
1T-MoS2 Coordinated Bimetal Atoms as Active Centers to Facilitate Hydrogen Generation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Density Functional Theory Calculations
2.2. Formation Energy
2.3. Reaction Free Energy
3. Results
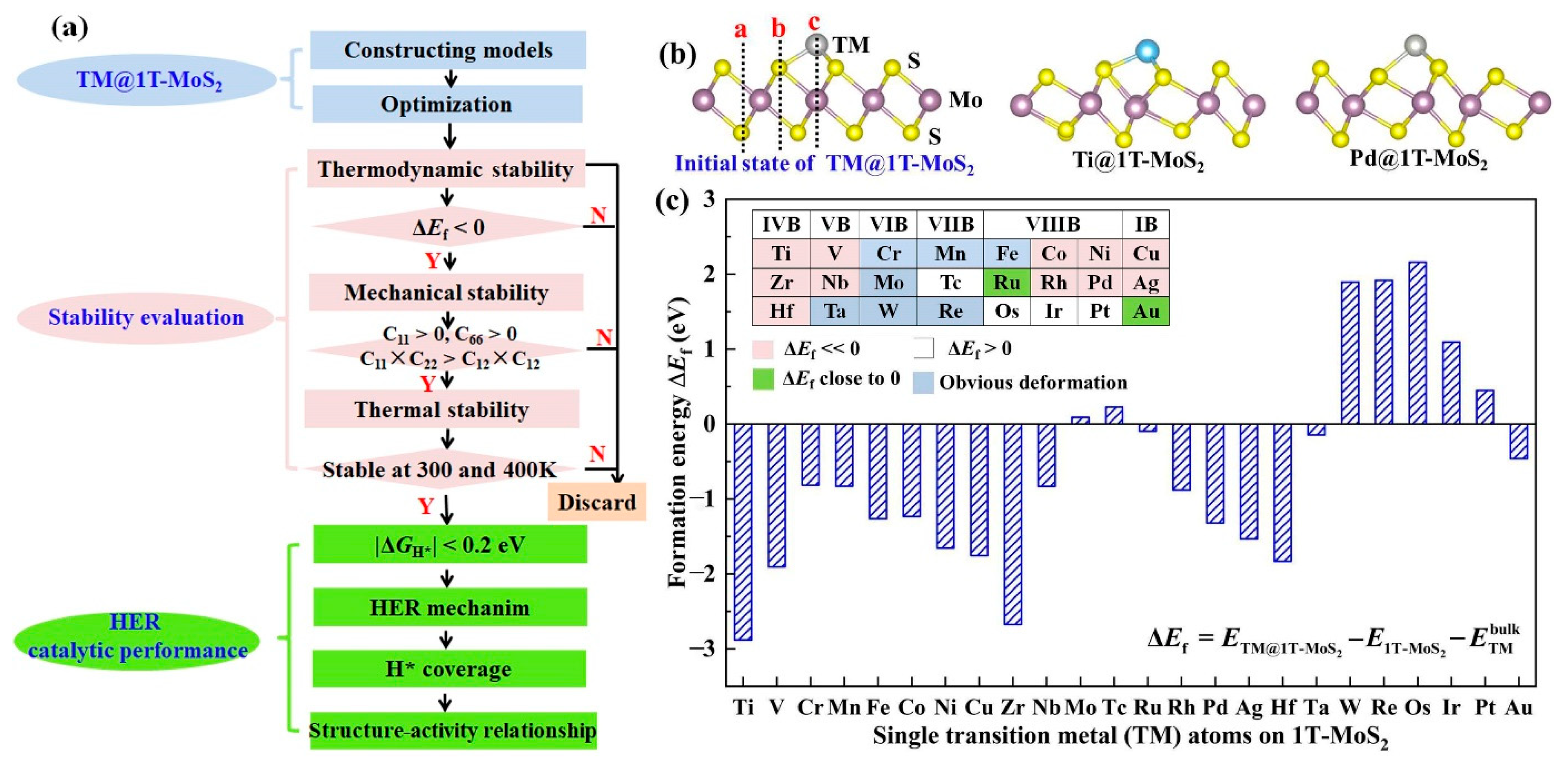
3.1. Structural Stability of Single Metal Atoms Mediated 1T-MoS2
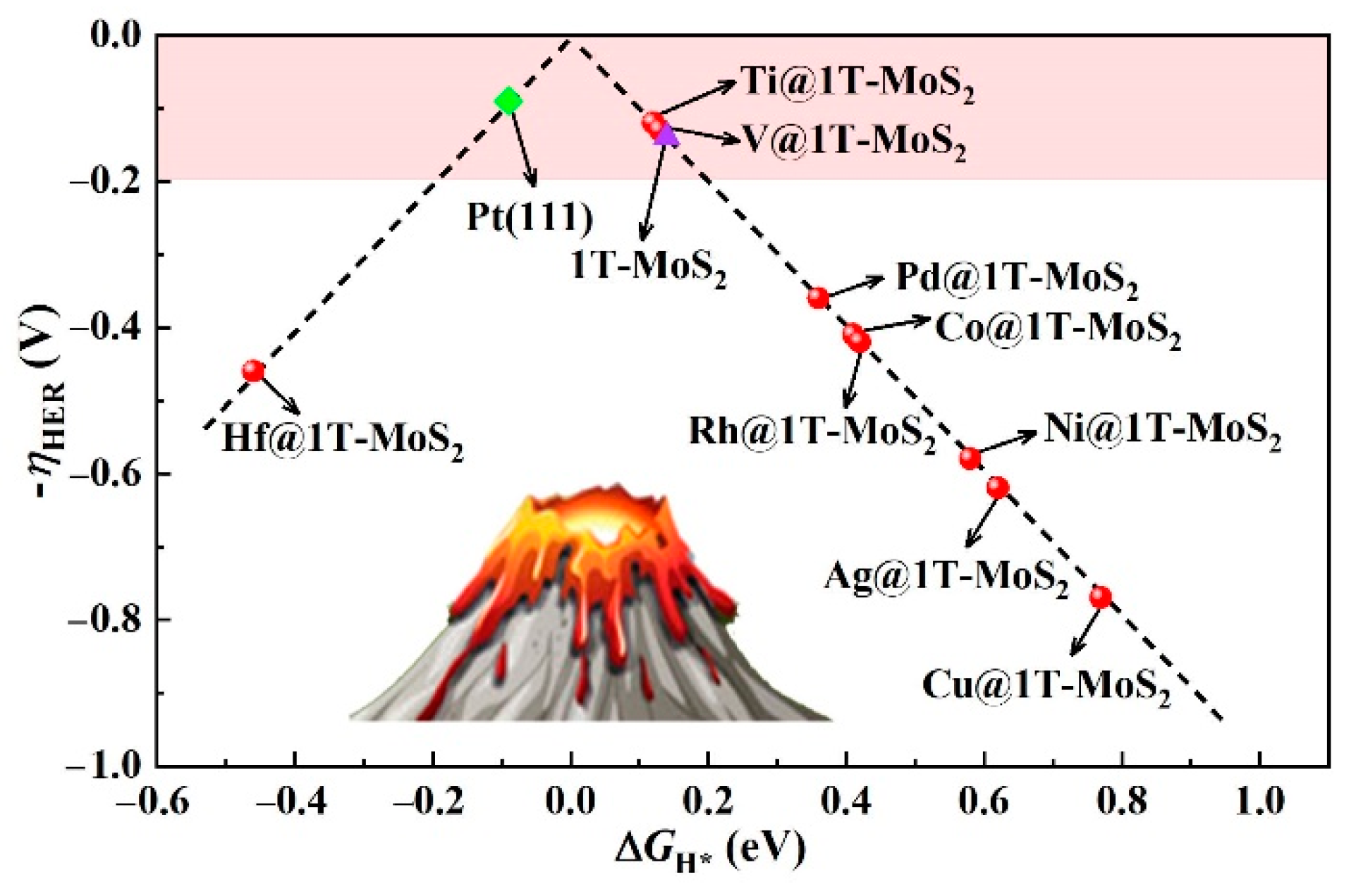
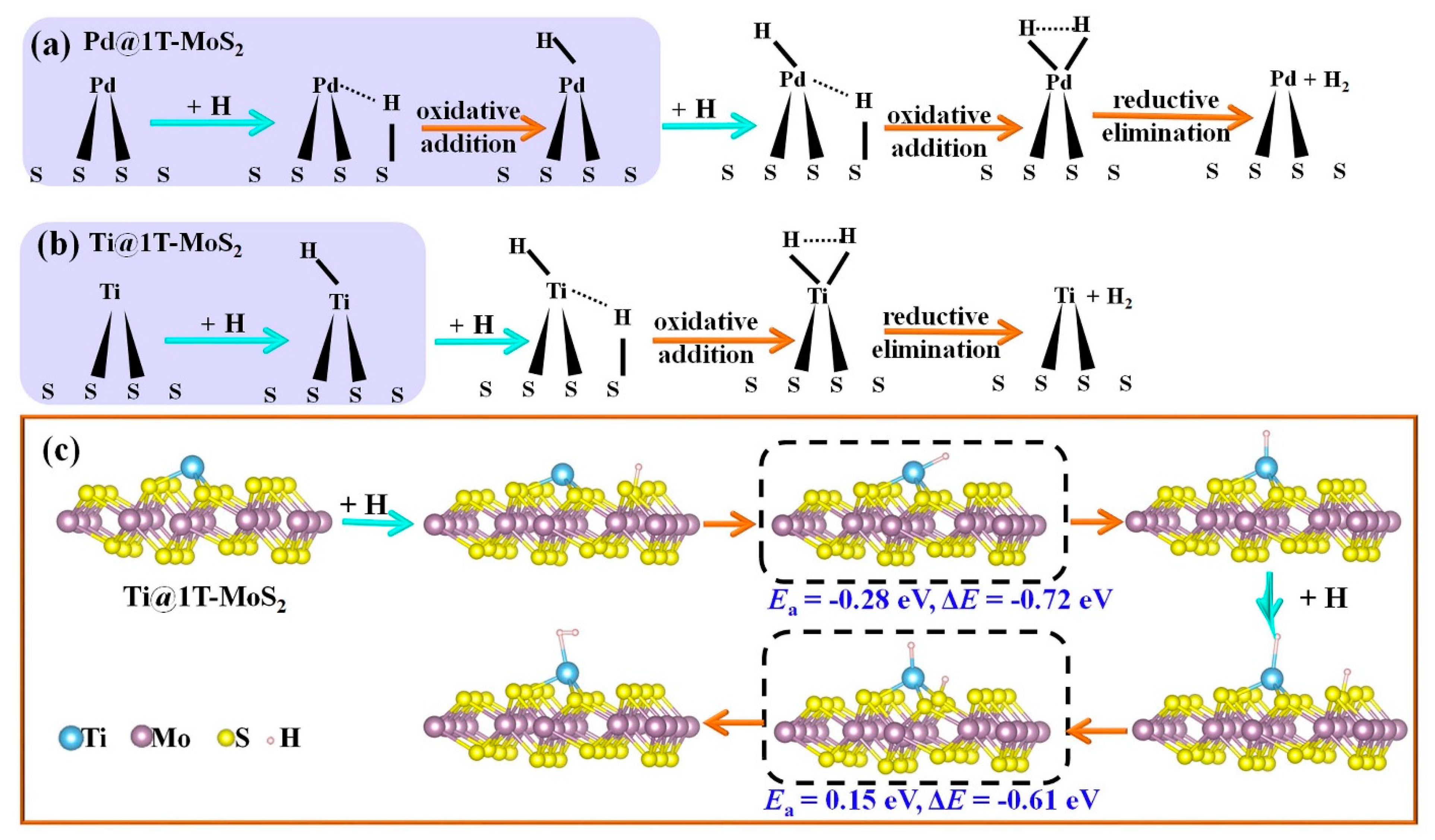
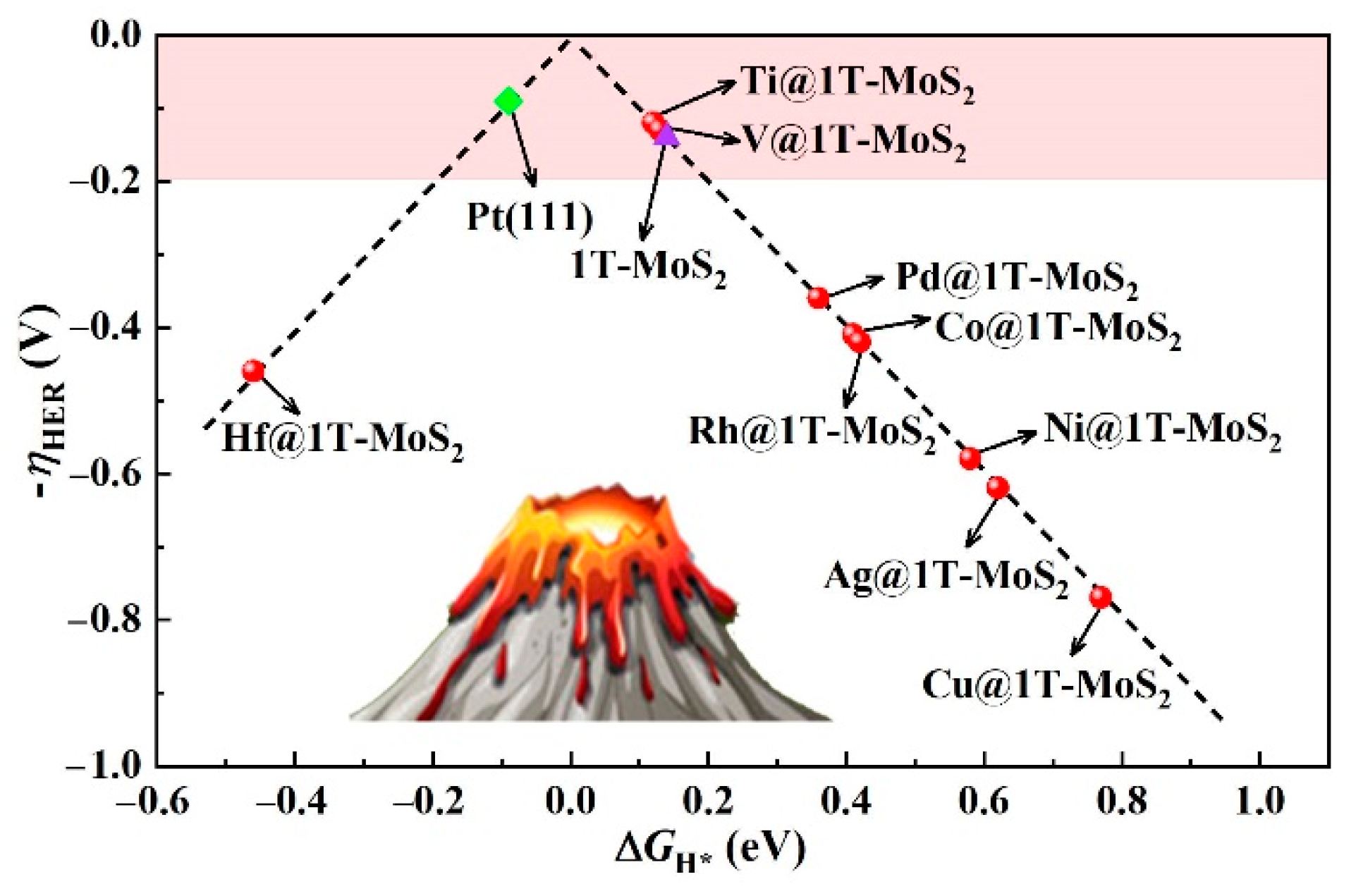
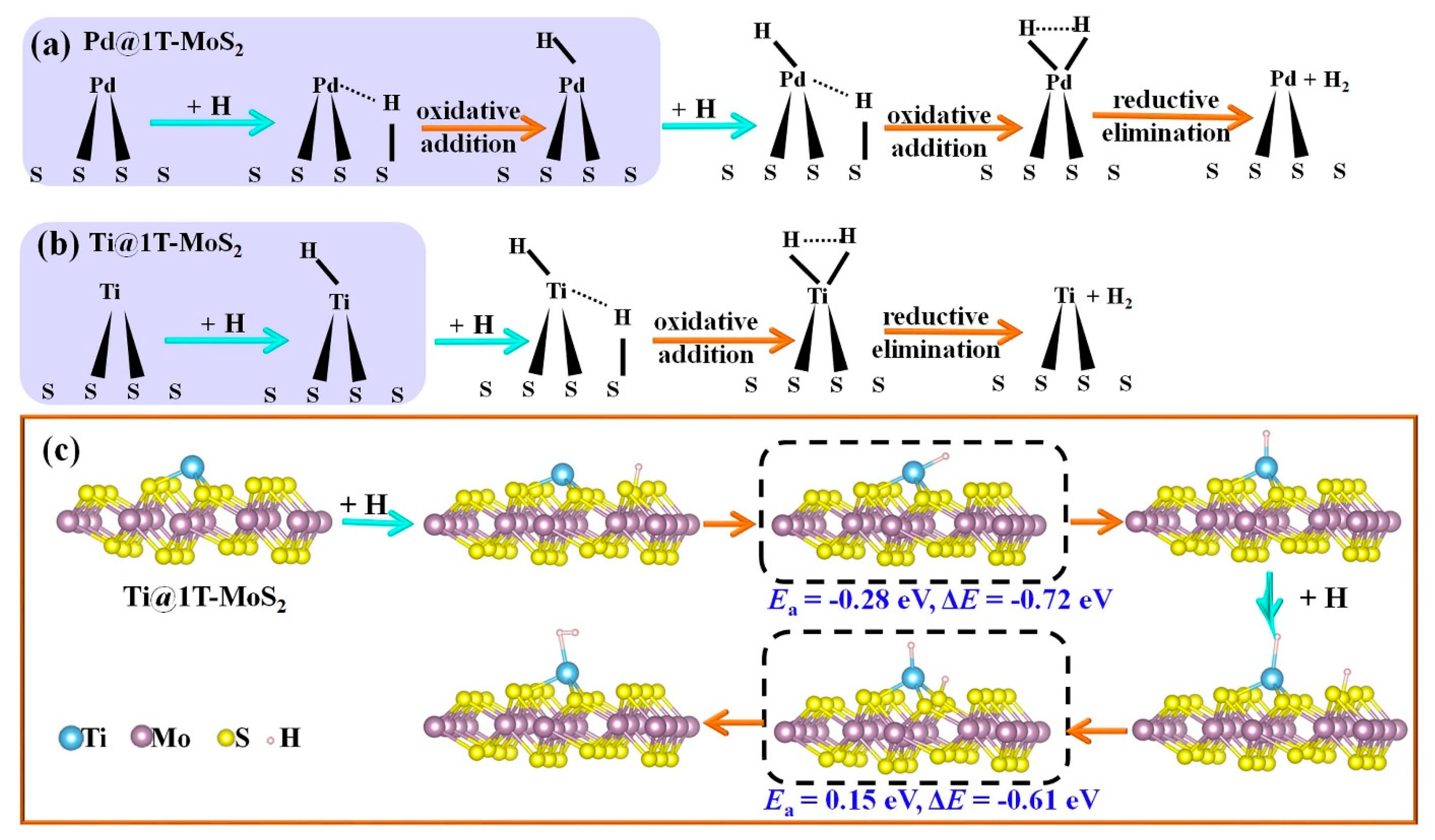
3.2. HER Activity and Kinetics of TM@1T-MoS2
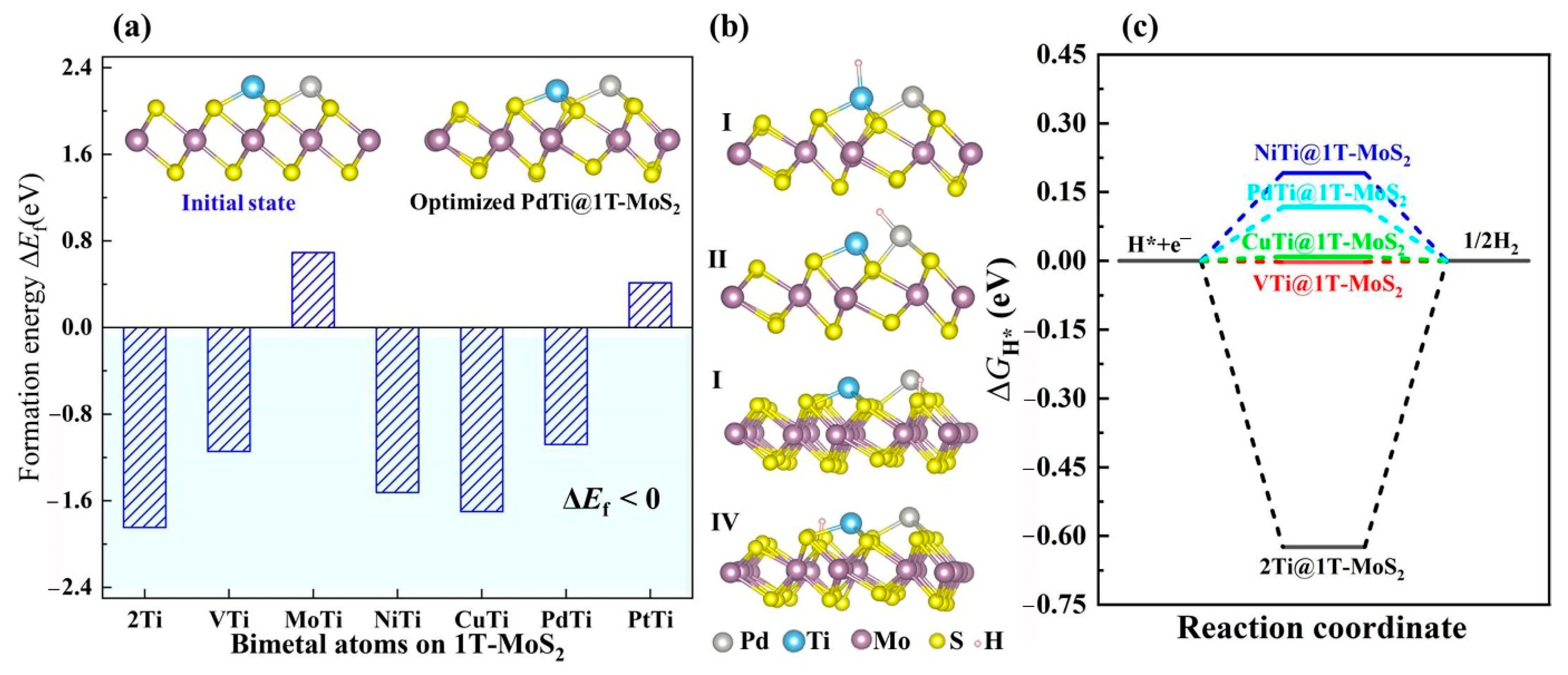
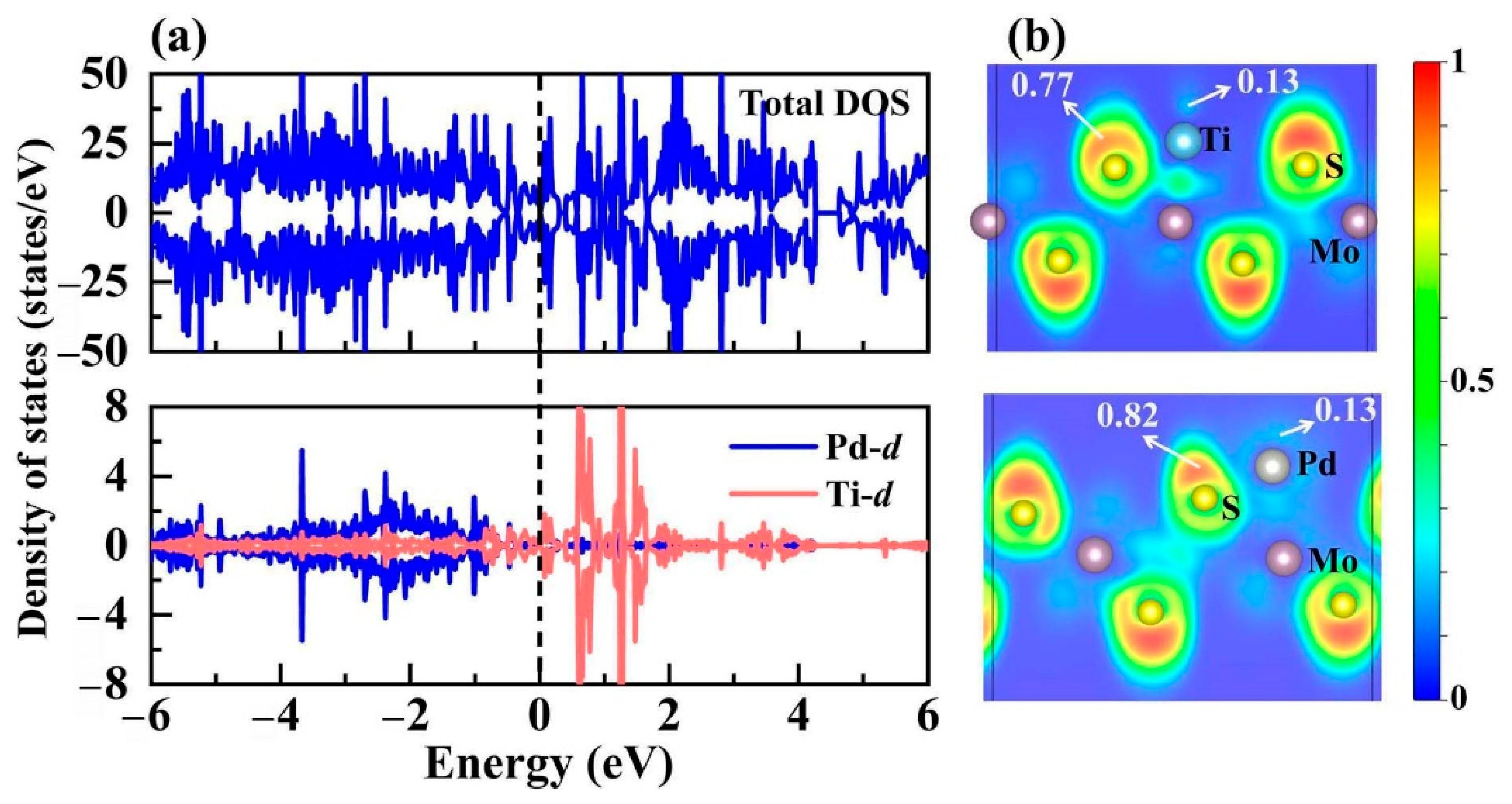
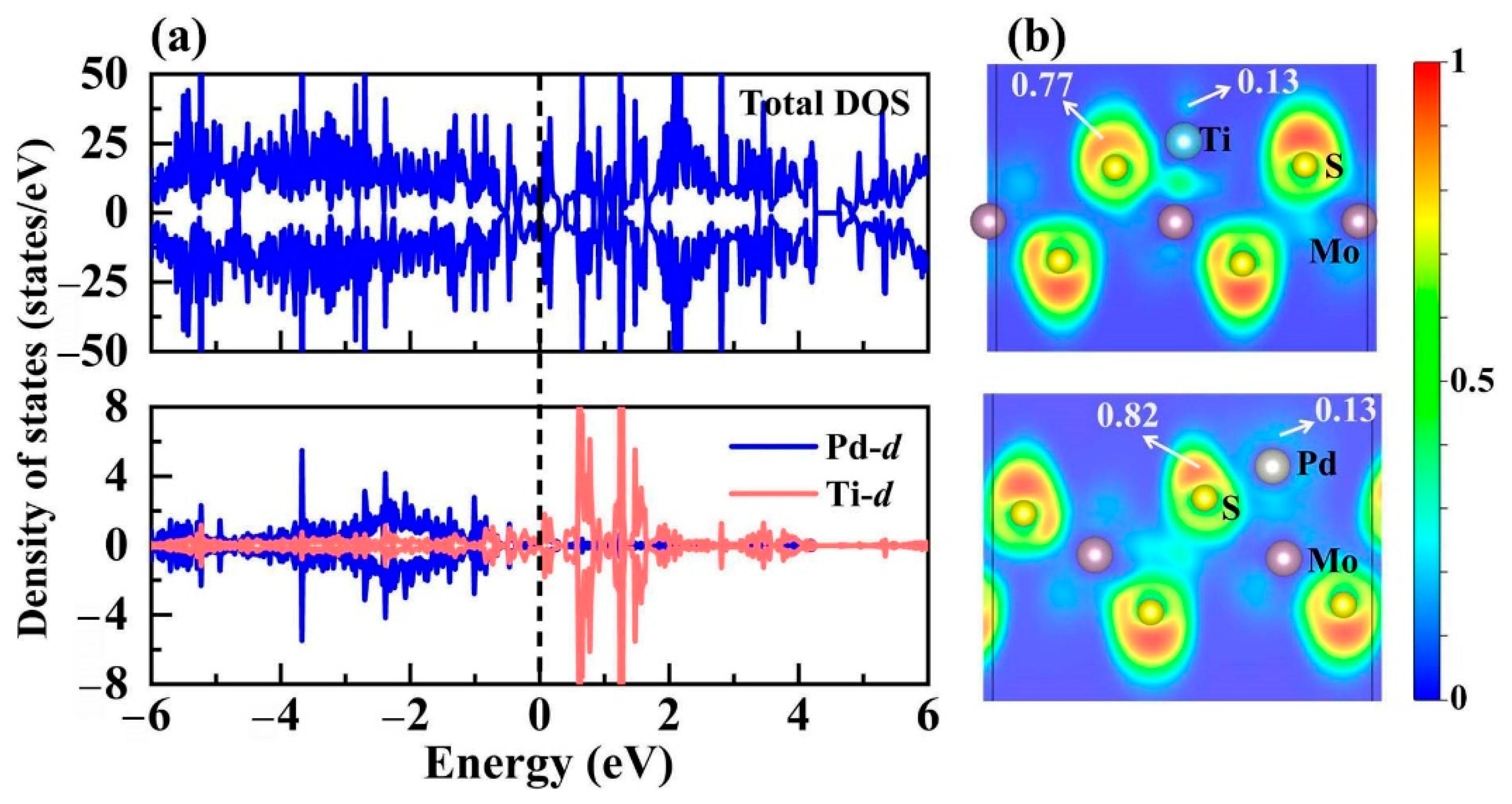
3.3. Bimetal Active Sites on TMTi@1T-MoS2
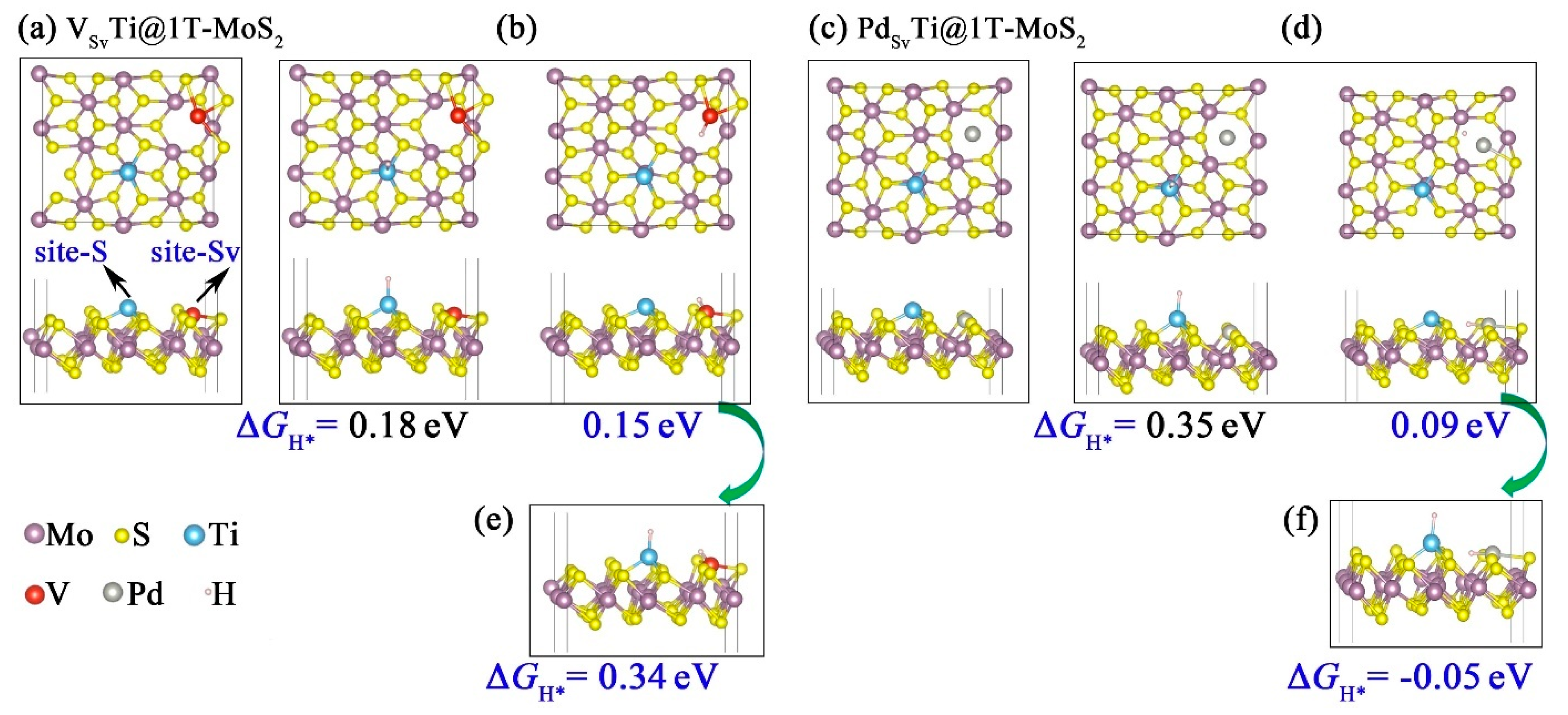
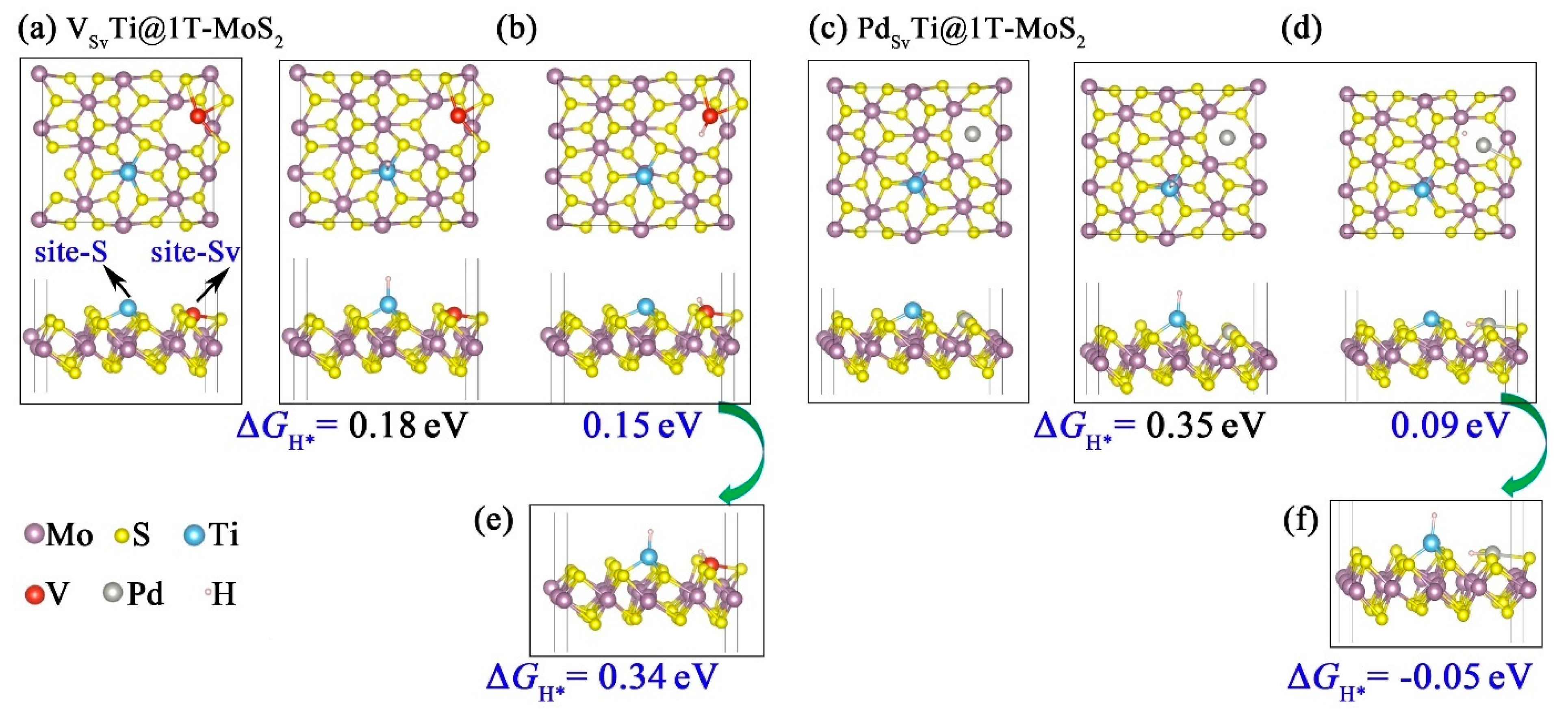
3.4. Sulfur Vacancy-Mediated Hydrogen Adsorption on TMTi@1T-MoS2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Q.; Feng, X. Support and interface effects in water-splitting electrocatalysts. Adv. Mater. 2019, 31, 1808167. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.A.; Puente Santiago, A.R.; Hong, Y.; Zhang, N.; Cano, M.; Rodriguez-Castellon, E.; Echegoyen, L.; Sreenivasan, S.T.; Noveron, J.C. Tuning of trifunctional NiCu bimetallic nanoparticles confined in a porous carbon network with surface composition and local structural distortions for the electrocatalytic oxygen reduction, oxygen and hydrogen evolution reactions. J. Am. Chem. Soc. 2020, 142, 14688–14701. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Luo, M.; Yang, Y.; Li, Y.; Guo, S. Advanced multifunctional electrocatalysts for energy conversion. ACS Energy Lett. 2019, 4, 1672–1680. [Google Scholar] [CrossRef]
- Hu, C.; Dai, L. Multifunctional carbon-based metal-free electrocatalysts for simultaneous oxygen reduction, oxygen evolution, and hydrogen evolution. Adv. Mater. 2017, 29, 1604942. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Gong, Y.; Lin, J.; Li, B.; He, Y.; Pantelides, S.T.; Zhou, W.; Vajtai, R.; Ajayan, P.M. Defects engineered monolayer MoS2 for improved hydrogen evolution reaction. Nano Lett. 2016, 16, 1097–1103. [Google Scholar] [CrossRef]
- Ilyas, T.; Raziq, F.; Ali, S.; Zada, A.; Ilyas, N.; Shaha, R.; Wang, Y.; Qiao, L. Facile synthesis of MoS2/Cu as trifunctional catalyst for electrochemical overall water splitting and photocatalytic CO2 conversion. Mater. Des. 2021, 204, 109674. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, B.; Peng, Q.; Zhou, J.; Sun, Z. Mo2B2 MBene-supported single-atom catalysts as bifunctional HER/OER and OER/ORR electrocatalysts. J. Mater. Chem. A 2021, 9, 433–441. [Google Scholar] [CrossRef]
- Ling, T.; Zhang, T.; Ge, B.; Han, L.; Zheng, L.; Lin, F.; Xu, Z.; Hu, W.-B.; Du, X.-W.; Davey, K.; et al. Well-dispersed nickel- and zinc-tailored electronic structure of a transition metal oxide for highly active alkaline hydrogen evolution reaction. Adv. Mater. 2019, 31, 1807771. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, B. Recent advances in transition metal phosphide nanomaterials: Synthesis and applications in hydrogen evolution reaction. Chem. Soc. Rev. 2016, 45, 1529–1541. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, Z.; Liu, Y.; Xia, M.; Wang, R.; Li, N. Heterogeneous nanostructure based on 1T-phase MoS2 for enhanced electrocatalytic hydrogen evolution. ACS Appl. Mater. Interfaces 2017, 9, 25291–25297. [Google Scholar] [CrossRef]
- Tang, Q.; Jiang, D.-E. Mechanism of hydrogen evolution reaction on 1T-MoS2 from first principles. ACS Catal. 2016, 6, 4953–4961. [Google Scholar] [CrossRef]
- Voiry, D.; Salehi, M.; Silva, R.; Fujita, T.; Chen, M.; Asefa, T.; Shenoy, V.B.; Eda, G.; Chhowalla, M. Conducting MoS2 nanosheets as catalysts for hydrogen evolution reaction. Nano Lett. 2013, 13, 6222–6227. [Google Scholar] [CrossRef]
- Lukowski, M.A.; Daniel, A.S.; Meng, F.; Forticaux, A.; Li, L.; Jin, S. Enhanced hydrogen evolution catalysis from chemically exfoliated metallic MoS2 nanosheets. J. Am. Chem. Soc. 2013, 135, 10274–10277. [Google Scholar] [CrossRef] [PubMed]
- Pattengale, B.; Huang, Y.; Yan, X.; Yang, S.; Younan, S.; Hu, W.; Li, Z.; Lee, S.; Pan, X.; Gu, J.; et al. Dynamic evolution and reversibility of single-atom Ni(II) active site in 1T-MoS2 electrocatalysts for hydrogen evolution. Nat. Commun. 2020, 11, 4114. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Cai, J.; Wang, G. Two-dimensional MoS2 for hydrogen evolution reaction catalysis: The electronic structure regulation. Nano Res. 2021, 14, 1985–2002. [Google Scholar] [CrossRef]
- Vinnik, D.A.; Kokovkin, V.V.; Volchek, V.V.; Zhivulin, V.E.; Abramov, P.A.; Cherkasova, N.A.; Sun, Z.; Sayyed, M.I.; Tishkevich, D.I.; Trukhanov, A.V. Electrocatalytic activity of various hexagonal ferrites in OER process. Mater. Chem. Phys. 2021, 270, 124818. [Google Scholar] [CrossRef]
- Zdorovets, M.; Kozlovskiy, A.; Tishkevich, D.; Zubar, T.; Trukhanov, A. The effect of doping of TiO2 thin films with low-energy O2+ ions on increasing the efficiency of hydrogen evolution in photocatalytic reactions of water splitting. J. Mater. Sci. Mater. Electron. 2020, 31, 21142–21153. [Google Scholar] [CrossRef]
- Tishkevich, D.I.; Vorobjova, A.I.; Vinnik, D.A. Template assisted Ni nanowires fabrication. Mater. Sci. Forum 2019, 946, 235–241. [Google Scholar] [CrossRef]
- Peng, Q.; Zhou, J.; Chen, J.; Zhang, T.; Sun, Z. Cu single atoms on Ti2CO2 as a highly efficient oxygen reduction catalyst in a proton exchange membrane fuel cell. J. Mater. Chem. A 2019, 7, 26062–26070. [Google Scholar] [CrossRef]
- Huang, Y.; Sun, Y.; Zheng, X.; Aoki, T.; Pattengale, B.; Huang, J.; He, X.; Bian, W.; Younan, S.; Williams, N.; et al. Atomically engineering activation sites onto metallic 1T-MoS2 catalysts for enhanced electrochemical hydrogen evolution. Nat. Commun. 2019, 10, 982. [Google Scholar] [CrossRef]
- Qi, K.; Cui, X.; Gu, L.; Yu, S.; Fan, X.; Luo, M.; Xu, S.; Li, N.; Zheng, L.; Zhang, Q.; et al. Single-atom cobalt array bound to distorted 1T MoS2 with ensemble effect for hydrogen evolution catalysis. Nat. Commun. 2019, 10, 5231. [Google Scholar] [CrossRef]
- Lau, T.H.M.; Wu, S.; Kato, R.; Wu, T.-S.; Kulhavý, J.; Mo, J.; Zheng, J.; Foord, J.S.; Soo, Y.-L.; Suenaga, K.; et al. Engineering monolayer 1T-MoS2 into a bifunctional electrocatalyst via sonochemical doping of isolated transition metal atoms. ACS Catal. 2019, 9, 7527–7534. [Google Scholar] [CrossRef]
- Chen, B.W.J.; Xu, L.; Mavrikakis, M. Computational methods in heterogeneous catalysis. Chem. Rev. 2021, 121, 1007–1048. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Wang, G.J.; Peng, L.Y.; Li, K.Q.; Zhu, L.G.; Zhou, J.; Miao, N.H.; Sun, Z.M. ALKEMIE: An intelligent computational platform for accelerating materials discovery and design. Comput. Mater. Sci. 2021, 186, 11. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Cheng, D.; Cao, D.; Zeng, X.C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. [Google Scholar] [CrossRef]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar] [CrossRef]
- Wang, V.; Geng, W.T. Lattice defects and the mechanical anisotropy of borophene. J. Phys. Chem. C 2017, 121, 10224–10232. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Han, L.; Lian, J.; Wen, X.; Liu, S.; Chen, Z.; Koratkar, N.; De, S. Mechanical degradation of graphene by epoxidation: Insights from first-principles calculations. Phys. Chem. Chem. Phys. 2015, 17, 19484–19490. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; De, S. Outstanding mechanical properties of monolayer MoS2 and its application in elastic energy storage. Phys. Chem. Chem. Phys. 2013, 15, 19427–19437. [Google Scholar] [CrossRef]
- Maździarz, M. Comment on ‘The computational 2D materials database: High-throughput modeling and discovery of atomically thin crystals’. 2D Mater. 2019, 6, 048001. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Shang, C.; Zhou, S.; Zhao, J. MBenes: Emerging 2D materials as efficient electrocatalysts for the nitrogen reduction reaction. Nanoscale Horiz. 2020, 5, 1106–1115. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, T.; Pohl, D.; Rellinghaus, B.; Dong, R.; Liu, S.; Zhuang, X.; Feng, X. Interface engineering of MoS2/Ni3S2 heterostructures for highly enhanced electrochemical overall-water-splitting activity. Angew. Chem. Int. Ed. 2016, 55, 6702–6707. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Wang, L.; Zhang, Z.; Li, Y. Activating the MoS2 basal planes for electrocatalytic hydrogen evolution by 2H/1T′ structural interfaces. ACS Appl. Mater. Interfaces 2019, 11, 42014–42020. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, J.; Sun, Z. Novel 2D transition-metal carbides: Ultrahigh performance electrocatalysts for overall water splitting and oxygen reduction. Adv. Funct. Mater. 2020, 30, 2000570. [Google Scholar] [CrossRef]
- Henkelman, G. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Shui, J.; Liu, X.; Liu, Q.; Li, Y.; Shang, J.; Zheng, L.; Yu, R. Single-atom to single-atom grafting of Pt1 onto Fe-N4 center: Pt1@Fe-N-C multifunctional electrocatalyst with significantly enhanced properties. Adv. Energy Mater. 2018, 8, 1701345. [Google Scholar] [CrossRef]
- Chao, T.; Luo, X.; Chen, W.; Jiang, B.; Ge, J.; Lin, Y.; Wu, G.; Wang, X.; Hu, Y.; Zhuang, Z.; et al. Atomically dispersed copper–platinum dual sites alloyed with palladium nanorings catalyze the hydrogen evolution reaction. Angew. Chem. Int. Ed. 2017, 129, 16263–16267. [Google Scholar] [CrossRef]
- Pető, J.; Ollár, T.; Vancsó, P.; Popov, Z.I.; Magda, G.Z.; Dobrik, G.; Hwang, C.; Sorokin, P.B.; Tapasztó, L. Spontaneous doping of the basal plane of MoS2 single layers through oxygen substitution under ambient conditions. Nat. Chem. 2018, 10, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Zhu, C.; Fu, S.; Shi, Q.; Du, D.; Lin, Y. Single-Atom Electrocatalysts. Angew. Chem. Int. Ed. 2017, 56, 13944–13960. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Q.; Qi, X.; Gong, X.; Chen, Y. 1T-MoS2 Coordinated Bimetal Atoms as Active Centers to Facilitate Hydrogen Generation. Materials 2021, 14, 4073. https://doi.org/10.3390/ma14154073
Peng Q, Qi X, Gong X, Chen Y. 1T-MoS2 Coordinated Bimetal Atoms as Active Centers to Facilitate Hydrogen Generation. Materials. 2021; 14(15):4073. https://doi.org/10.3390/ma14154073
Chicago/Turabian StylePeng, Qiong, Xiaosi Qi, Xiu Gong, and Yanli Chen. 2021. "1T-MoS2 Coordinated Bimetal Atoms as Active Centers to Facilitate Hydrogen Generation" Materials 14, no. 15: 4073. https://doi.org/10.3390/ma14154073
APA StylePeng, Q., Qi, X., Gong, X., & Chen, Y. (2021). 1T-MoS2 Coordinated Bimetal Atoms as Active Centers to Facilitate Hydrogen Generation. Materials, 14(15), 4073. https://doi.org/10.3390/ma14154073