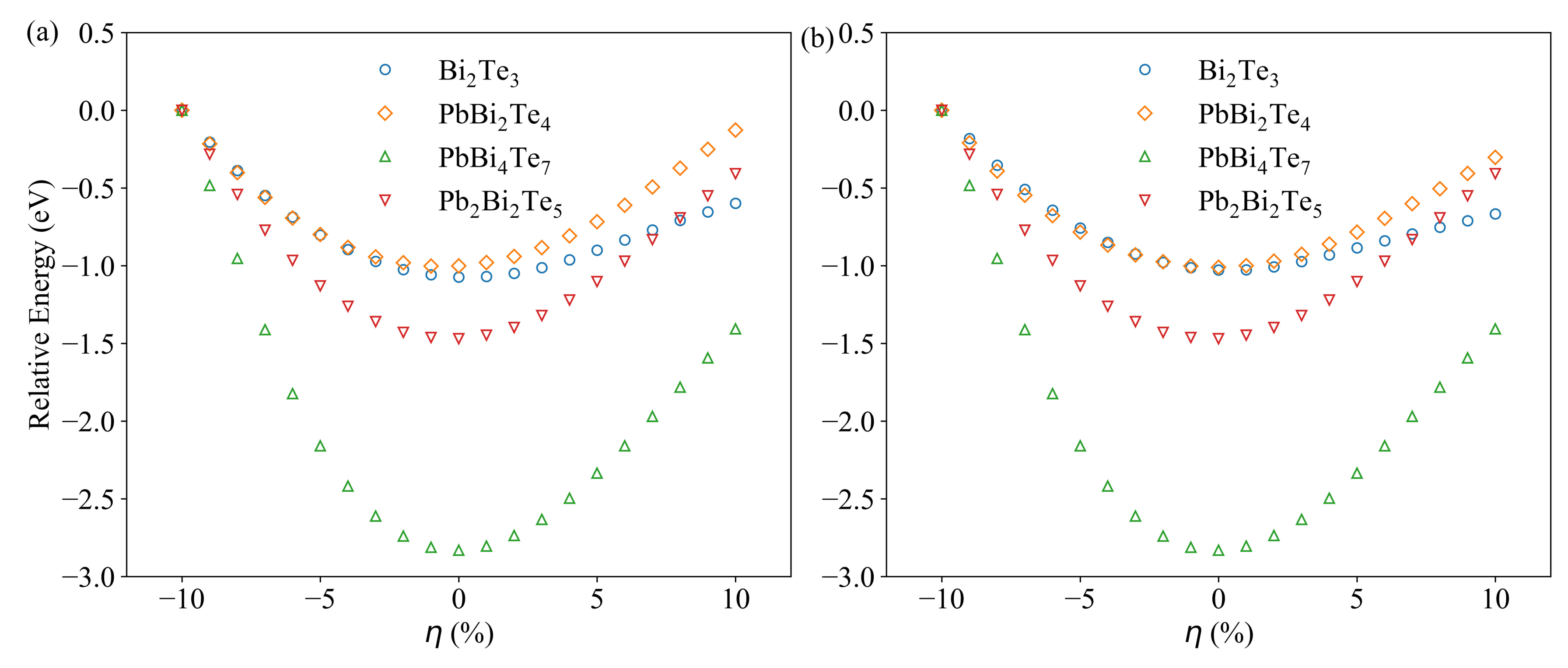
3.1. Compounds’ Structural Information
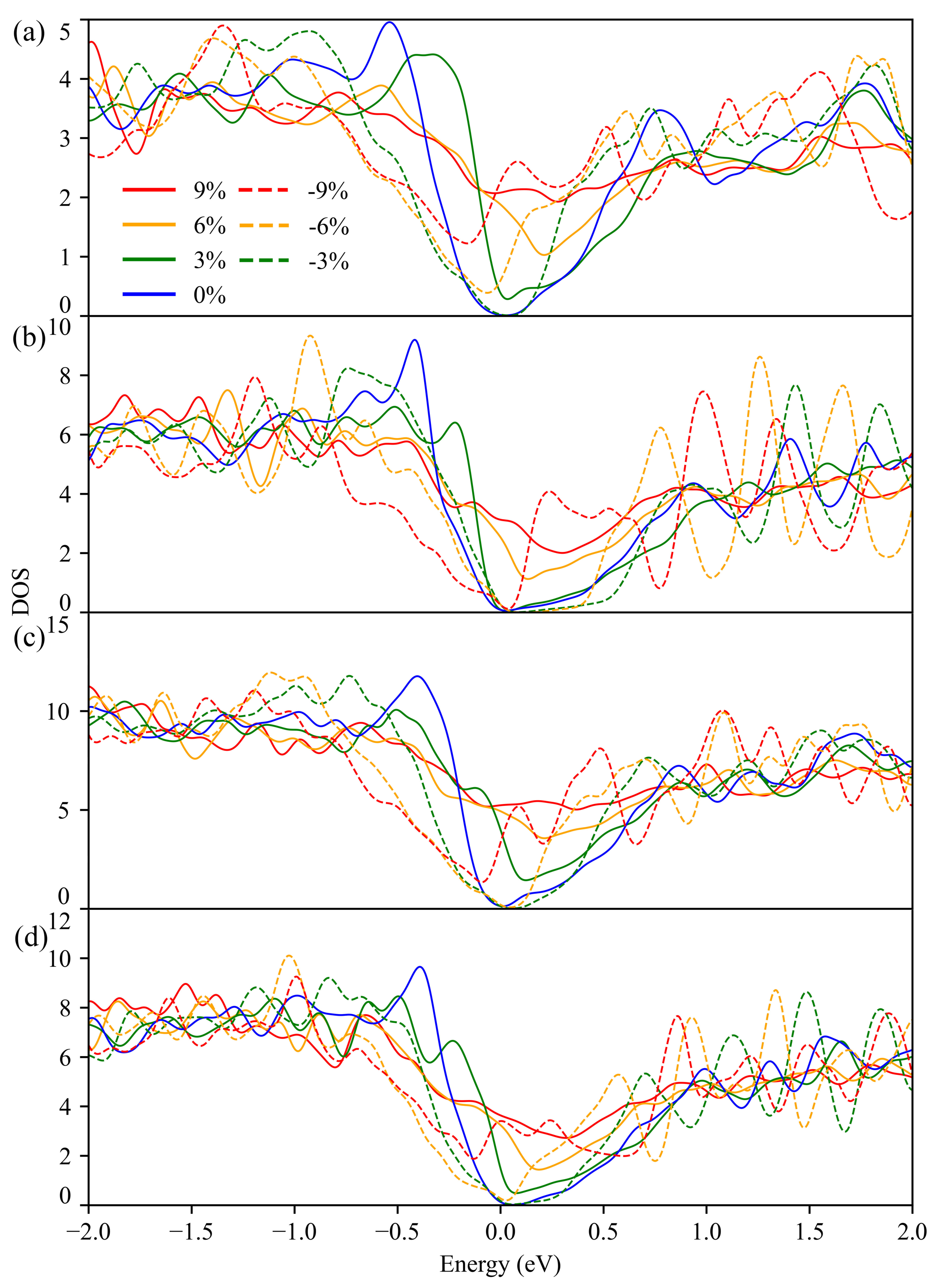
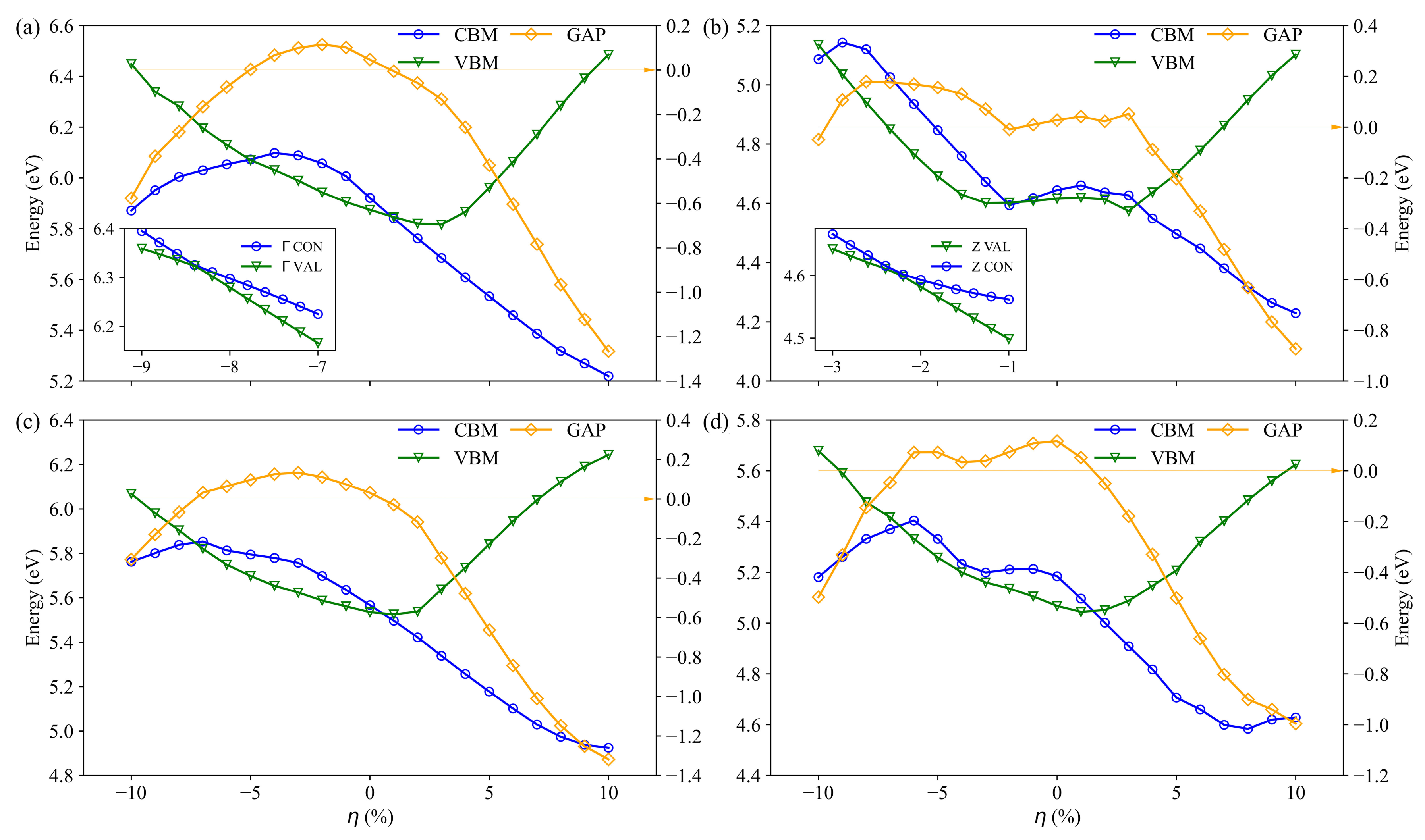
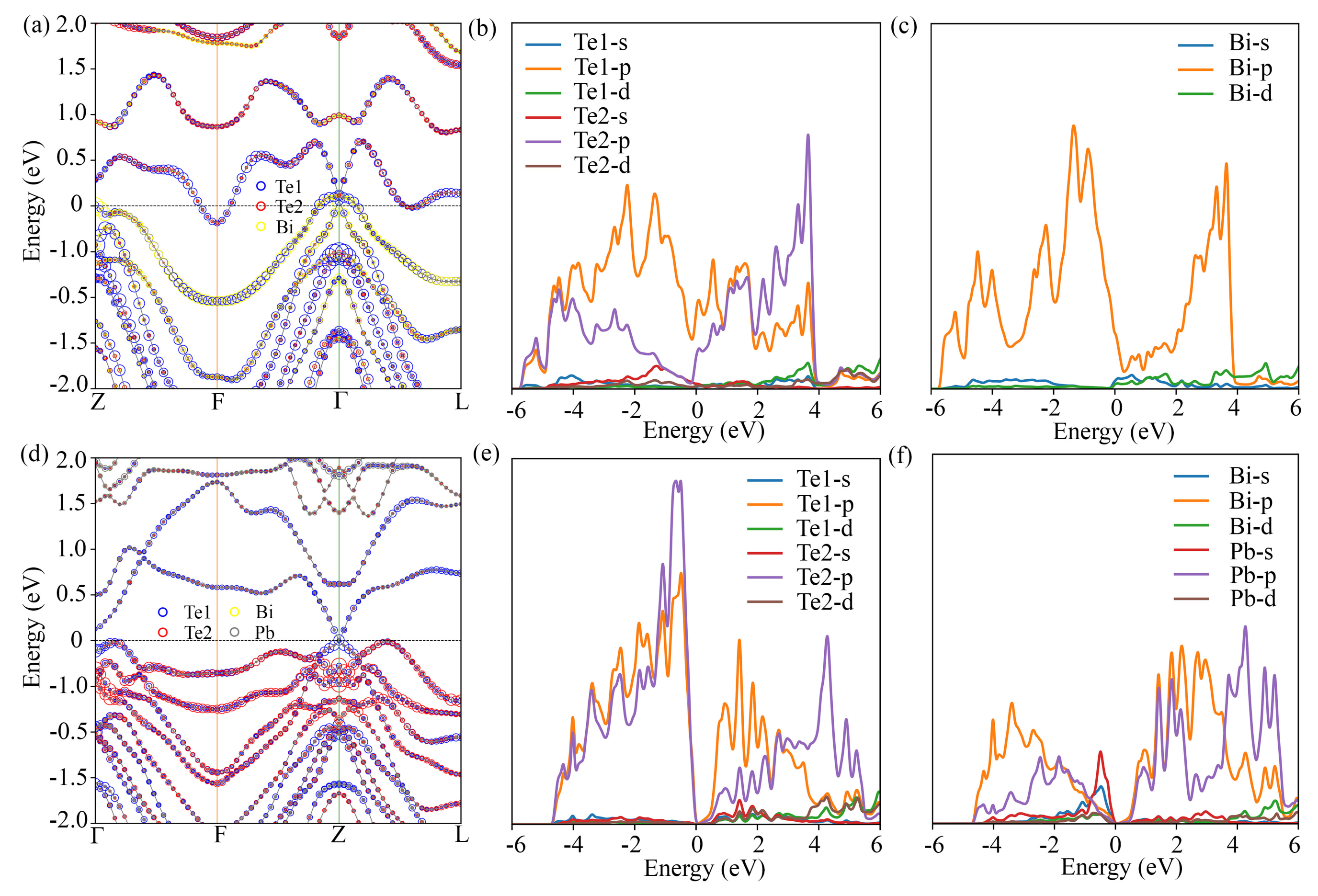
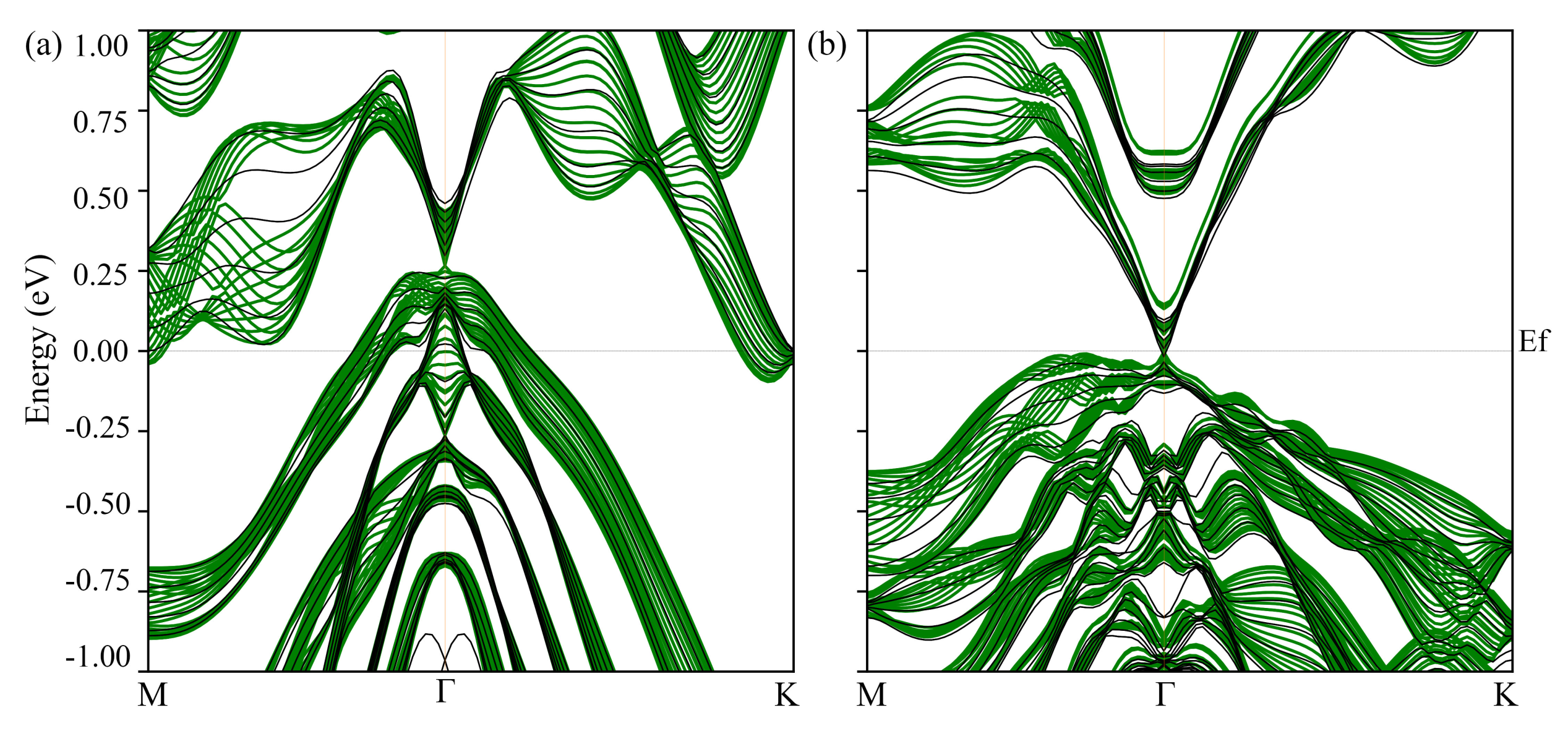
3.2. Electronic Structure
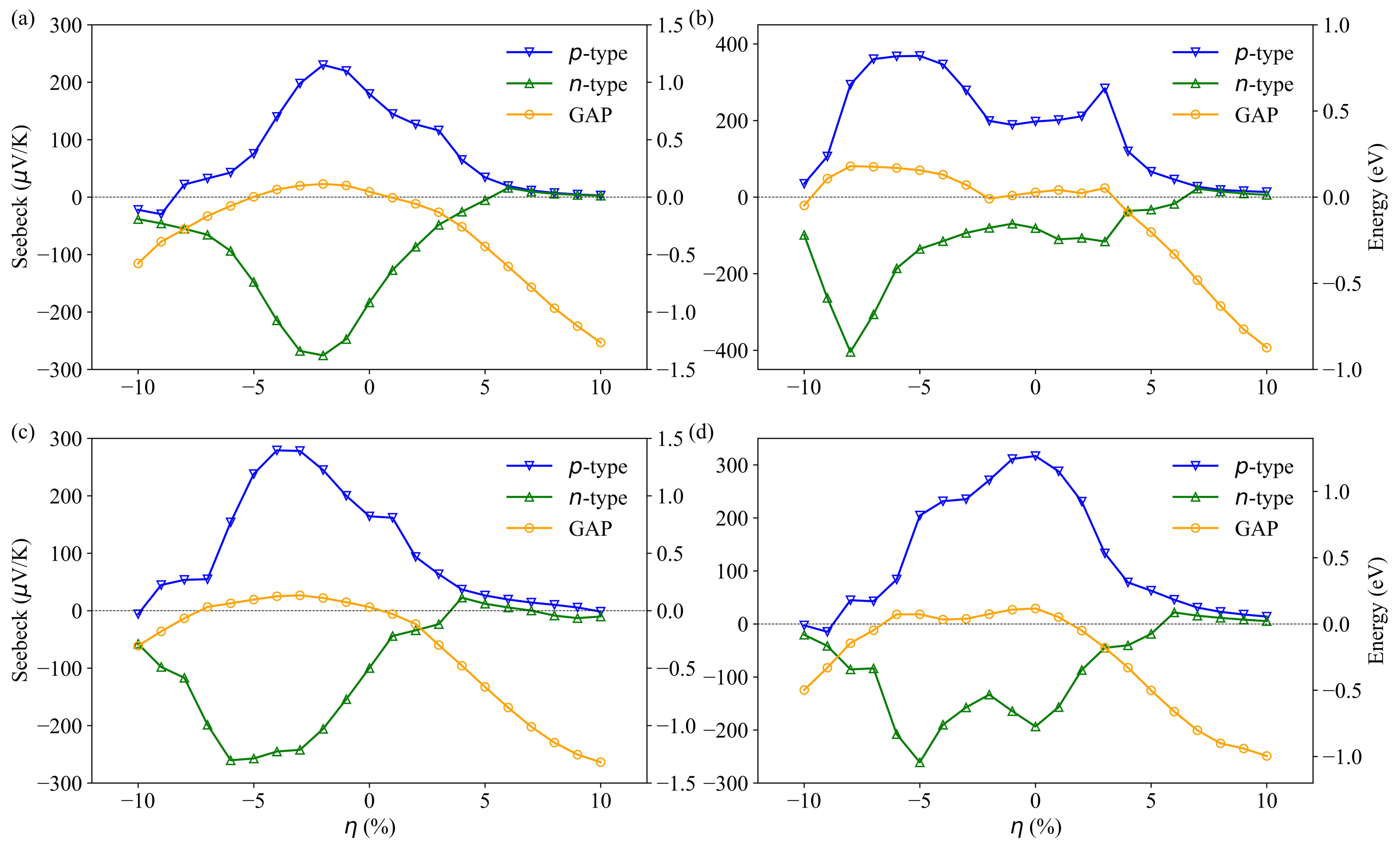
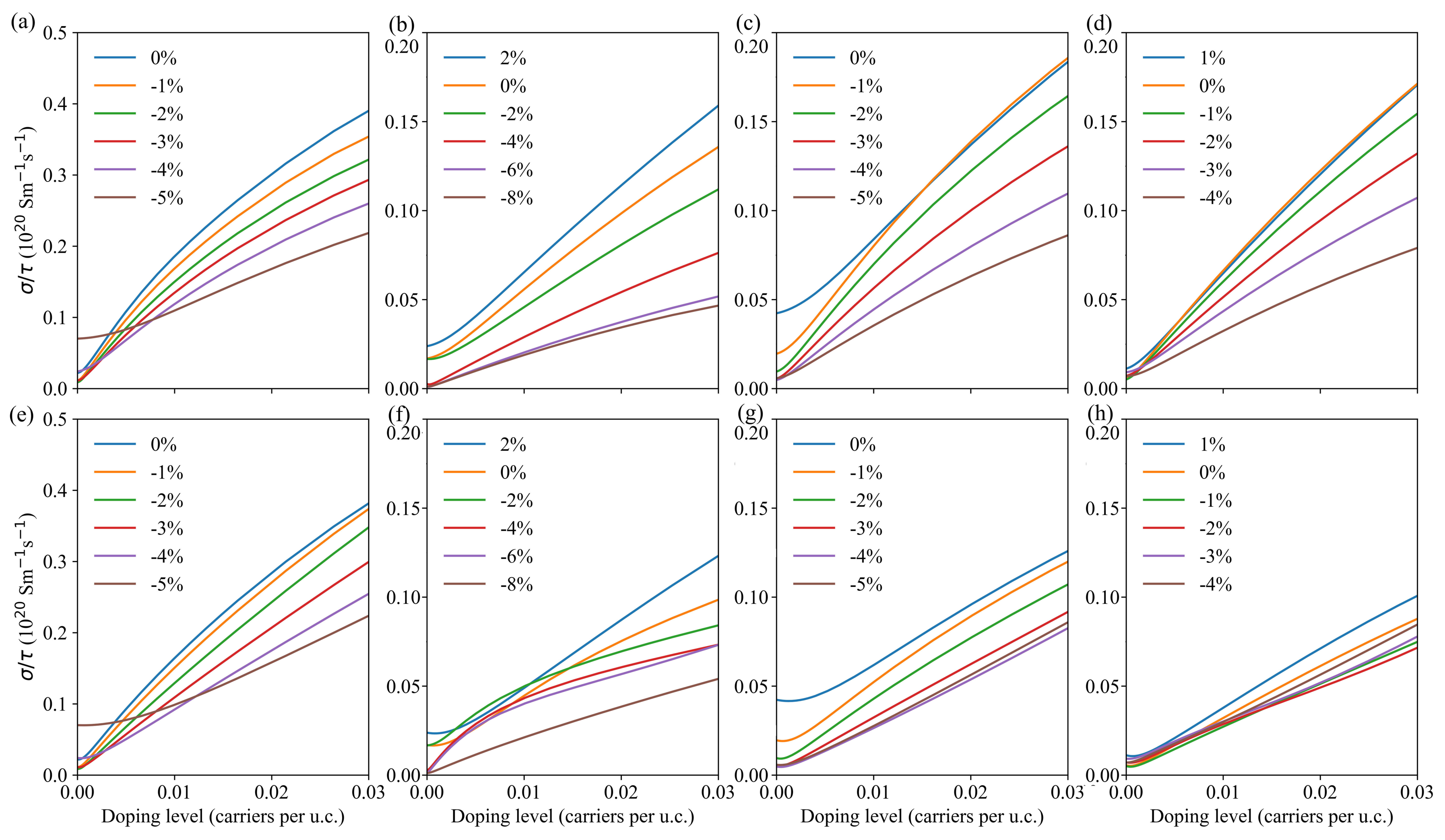
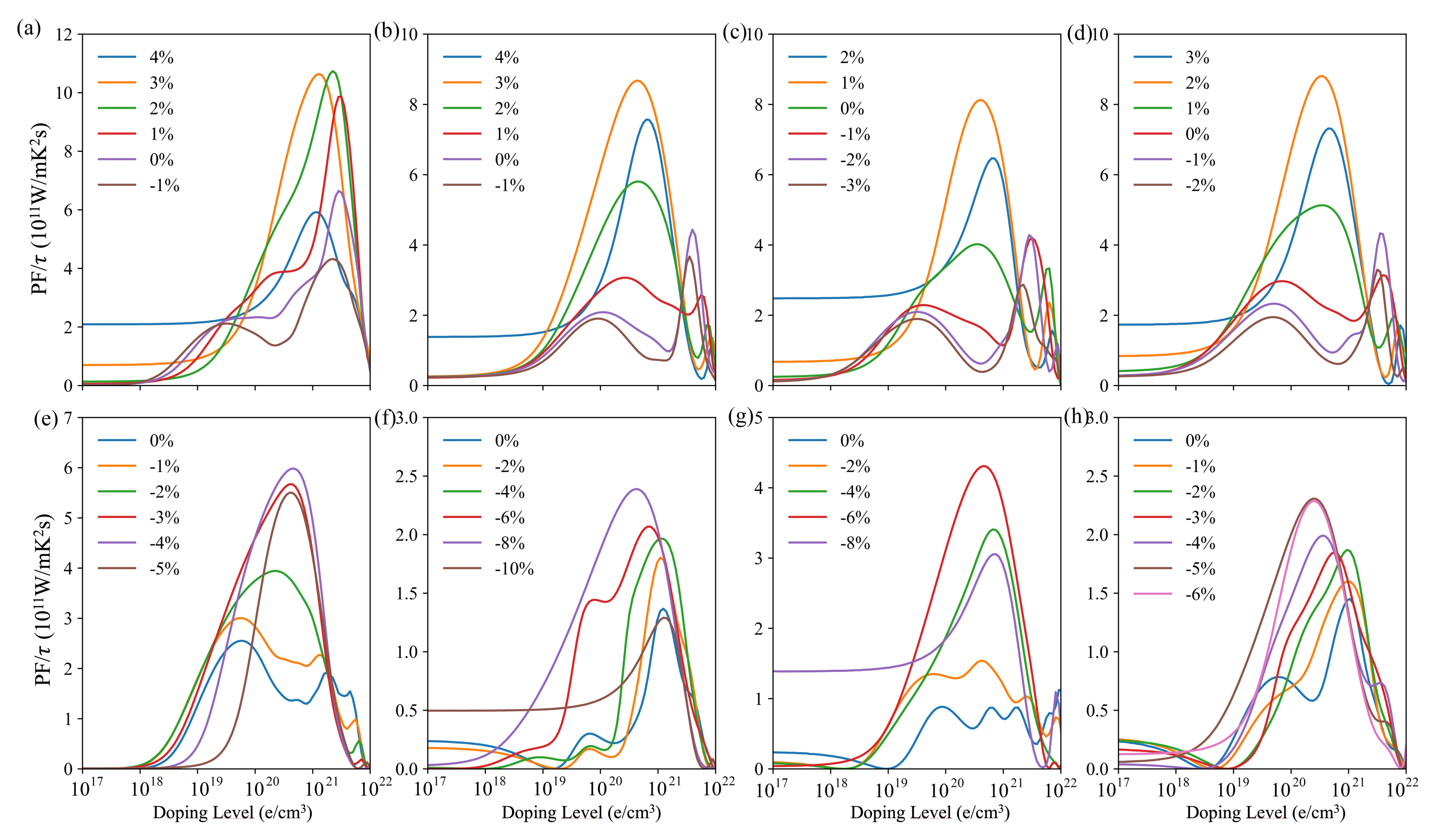
3.3. Transport Properties
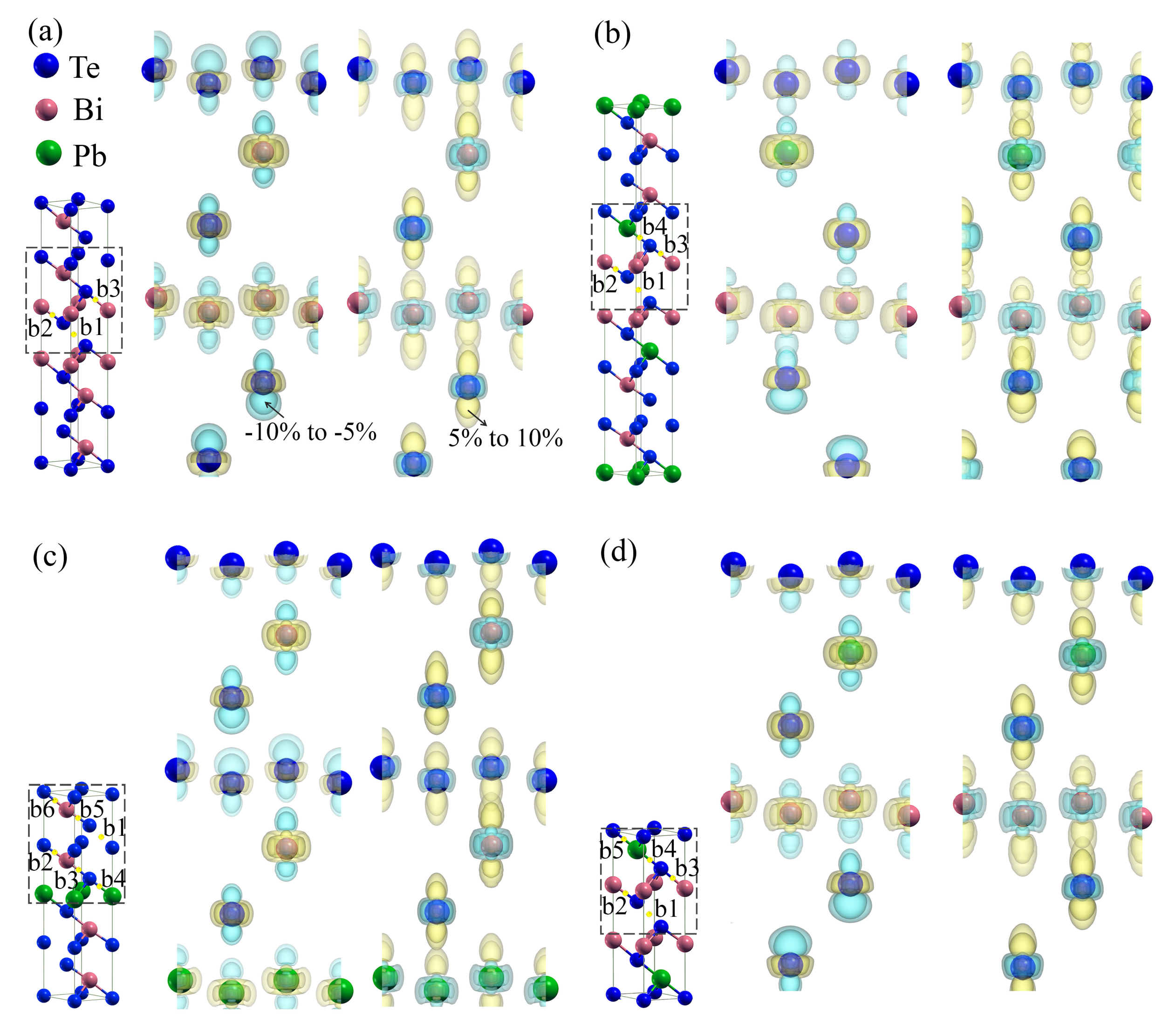
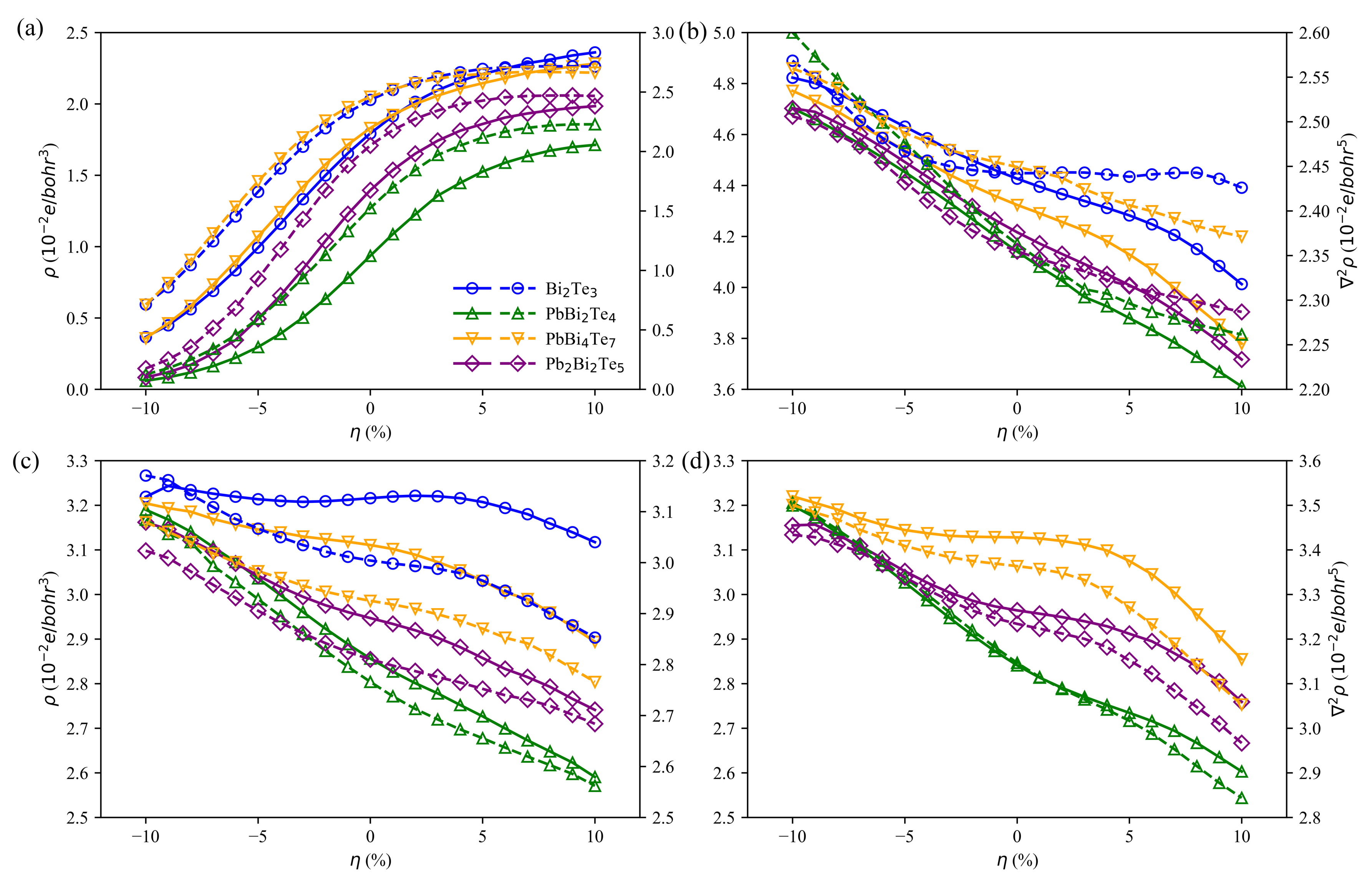
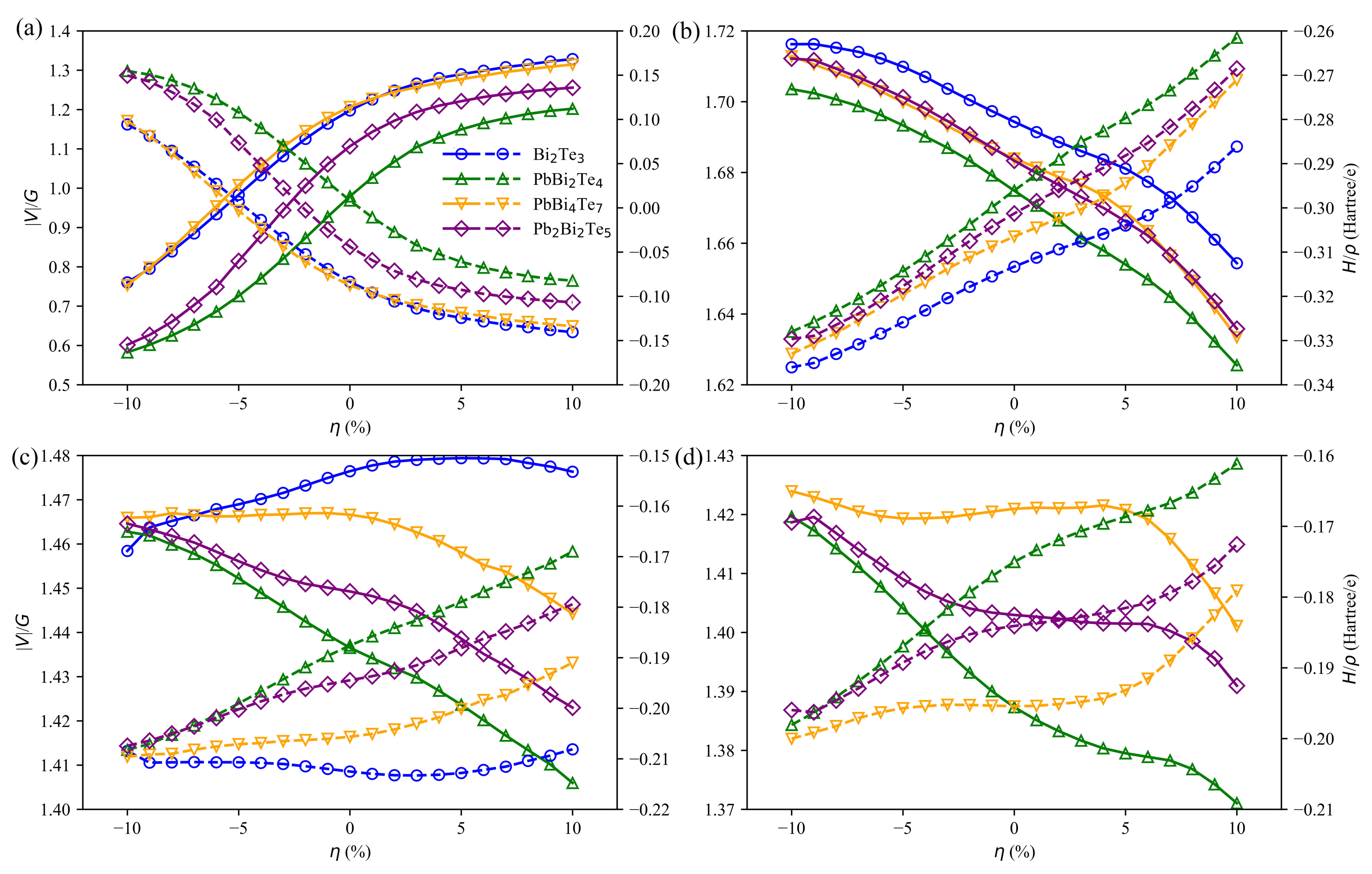
3.4. Charge and Bonding Evolution under Strains
3.5. Topological Properties of BiTe and PbBiTe under Peculiar Strains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TE | Thermoelectric |
| DFT | Density functional theory |
| QTAIM | Quantum theory of atoms in molecules |
| TEG | Thermoelectric generators |
| PBE | Perdew-Burke-Ernzerhof |
| DOS | Density of states |
| RMT | Radius of Muffin Tin |
| SOC | Spin-orbit coupling |
| CBM | Conduction band minimum |
| VBM | Valence band maximum |
References
- Slack, G.A.; Rowe, D. CRC Handbook of Thermoelectrics, 1st ed.; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar] [CrossRef]
- Balout, H.; Boulet, P.; Record, M.C. Strain-Induced Electronic Band Convergence: Effect on the Seebeck Coefficient of Mg2Si for Thermoelectric Applications. J. Mol. Mod. 2017, 23, 130. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Hossain, M.A. First-Principles Study of Elastic, Electronic, Thermodynamic, and Thermoelectric Transport Properties of TaCoSn. Results Phys. 2018, 10, 458–465. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, Y.M.; Mun, H.; Kim, J.; Park, J.; Borisevich, A.Y.; Lee, K.H.; Kim, S.W. Direct Observation of Inherent Atomic-Scale Defect Disorders Responsible for High-Performance Ti1−xHfxNiSn1−ySby Half-Heusler Thermoelectric Alloys. Adv. Mater. 2017, 29, 1702091. [Google Scholar] [CrossRef] [PubMed]
- Zeier, W.G.; Schmitt, J.; Hautier, G.; Aydemir, U.; Gibbs, Z.M.; Felser, C.; Snyder, G.J. Engineering Half-Heusler Thermoelectric Materials Using Zintl Chemistry. Nat. Rev. Mater. 2016, 1, 16032. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, Q.; Yuan, B.; Lai, X.; Yan, X.; Ren, Z. Recent Progress in Half-Heusler Thermoelectric Materials. Mater. Res. Bull. 2016, 76, 107–112. [Google Scholar] [CrossRef]
- Kuznetsova, L.; Kuznetsov, V.; Rowe, D. Thermoelectric Properties and Crystal Structure of Ternary Compounds in the Ge(Sn,Pb)Te–Bi2Te3 Systems. J. Phys. Chem. Solids 2000, 61, 1269–1274. [Google Scholar] [CrossRef]
- Sootsman, J.R.; Chung, D.Y.; Kanatzidis, M.G. New and Old Concepts in Thermoelectric Materials. Angew. Chem. Int. Ed. 2009, 48, 8616–8639. [Google Scholar] [CrossRef]
- Sharma, S.; Schwingenschlögl, U. Thermoelectric Response in Single Quintuple Layer Bi2Te3. ACS Energy Lett. 2016, 1, 875–879. [Google Scholar] [CrossRef]
- Xu, B.; Feng, T.; Li, Z.; Zhou, L.; Pantelides, S.T.; Wu, Y. Creating Zipper-Like van Der Waals Gap Discontinuity in Low-Temperature-Processed Nanostructured PbBi2nTe1+3n: Enhanced Phonon Scattering and Improved Thermoelectric Performance. Angew. Chem. Int. Ed. 2018, 57, 10938–10943. [Google Scholar] [CrossRef] [PubMed]
- Nolas, G.S.; Poon, J.; Kanatzidis, M. Recent Developments in Bulk Thermoelectric Materials. MRS Bull. 2006, 31, 199–205. [Google Scholar] [CrossRef]
- Tritt, T. Recent Trends in Thermoelectric Materials Research: Part Two; Number 69 in Semiconductors and Semimetals; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Zhang, Q.; Zhou, Z.; Dylla, M.; Agne, M.T.; Pei, Y.; Wang, L.; Tang, Y.; Liao, J.; Li, J.; Bai, S.; et al. Realizing High-Performance Thermoelectric Power Generation through Grain Boundary Engineering of Skutterudite-Based Nanocomposites. Nano Energy 2017, 41, 501–510. [Google Scholar] [CrossRef]
- Rull-Bravo, M.; Moure, A.; Fernández, J.F.; Martín-González, M. Skutterudites as Thermoelectric Material: Revisited. RSC Adv. 2015, 5, 41653–41667. [Google Scholar] [CrossRef]
- Iversen, B.B.; Palmqvist, A.E.; Cox, D.E.; Nolas, G.S.; Stucky, G.D.; Blake, N.P.; Metiu, H. Why Are Clathrates Good Candidates for Thermoelectric Materials? J. Solid State Chem. 2000, 149, 455–458. [Google Scholar] [CrossRef]
- Dolyniuk, J.A.; Owens-Baird, B.; Wang, J.; Zaikina, J.V.; Kovnir, K. Clathrate Thermoelectrics. Mater. Sci. Eng. R Rep. 2016, 108, 1–46. [Google Scholar] [CrossRef]
- Christensen, M.; Abrahamsen, A.B.; Christensen, N.B.; Juranyi, F.; Andersen, N.H.; Lefmann, K.; Andreasson, J.; Bahl, C.R.H.; Iversen, B.B. Avoided Crossing of Rattler Modes in Thermoelectric Materials. Nat. Mater. 2008, 7, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Jia, X.; Zhao, J.; Li, Y.; Liu, H.; Ma, H. Effects of Pressure on the Microstructure and Simultaneous Optimization of the Electrical and Thermal Transport Properties of Yb0.5Ba7.5Ga16Ge30. Inorg. Chem. 2018, 57, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Gascoin, F.; Ottensmann, S.; Stark, D.; Haïle, S.M.; Snyder, G.J. Zintl Phases as Thermoelectric Materials: Tuned Transport Properties of the Compounds CaxYb1−x-Zn2Sb2. Adv. Funct. Mater. 2005, 15, 1860–1864. [Google Scholar] [CrossRef]
- Shuai, J.; Mao, J.; Song, S.; Zhang, Q.; Chen, G.; Ren, Z. Recent Progress and Future Challenges on Thermoelectric Zintl Materials. Mater. Today Phys. 2017, 1, 74–95. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Chauhan, N.; Goel, S.; Singh, V.; Pulikkotil, J.; Senguttuvan, T.; Misra, D. Tuning the Carrier Concentration Using Zintl Chemistry in Mg3Sb2, and Its Implications for Thermoelectric Figure-of-Merit. Phys. Chem. Chem. Phys. 2016, 18, 6191–6200. [Google Scholar] [CrossRef]
- Toberer, E.S.; Cox, C.A.; Brown, S.R.; Ikeda, T.; May, A.F.; Kauzlarich, S.M.; Snyder, G.J. Traversing the Metal-Insulator Transition in a Zintl Phase: Rational Enhancement of Thermoelectric Efficiency in Yb14Mn1−x AlxSb11. Adv. Funct. Mater. 2008, 18, 2795–2800. [Google Scholar] [CrossRef]
- Ma, T.; Lin, C.T.; Wang, Y. The Dimensionality Effect on Phonon Localization in Graphene/Hexagonal Boron Nitride Superlattices. 2D Mater. 2020, 7, 035029. [Google Scholar] [CrossRef]
- Fu, B.; Tang, G.; Li, Y. Electron–Phonon Scattering Effect on the Lattice Thermal Conductivity of Silicon Nanostructures. Phys. Chem. Chem. Phys. 2017, 19, 28517–28526. [Google Scholar] [CrossRef]
- Diznab, M.R.; Maleki, I.; Vaez Allaei, S.M.; Xia, Y.; Naghavi, S.S. Achieving an Ultrahigh Power Factor in Sb2Te2Se Monolayers via Valence Band Convergence. ACS Appl. Mater. Inter. 2019, 11, 46688–46695. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, M.; Xiao, H.; Gong, H.; Liu, Z.; Zu, X.; Qiao, L. Optimizing the Thermoelectric Transport Properties of Bi2O2Se Monolayer via Biaxial Strain. Phys. Chem. Chem. Phys. 2019, 21, 15097–15105. [Google Scholar] [CrossRef] [PubMed]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APW+lo Program for Calculating the Properties of Solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef] [PubMed]
- Eglitis, R.I. Ab initio hybrid DFT calculations of BaTiO3, PbTiO3, SrZrO3 and PbZrO3 (111) surfaces. Appl. Surf. Sci. 2015, 358, 556–562. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Jia, R. Comparative hybrid Hartree-Fock-DFT calculations of WO2-terminated cubic WO3 as well as SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001) surfaces. Crystals 2021, 11, 455. [Google Scholar] [CrossRef]
- Bader, R. Atoms in Molecules: A Quantum Theory; Oxford University: Oxford, UK, 1990. [Google Scholar] [CrossRef]
- Madsen, G.K.; Carrete, J.; Verstraete, M.J. BoltzTraP2, a Program for Interpolating Band Structures and Calculating Semi-Classical Transport Coefficients. Comput. Phys. Commun. 2018, 231, 140–145. [Google Scholar] [CrossRef]
- Petrov, I.I.; Imamov, R.M. Electron-Diffraction Analysis of PbTe-Bi2Te3 System Phases. Sov. Phys. Crystallogr. 1970, 14, 593–596. [Google Scholar]
- Chatterjee, A.; Biswas, K. Solution-Based Synthesis of Layered Intergrowth Compounds of the Homologous PbmBi2nTe3n+m Series as Nanosheets. Angew. Chem. Int. Ed. 2015, 54, 5623–5627. [Google Scholar] [CrossRef]
- Ma, W.; Record, M.C.; Tian, J.; Boulet, P. Influence of the Stacking Sequence on Layered-Chalcogenide Properties: First Principles Investigation of Pb2Bi2Te5. Phys. Chem. Chem. Phys. 2021, 23, 11300–11313. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Hamada, I. Van Der Waals Density Functional Made Accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Pei, Y.L.; Liu, Y. Electrical and Thermal Transport Properties of Pb-Based Chalcogenides: PbTe, PbSe, and PbS. J. Alloys Compd. 2012, 514, 40–44. [Google Scholar] [CrossRef]
- Francombe, M.H. Structure-Cell Data and Expansion Coefficients of Bismuth Telluride. Brit. J. Appl. Phys. 1958, 9, 415–417. [Google Scholar] [CrossRef]
- Shelimova, L.E.; Karpinskii, O.G.; Konstantinov, P.P.; Avilov, E.S.; Kretova, M.A.; Zemskov, V.S. Crystal Structures and Thermoelectric Properties of Layered Compounds in the ATe–Bi2Te3 (A = Ge, Sn, Pb) Systems. Inorg. Mater. 2004, 40, 451–460. [Google Scholar] [CrossRef]
- Yonezawa, S.; Muraoka, Y.; Ueda, Y.; Hiroi, Z. Epitaxial strain effects on the metal-insulator transition in V2O3 thin films. Solid State Comm. 2004, 129, 245–248. [Google Scholar] [CrossRef]
- Inamoto, T.; Takashiri, M. Experimental and First-Principles Study of the Electronic Transport Properties of Strained Bi2Te3 Thin Films on a Flexible Substrate. J. Appl. Phys. 2016, 120, 125105. [Google Scholar] [CrossRef]
- Shalímova, K. Fisica De Los Semiconductors; Energoatomizdat: Madrid, Spain, 1982. [Google Scholar]
- Korotaev, E.V.; Syrokvashin, M.M.; Filatova, I.Y.; Pelmenev, K.G.; Zvereva, V.V.; Peregudova, N.N. Seebeck Coefficient of Cation-Substituted Disulfides CuCr1−x-FexS2 and Cu1−x-FexCrS2. J. Electron. Mater. 2018, 47, 3392–3397. [Google Scholar] [CrossRef]
- Hinsche, N.F.; Yavorsky, B.Y.; Mertig, I.; Zahn, P. Influence of strain on anisotropic thermoelectric transport in Bi2Te3 and Sb2Te3. Phys. Rev. B 2011, 84, 165214. [Google Scholar] [CrossRef]
- Hinsche, N.F.; Mertig, I.; Zahn, P. Effect of strain on the thermoelectric properties of silicon: An ab initio study. J. Phys. Condens. Matter 2011, 23, 295502. [Google Scholar] [CrossRef][Green Version]
- Scheidemantel, T.J.; Ambrosch-Draxl, C.; Thonhauser, T.; Badding, J.V.; Sofo, J.O. Transport coefficients from first-principles calculations. Phys. Rev. B 2003, 68, 125210. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essén, H. The Characterization of Atomic Interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Dinda, S.; Samuelson, A.G. The Nature of Bond Critical Points in Dinuclear Copper(I) Complexes. Chem. Eur J. 2012, 18, 3032–3042. [Google Scholar] [CrossRef]
- Lawal, A.; Shaari, A.; Ahmed, R.; Jarkoni, N. First-Principles Investigations of Electron-Hole Inclusion Effects on Optoelectronic Properties of Bi2Te3, a Topological Insulator for Broadband Photodetector. Phys. B 2017, 520, 69–75. [Google Scholar] [CrossRef]
- Chen, Y.L.; Analytis, J.G.; Chu, J.H.; Liu, Z.K.; Mo, S.K.; Qi, X.L.; Zhang, H.J.; Lu, D.H.; Dai, X.; Fang, Z.; et al. Experimental Realization of a Three-Dimensional Topological Insulator, Bi2Te3. Science 2009, 325, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, K.; Miyahara, H.; Ye, M.; Eremeev, S.V.; Koroteev, Y.M.; Krasovskii, E.E.; Chulkov, E.V.; Hiramoto, S.; Moriyoshi, C.; Kuroiwa, Y.; et al. Experimental Verification of PbBi2Te4 as a 3D Topological Insulator. Phys. Rev. Lett. 2012, 108, 206803. [Google Scholar] [CrossRef]
- Vergniory, M.; Menshchikova, T.; Eremeev, S.; Chulkov, E. Bulk and Surface Electronic Structure of SnBi4Te7 Topological Insulator. Appl. Surf. Sci. 2013, 267, 146–149. [Google Scholar] [CrossRef]
- Moore, J.E.; Balents, L. Topological Invariants of Time-Reversal-Invariant Band Structures. Phys. Rev. B 2007, 75, 121306. [Google Scholar] [CrossRef]
- Roy, R. Topological Phases and the Quantum Spin Hall Effect in Three Dimensions. Phys. Rev. B 2009, 79, 195322. [Google Scholar] [CrossRef]
- Fu, L.; Kane, C.L.; Mele, E.J. Topological Insulators in Three Dimensions. Phys. Rev. Lett. 2007, 98, 106803. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LDA | PBE | Rev-vdW-DF2 | Exp. | ||
|---|---|---|---|---|---|
| PbTe | a | 6.38 | 6.57 | - | 6.46 [39] |
| BiTe | a | 4.36 | 4.46 | 4.35 | 4.38 [40] |
| c | 30.00 | 30.75 | 30.21 | 30.49 [40] | |
| Te1 | 0 | 0 | 0 | 0 [40] | |
| Te2 | 0.2029 | 0.2116 | 0.2086 | 0.212 [40] | |
| Bi | 0.3997 | 0.3991 | 0.4003 | 0.4 [40] | |
| PbBiTe | a | 4.45 | 4.55 | 4.52 | 4.34 [41] |
| c | 41.87 | 43.21 | 42.82 | 41.77 [41] | |
| Pb | 0 | 0 | 0 | ||
| Te1 | 0.1336 | 0.1304 | 01322 | ||
| Te2 | 0.2873 | 0.2881 | 0.2876 | ||
| Bi | 0.4271 | 0.4252 | 0.4263 | ||
| PbBiTe | a | 4.39 | 4.50 | 4.47 | 4.42 [7] |
| c | 22.88 | 23.30 | 23.24 | 24.04 [7] | |
| Te1 | 0 | 0 | 0 | ||
| Bi1 | 0.0849 | 0.0842 | 0.0846 | ||
| Te2 | 0.1582 | 0.1561 | 0.1568 | ||
| Te3 | 0.2602 | 0.2624 | 0.2616 | ||
| Bi2 | 0.3331 | 0.3337 | 0.3334 | ||
| Te4 | 0.4183 | 0.4182 | 0.4183 | ||
| Pb | 0.5 | 0.5 | 0.5 | ||
| PbBiTe | a | 4.43 | 4.55 | 4.50 | 4.42 [33] |
| c | 17.32 | 17.41 | 17.68 | 17.86 [33] | |
| Te1 | 0 | 0 | 0 | ||
| Pb | 0.1099 | 0.1097 | 0.1100 | ||
| Te2 | 0.2192 | 0.2194 | 0.2189 | ||
| Bi | 0.3326 | 0.3349 | 0.3323 | ||
| Te3 | 0.4299 | 0.4294 | 0.4261 |
| BiTe | −1 | −1 | −1 | −1 | −1 | −1 | −1 | −1 | 0 (0,0,0) |
| PbBiTe | −1 | −1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 (0,0.5,0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Record, M.-C.; Tian, J.; Boulet, P. Strain Effects on the Electronic and Thermoelectric Properties of n(PbTe)-m(Bi2Te3) System Compounds. Materials 2021, 14, 4086. https://doi.org/10.3390/ma14154086
Ma W, Record M-C, Tian J, Boulet P. Strain Effects on the Electronic and Thermoelectric Properties of n(PbTe)-m(Bi2Te3) System Compounds. Materials. 2021; 14(15):4086. https://doi.org/10.3390/ma14154086
Chicago/Turabian StyleMa, Weiliang, Marie-Christine Record, Jing Tian, and Pascal Boulet. 2021. "Strain Effects on the Electronic and Thermoelectric Properties of n(PbTe)-m(Bi2Te3) System Compounds" Materials 14, no. 15: 4086. https://doi.org/10.3390/ma14154086
APA StyleMa, W., Record, M.-C., Tian, J., & Boulet, P. (2021). Strain Effects on the Electronic and Thermoelectric Properties of n(PbTe)-m(Bi2Te3) System Compounds. Materials, 14(15), 4086. https://doi.org/10.3390/ma14154086