
The Physico-Chemical Properties and Exploratory Real-Time Cell Analysis of Hydroxyapatite Nanopowders Substituted with Ce, Mg, Sr, and Zn (0.5–5 at.%)

 , ,
, ,  ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
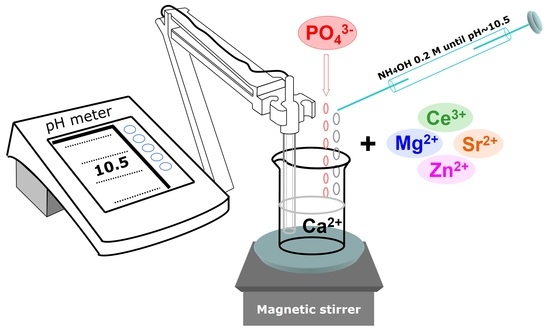
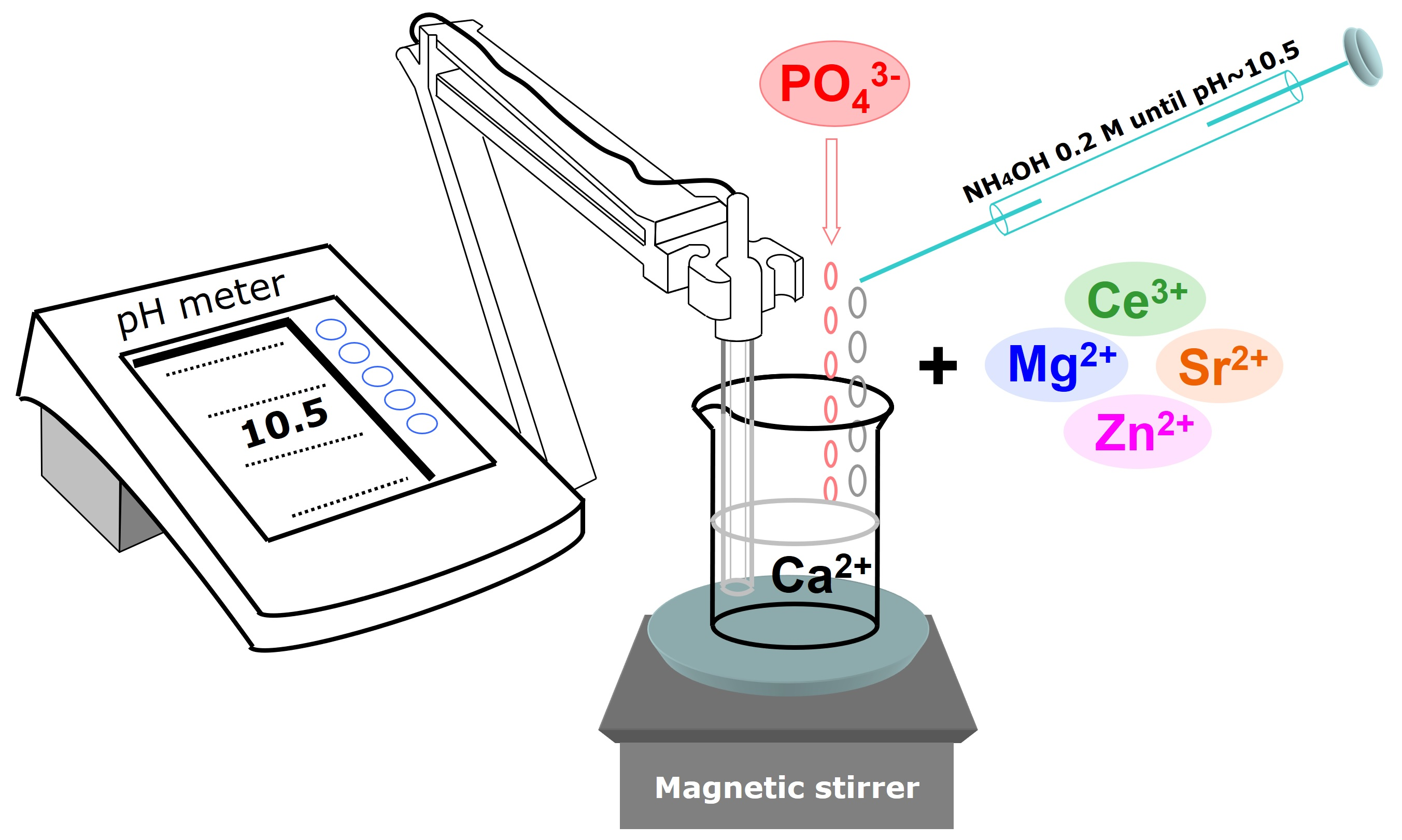
2.1. Preparation of the HA-Based Materials: From Synthesis to Optimisation
2.2. Physico-Chemical Characterisation Techniques
2.2.1. Morphological Examination
2.2.2. Compositional Analysis
2.2.3. Chemical State Analysis
2.2.4. Structural Investigation
2.3. In Vitro Biological Assays
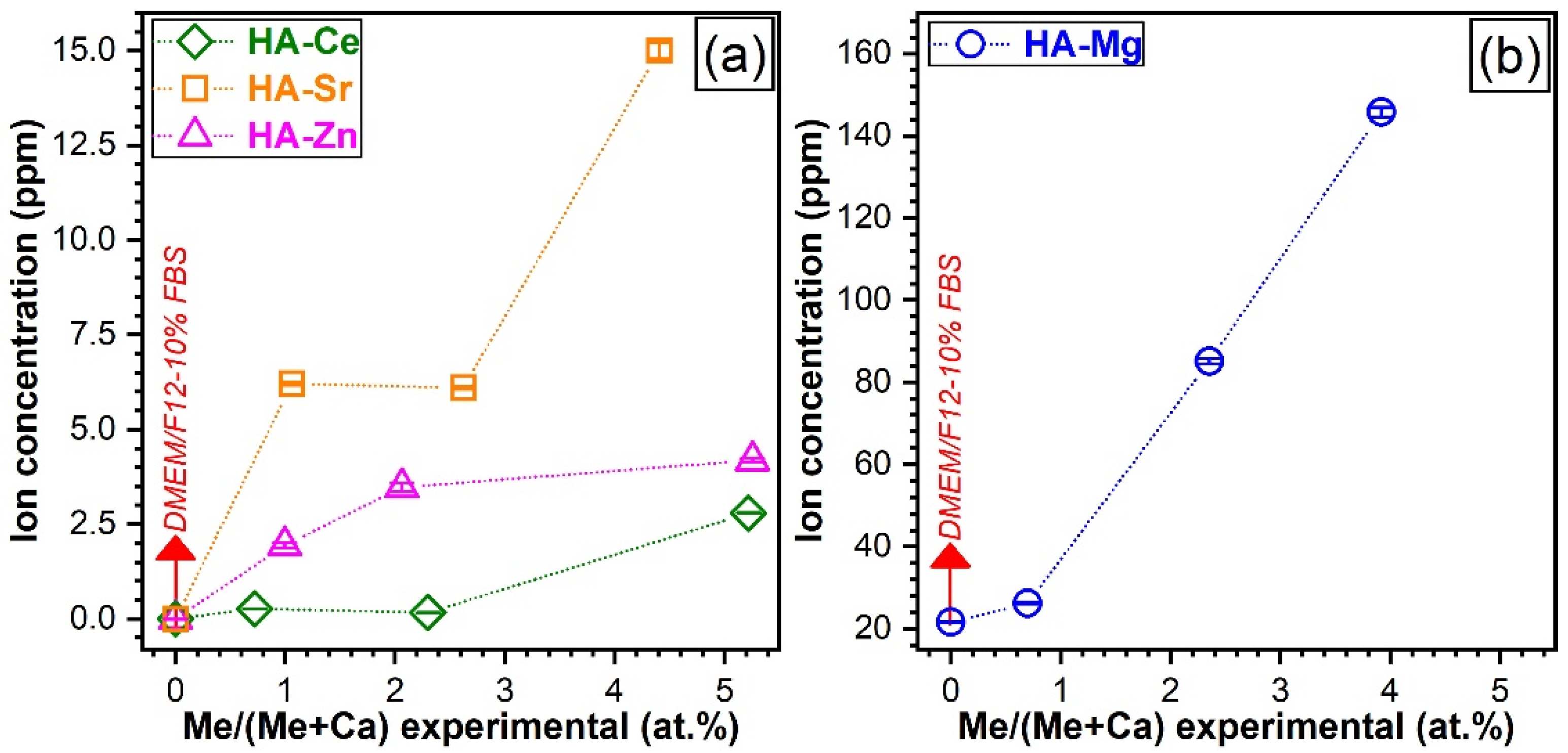
2.3.1. Cation Release Determinations
2.3.2. Cell Culture and Cell Treatments
2.3.3. Cell Culture Biological Assessments
2.3.4. Statistical Analysis
3. Results
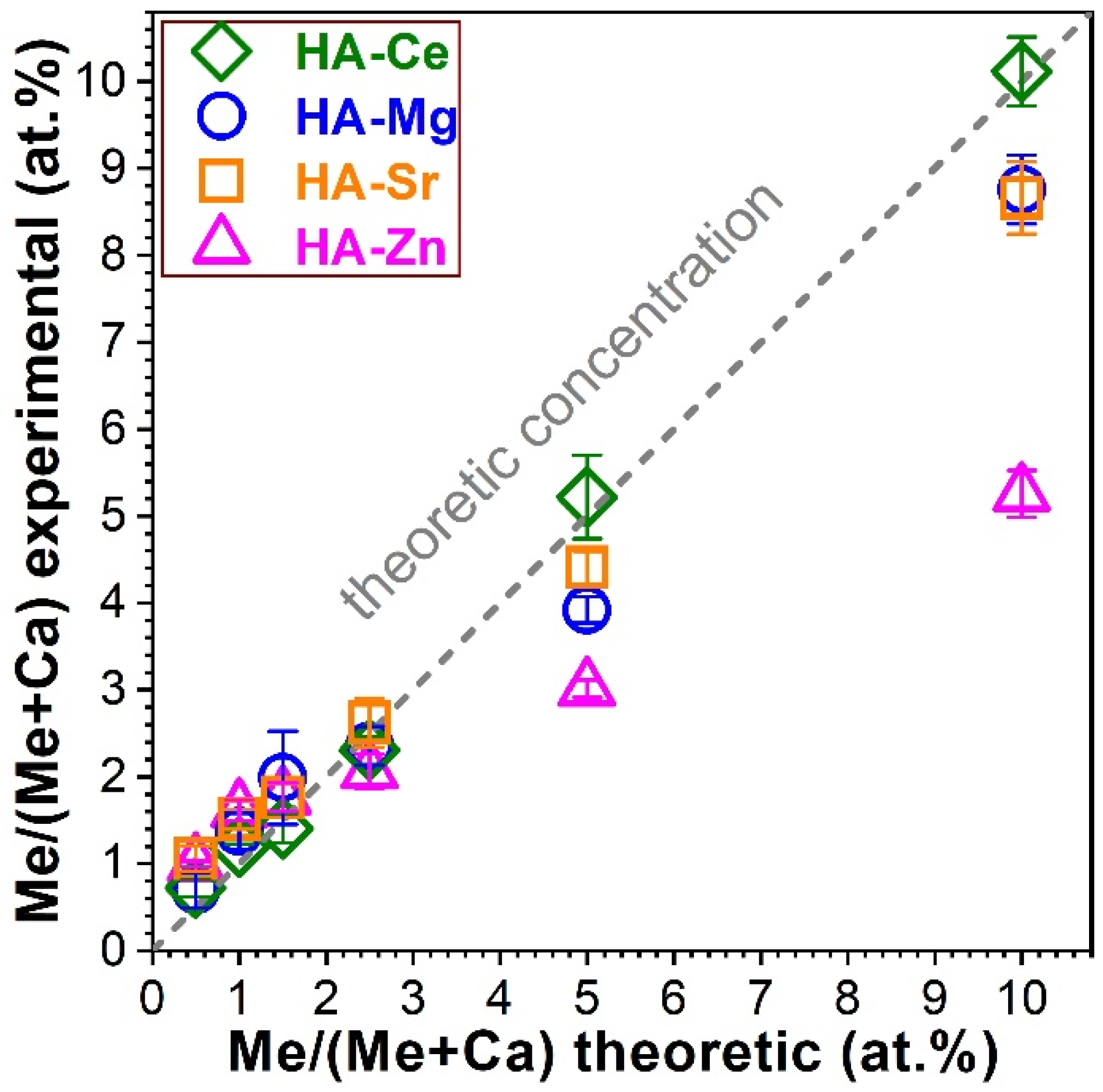
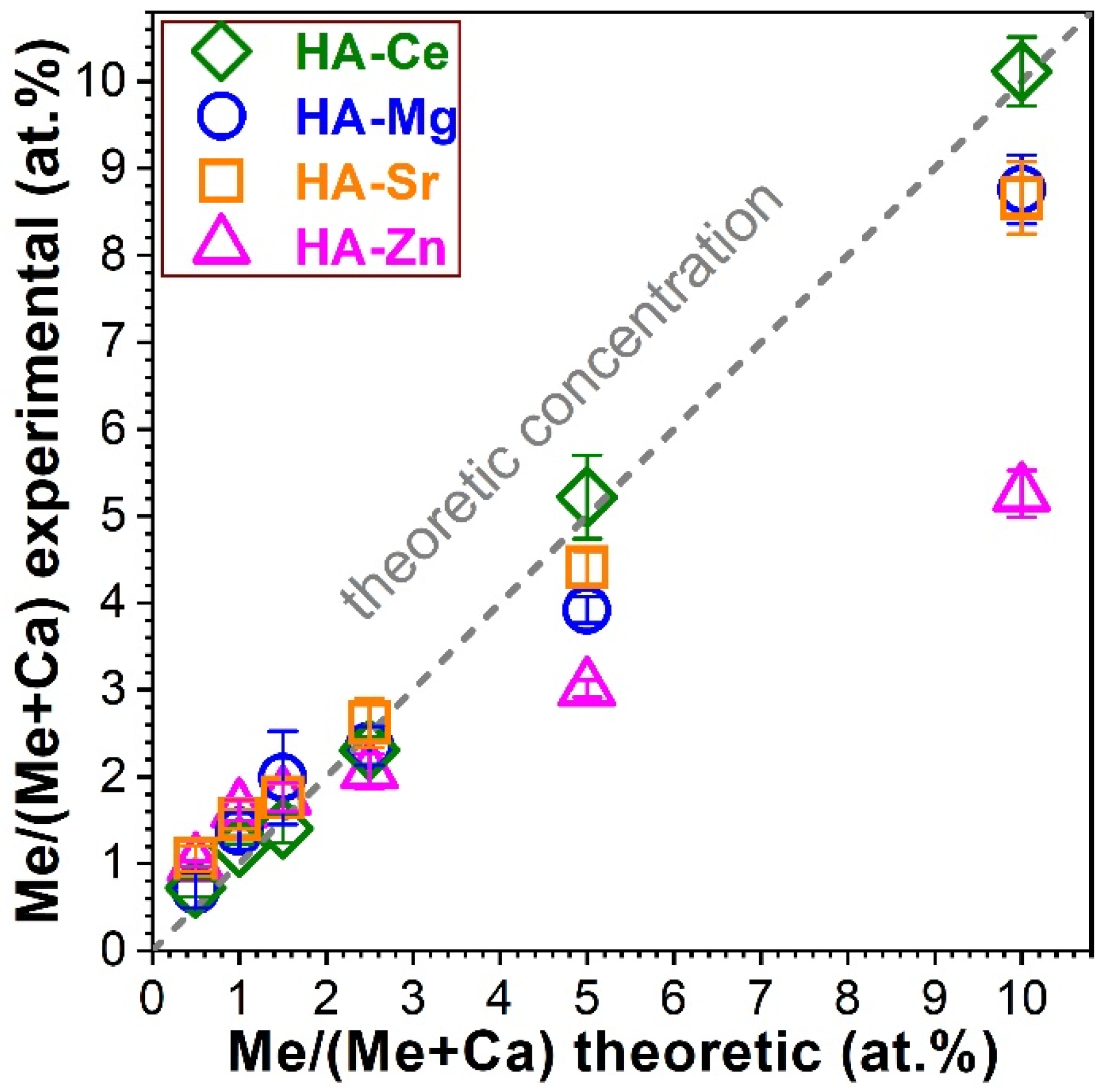
3.1. Composition Analysis via EDXS
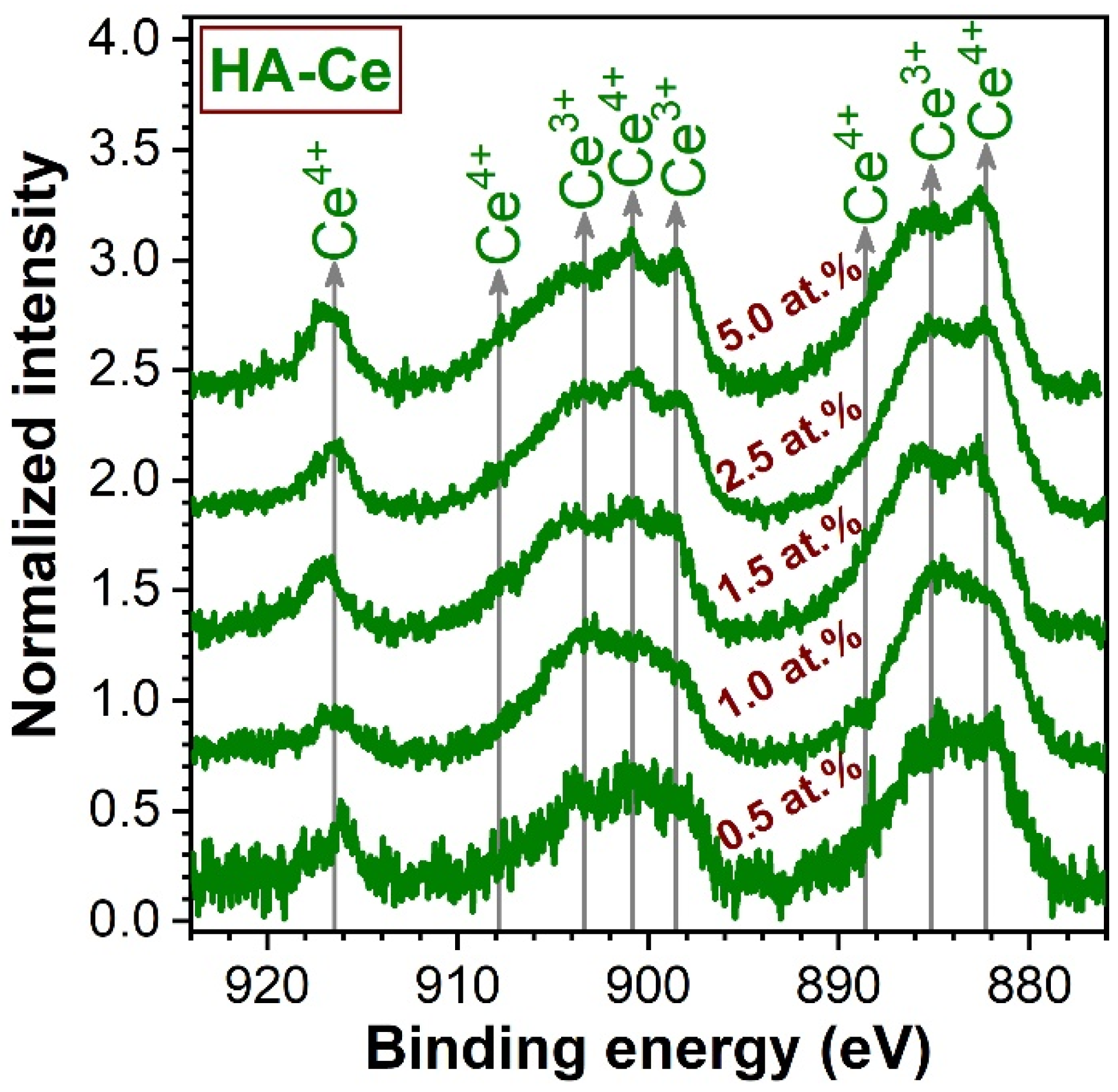
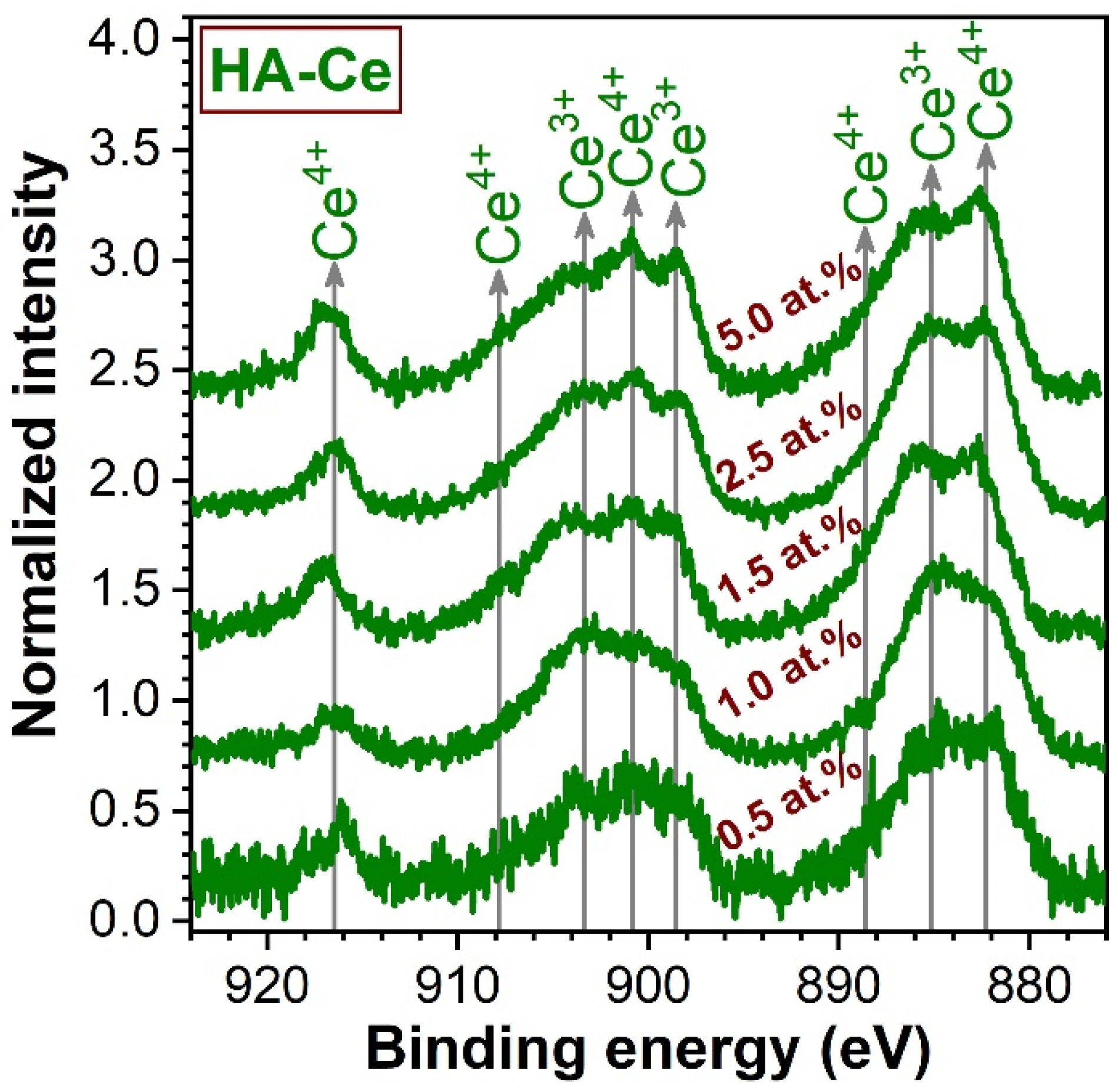
3.2. Chemical State Analysis of Cerium by XPS
3.3. Morphological Investigation
3.4. Structural Characterisation
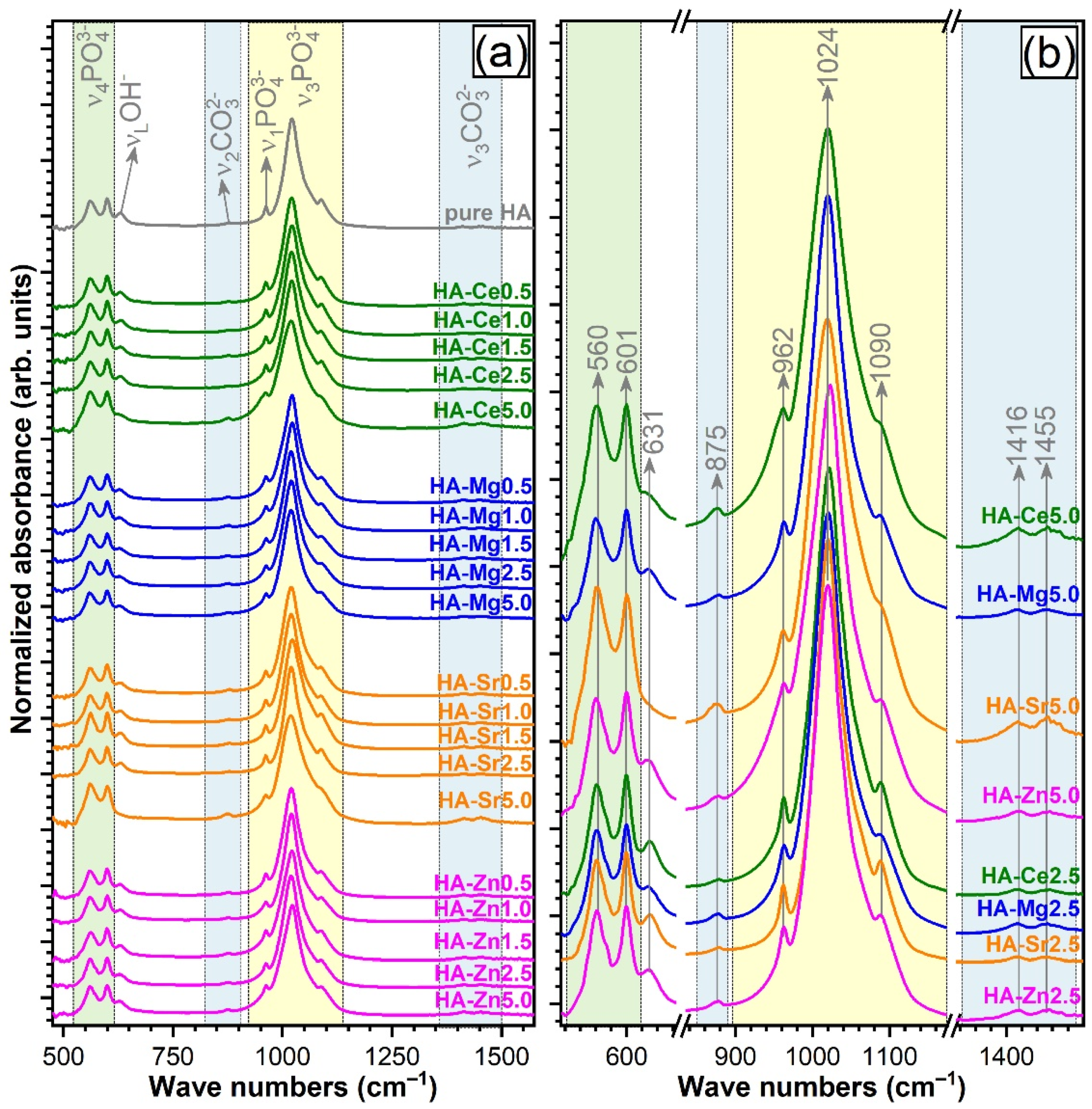
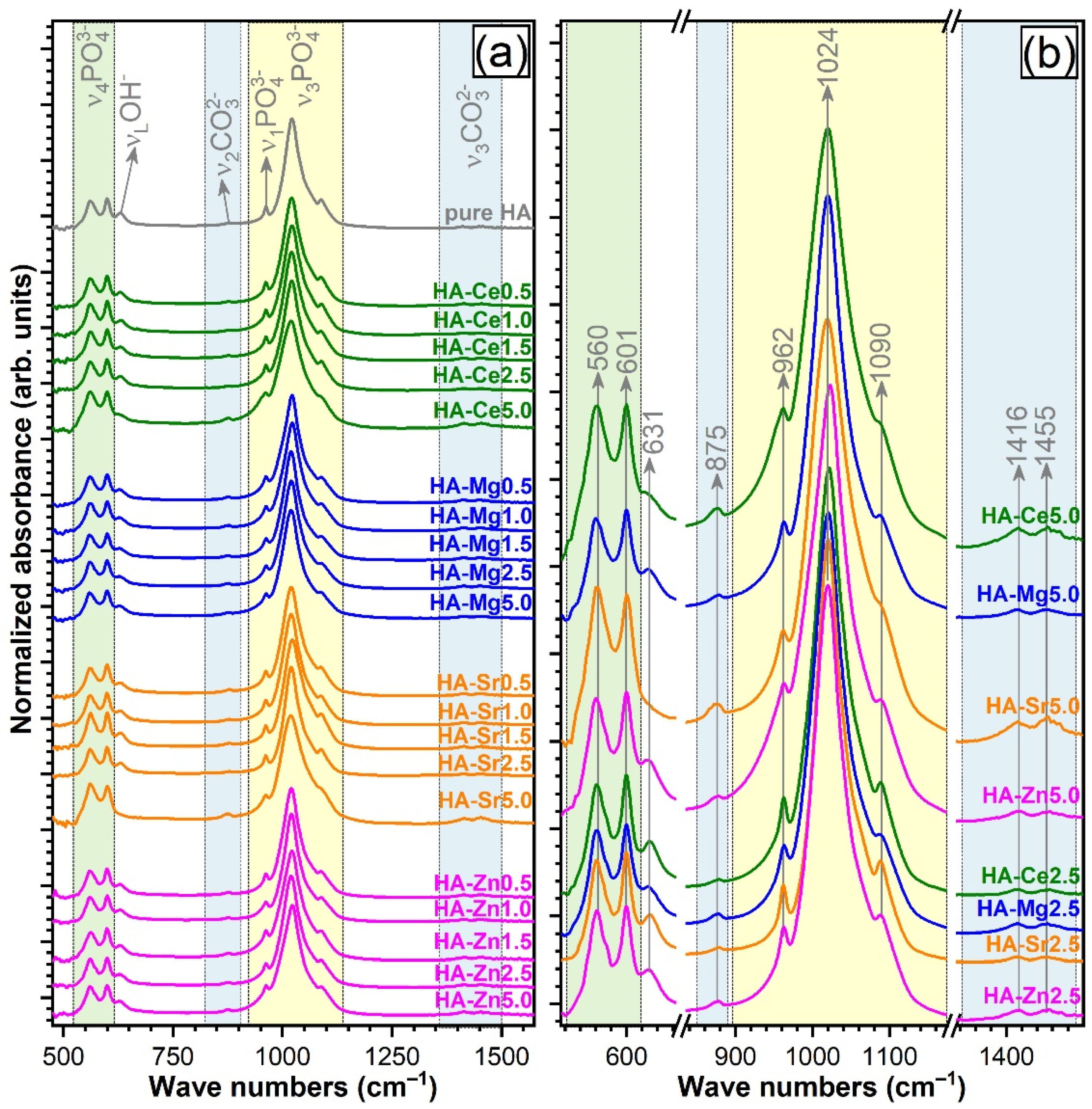
3.4.1. FTIR Spectroscopy
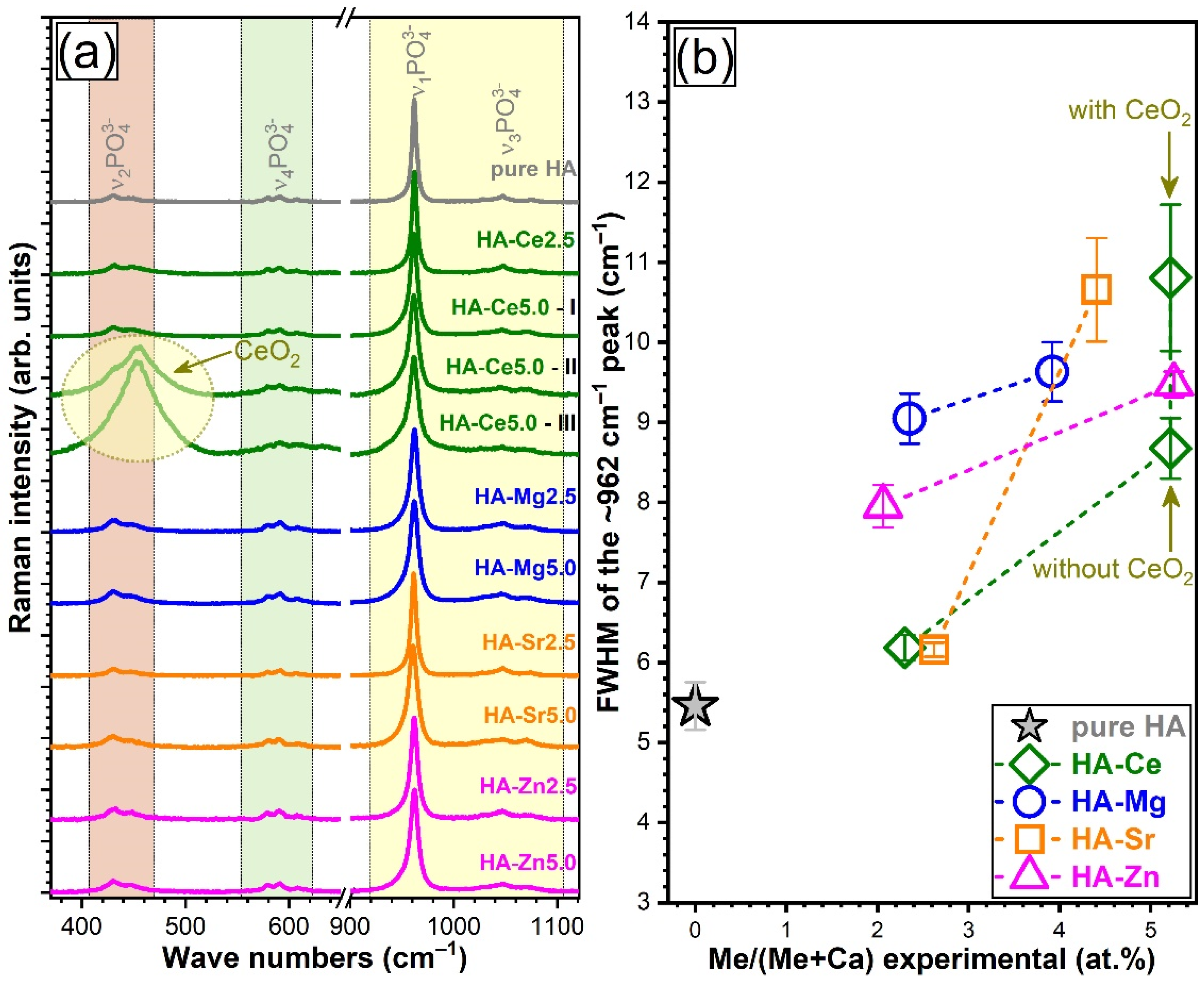
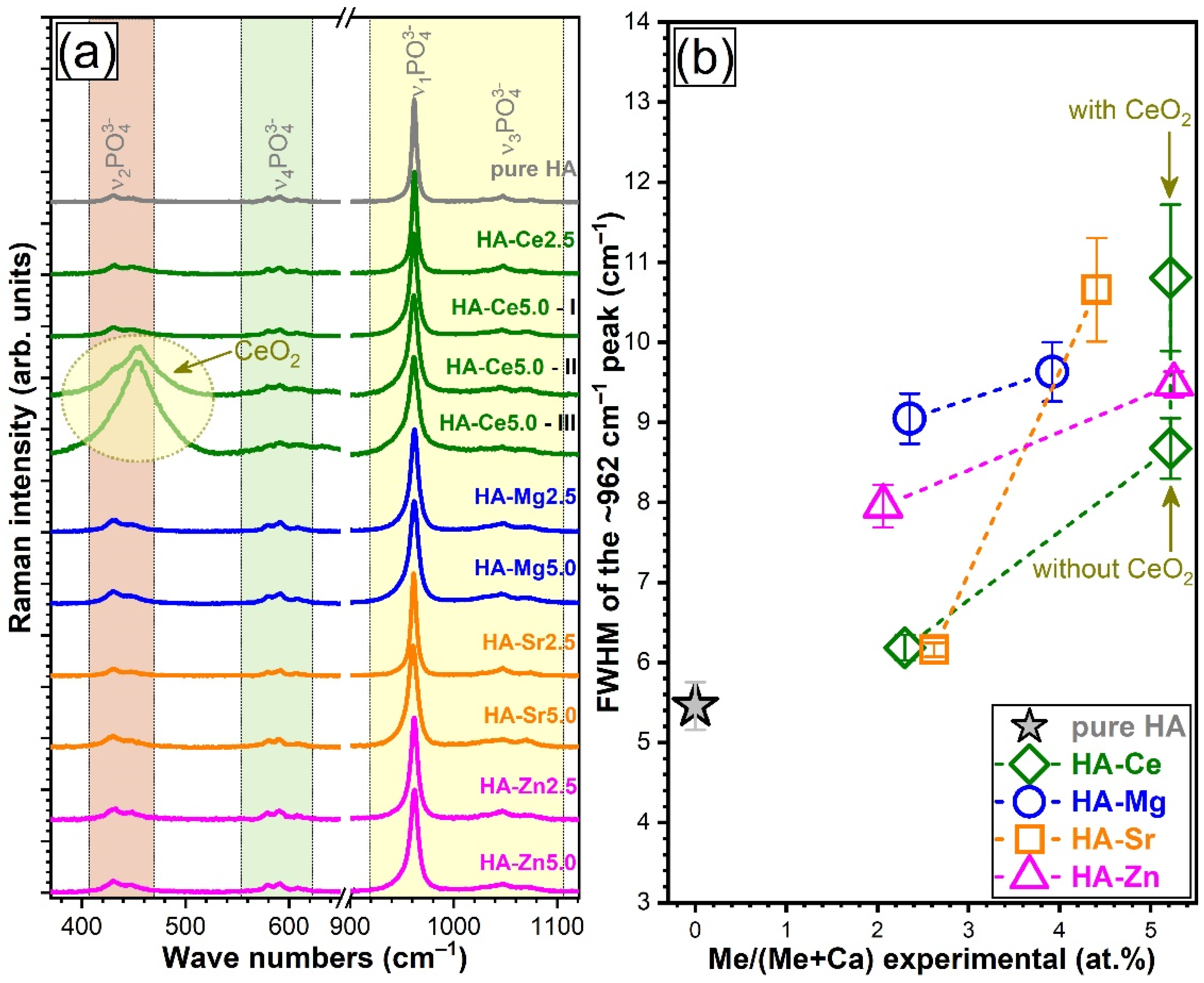
3.4.2. Micro-Raman Spectroscopy
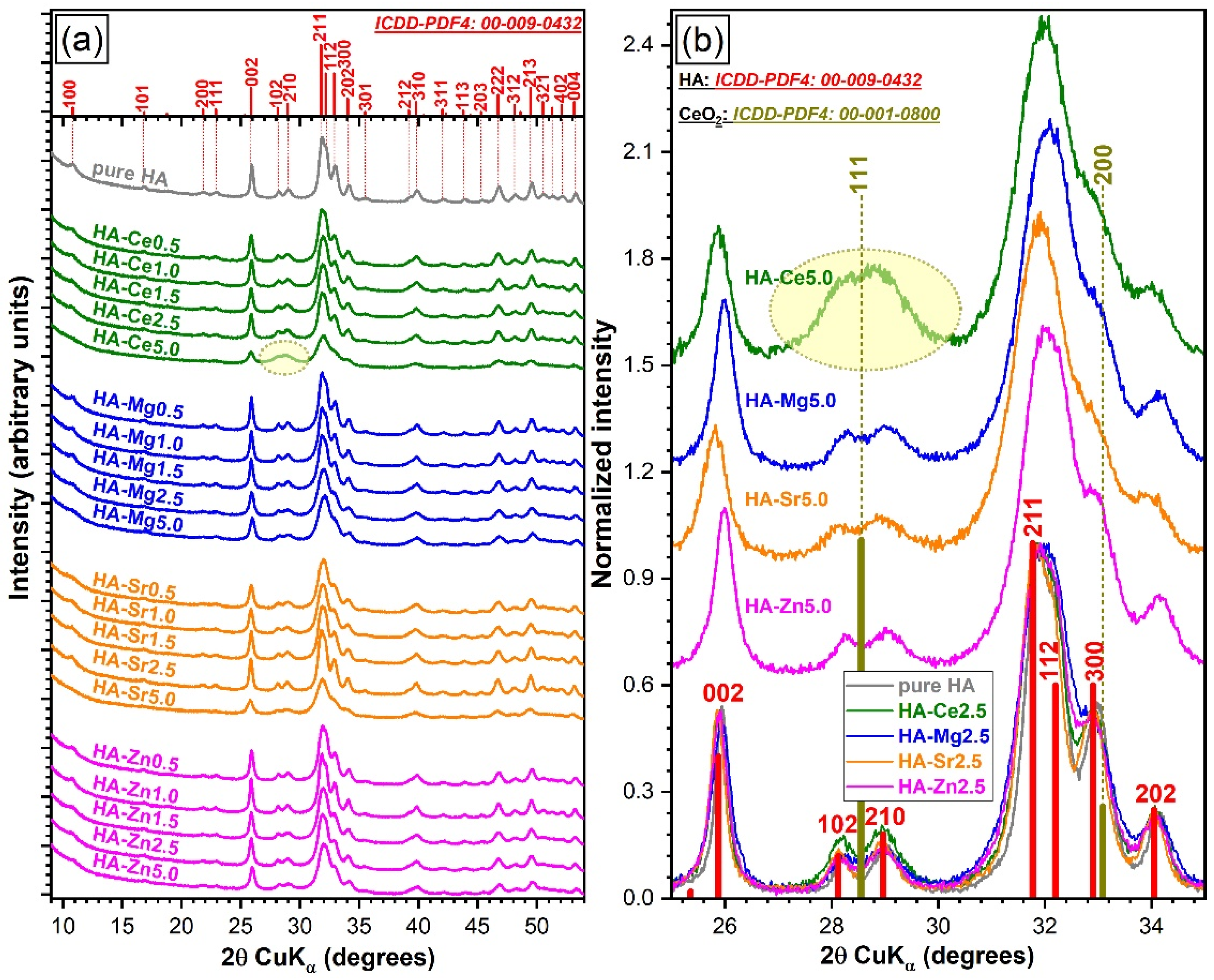
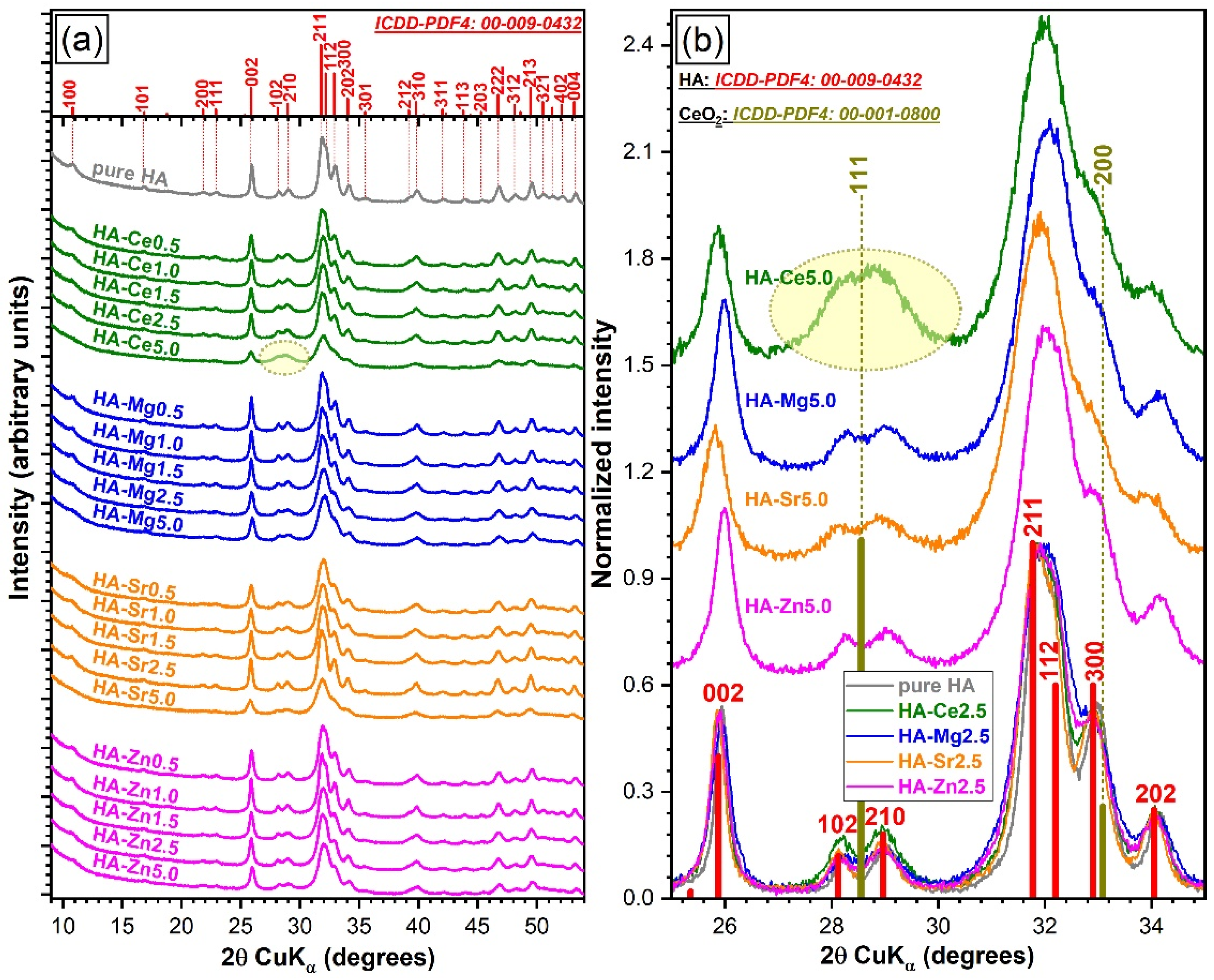
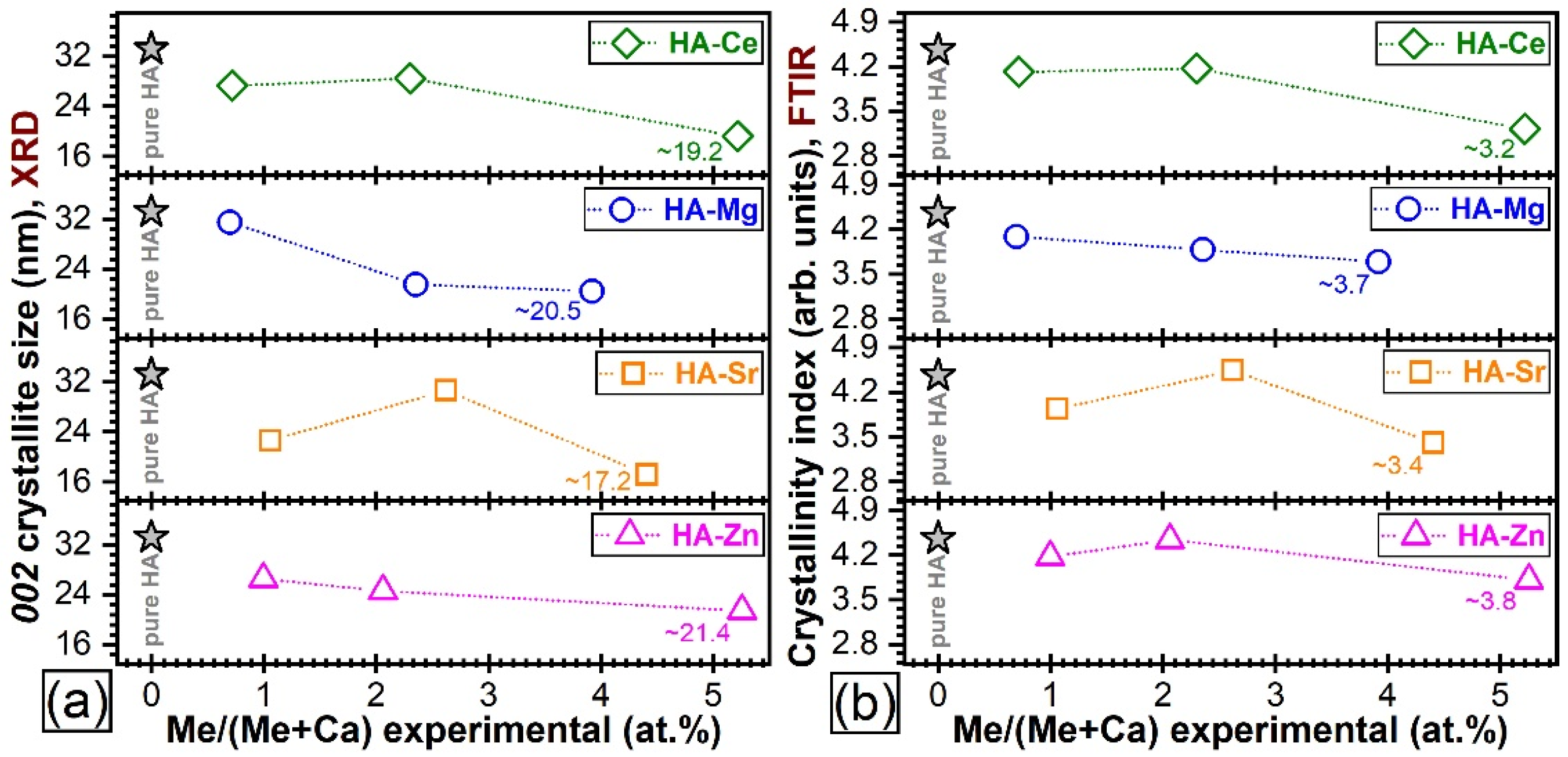
3.4.3. X-Ray Diffraction
Qualitative Assessment
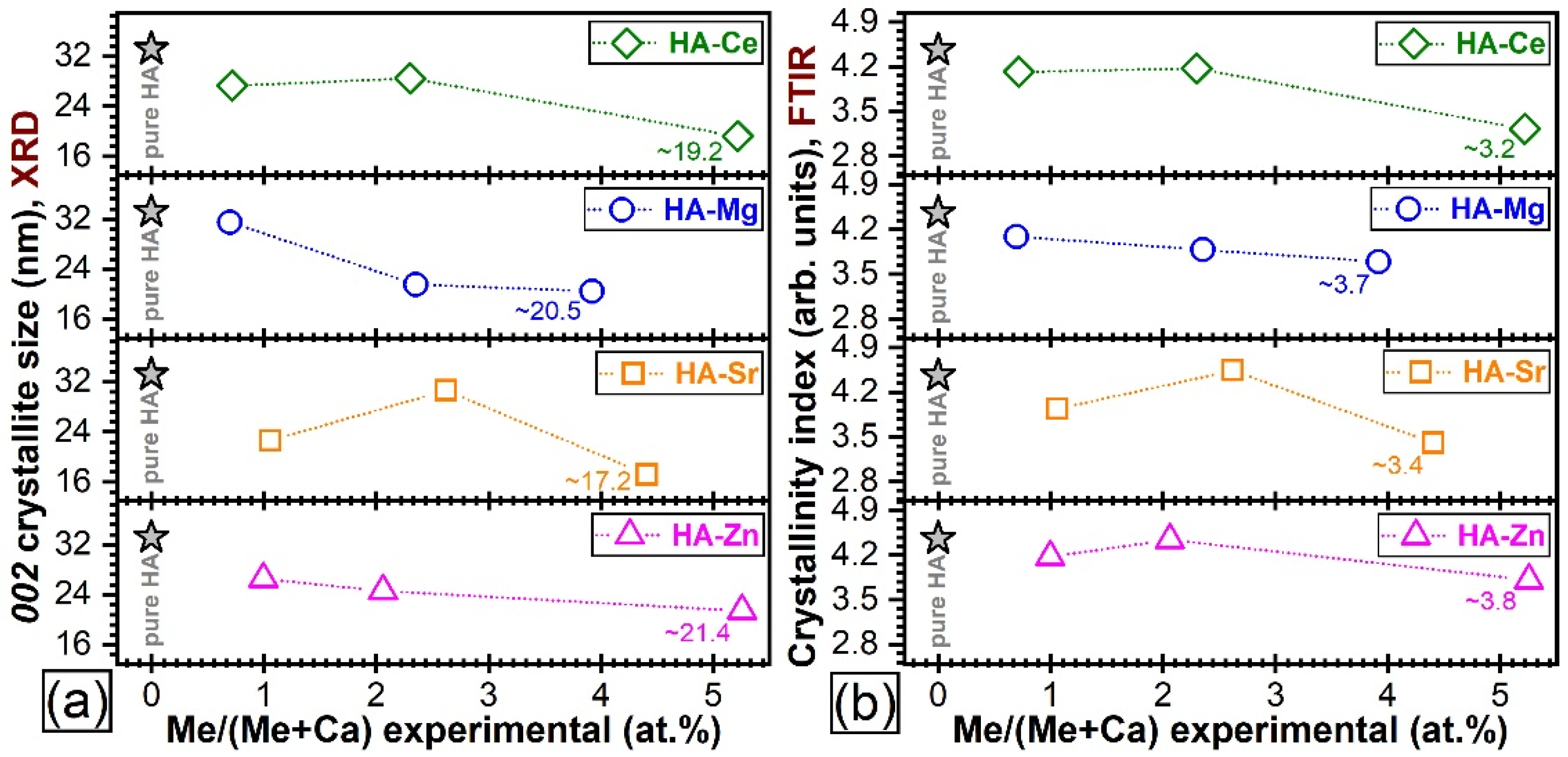
Quantitative Assessment of Crystalline Quality
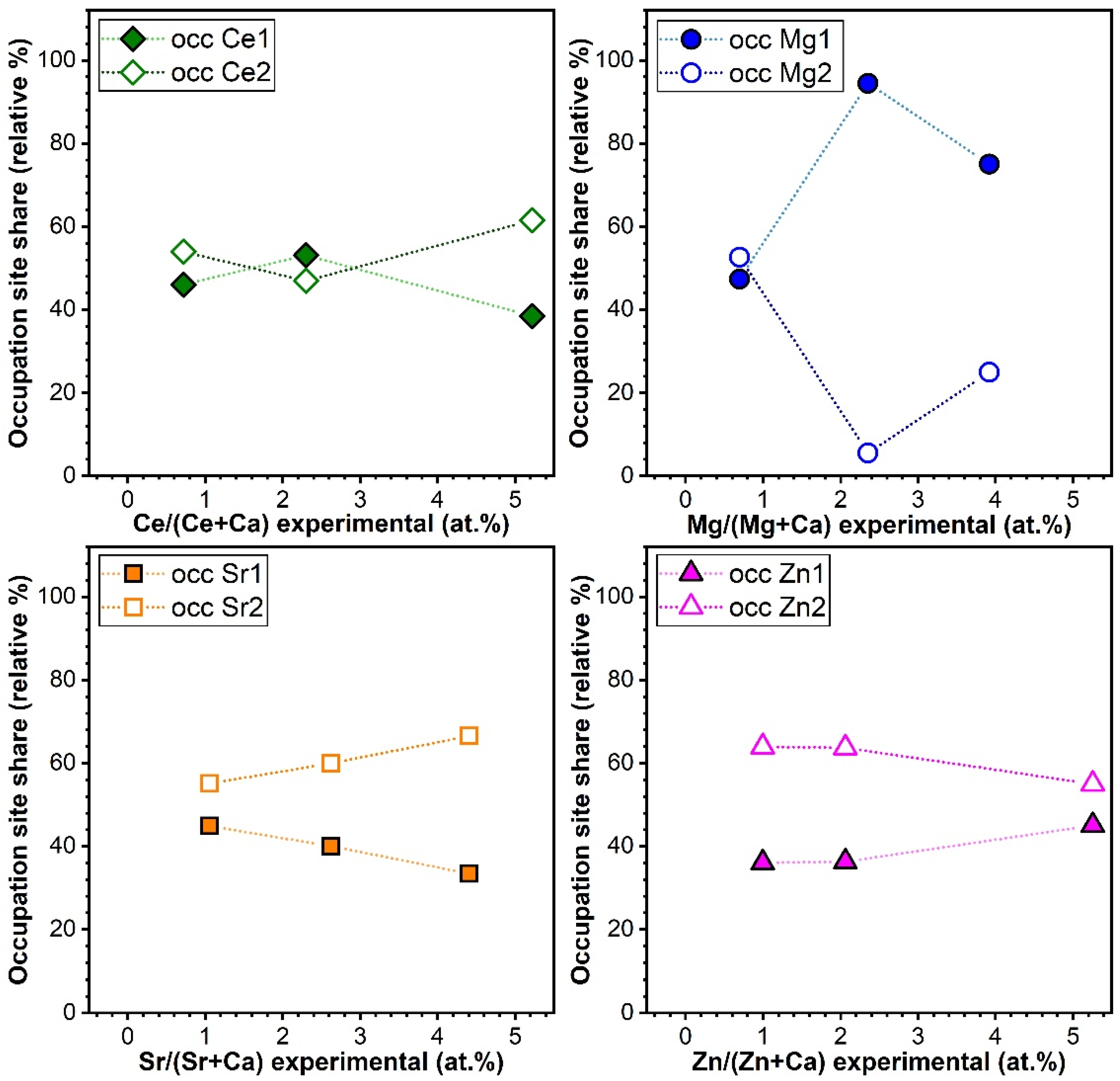
Variation of Cation Occupation with Substituent Concentration
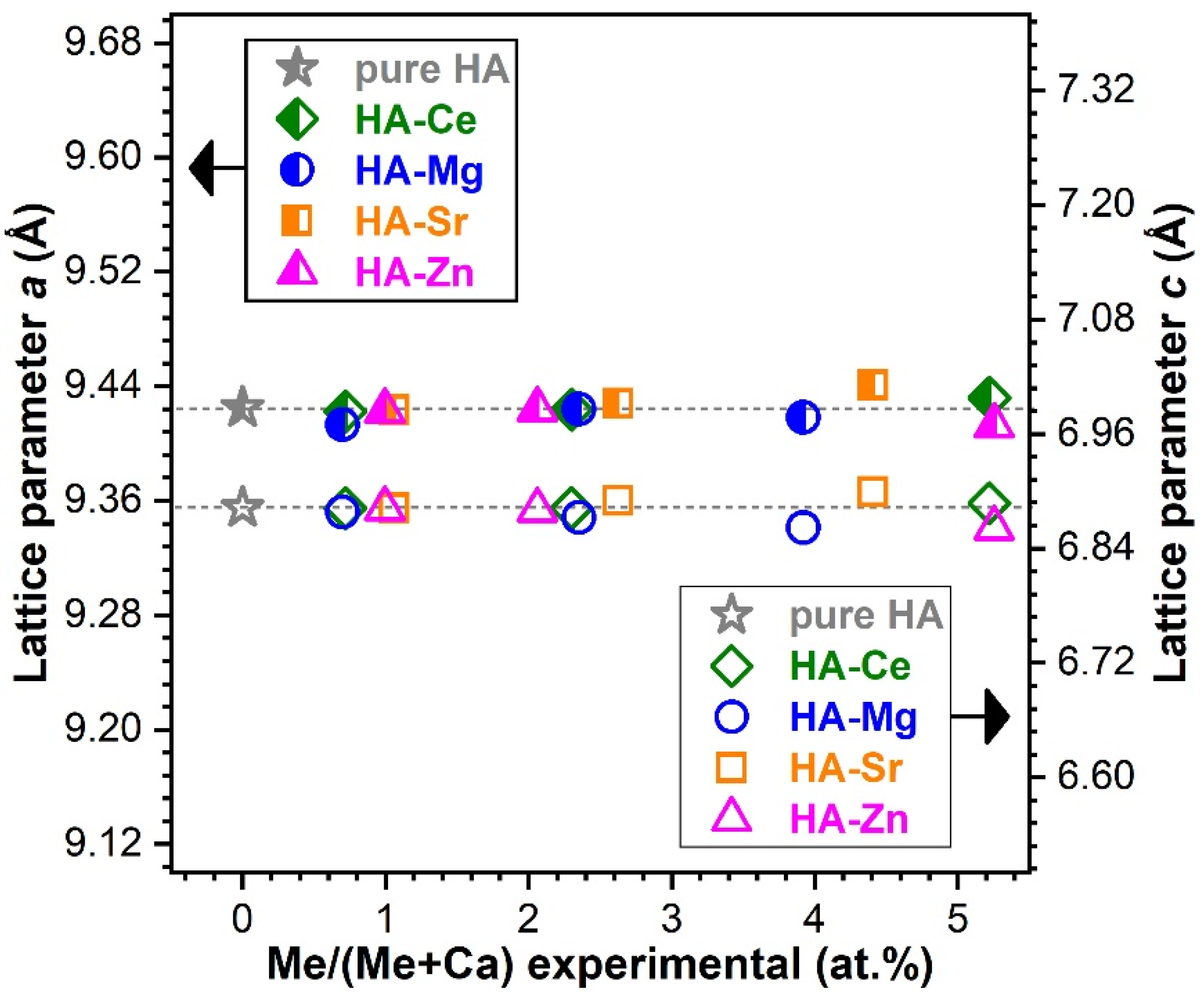
Modification of the Lattice Parameters
3.5. Biological Assays
3.5.1. Dopant Release Determinations
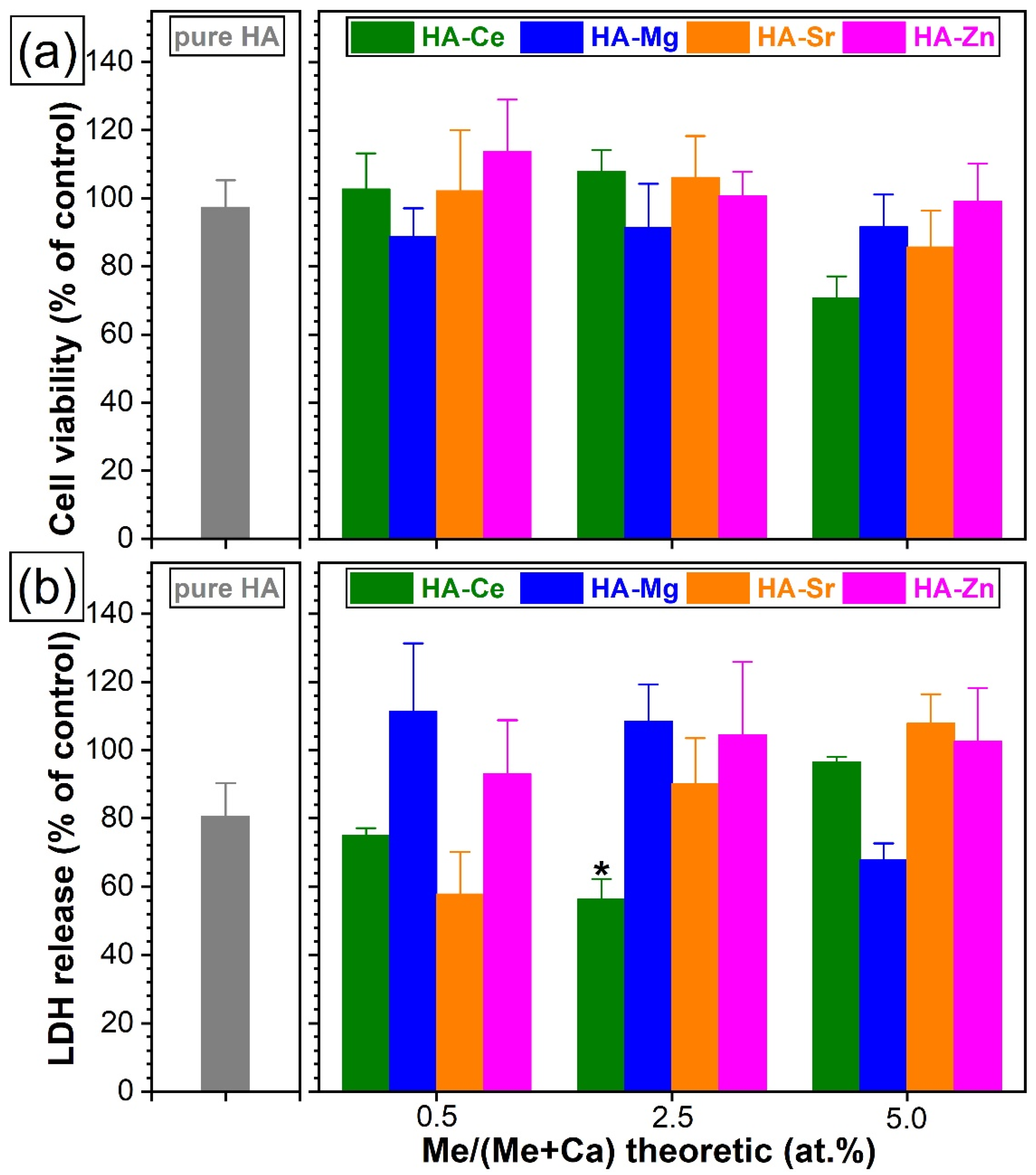
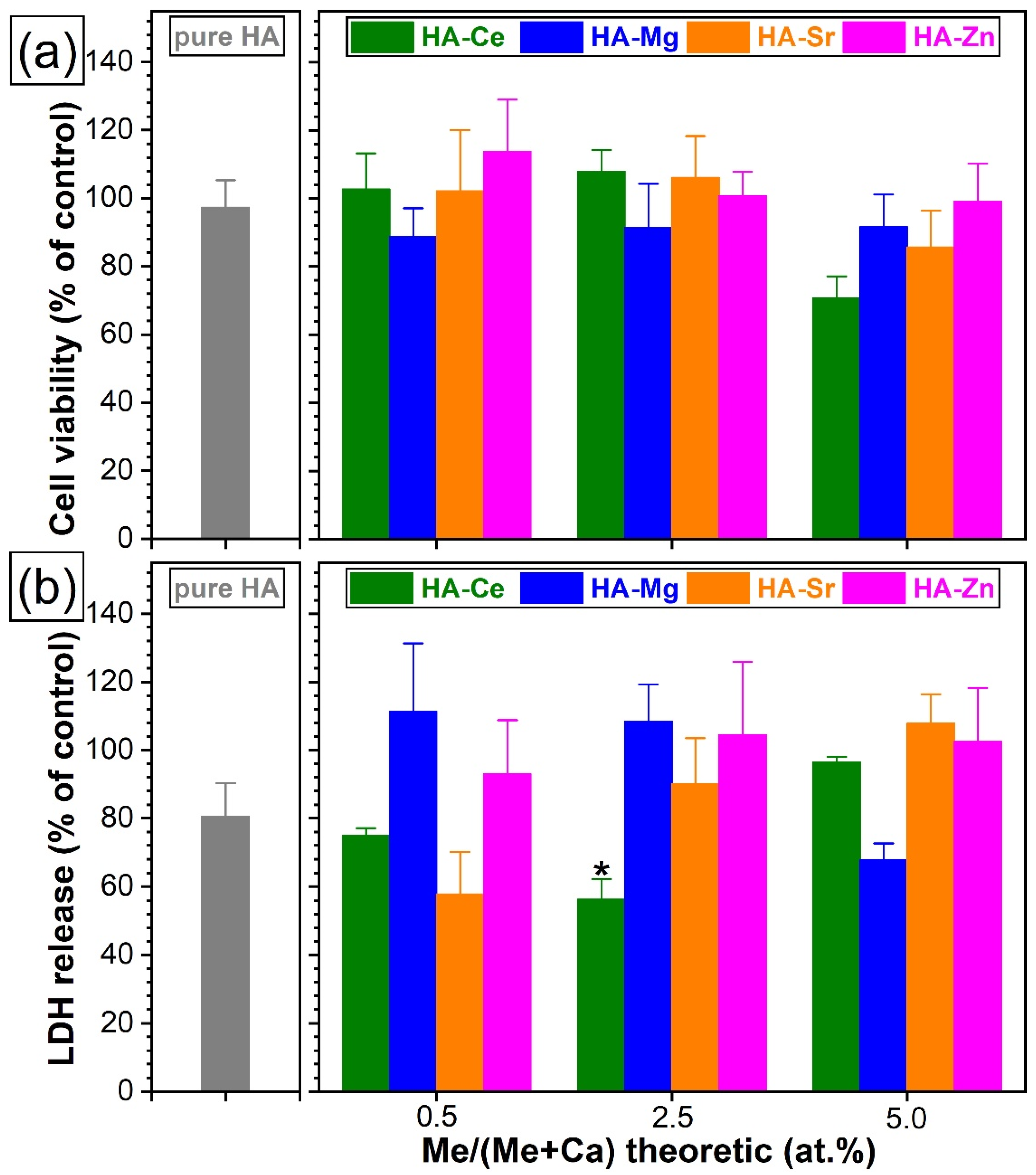
3.5.2. Cytotoxic Evaluation with Standard Colorimetric Assays
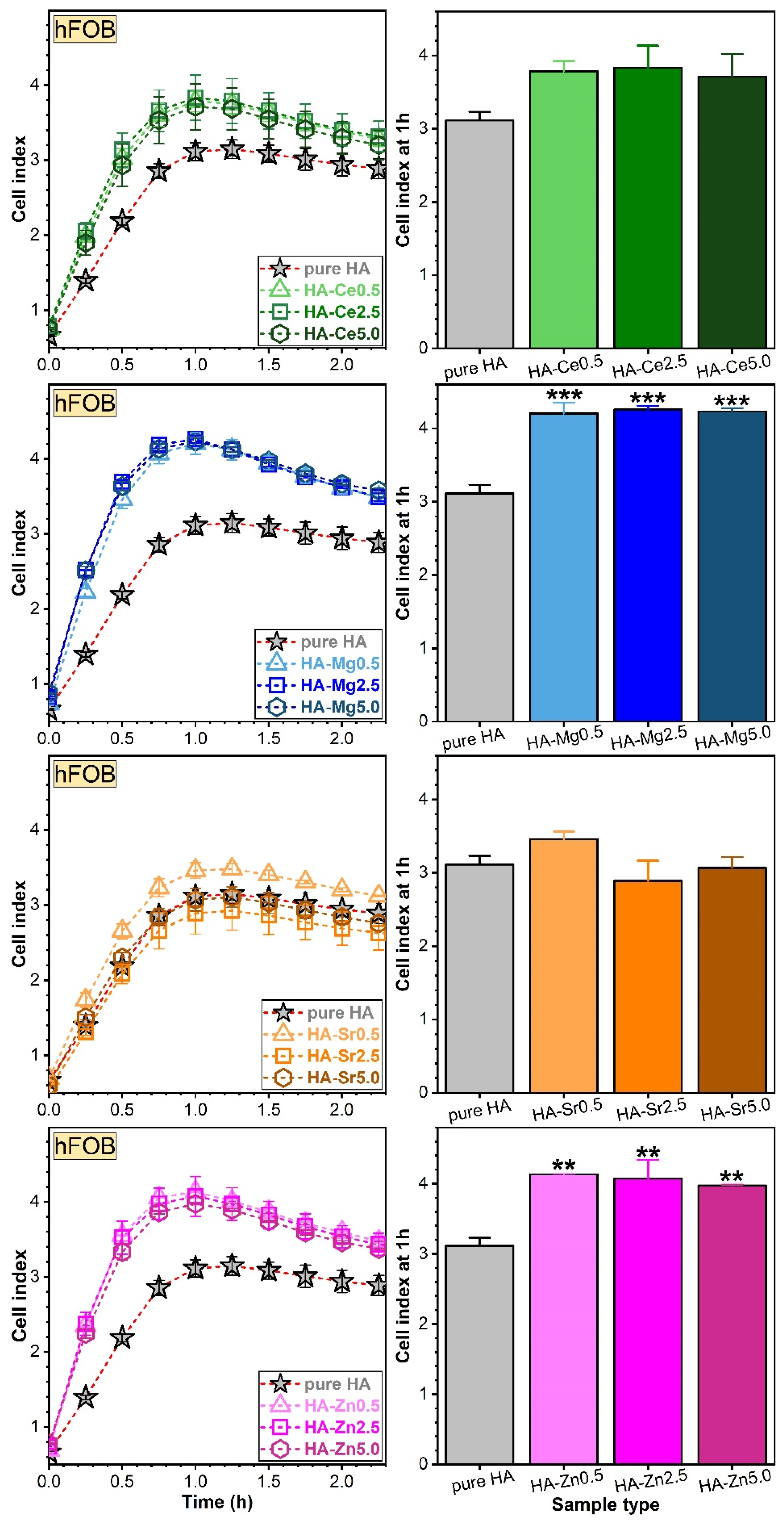
3.5.3. Measurement of Cell Adhesion and Proliferation via Real-Time Impedance Readings
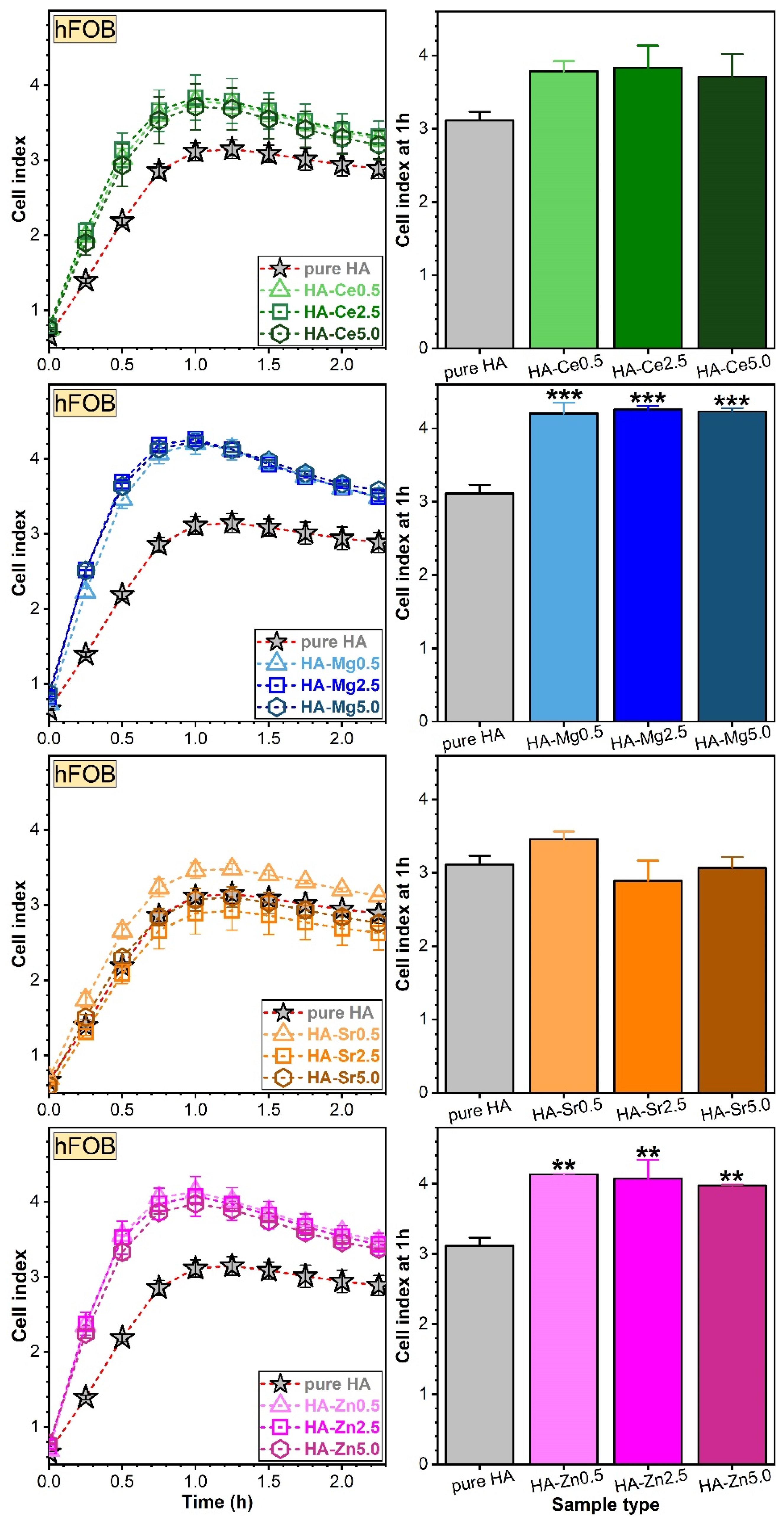
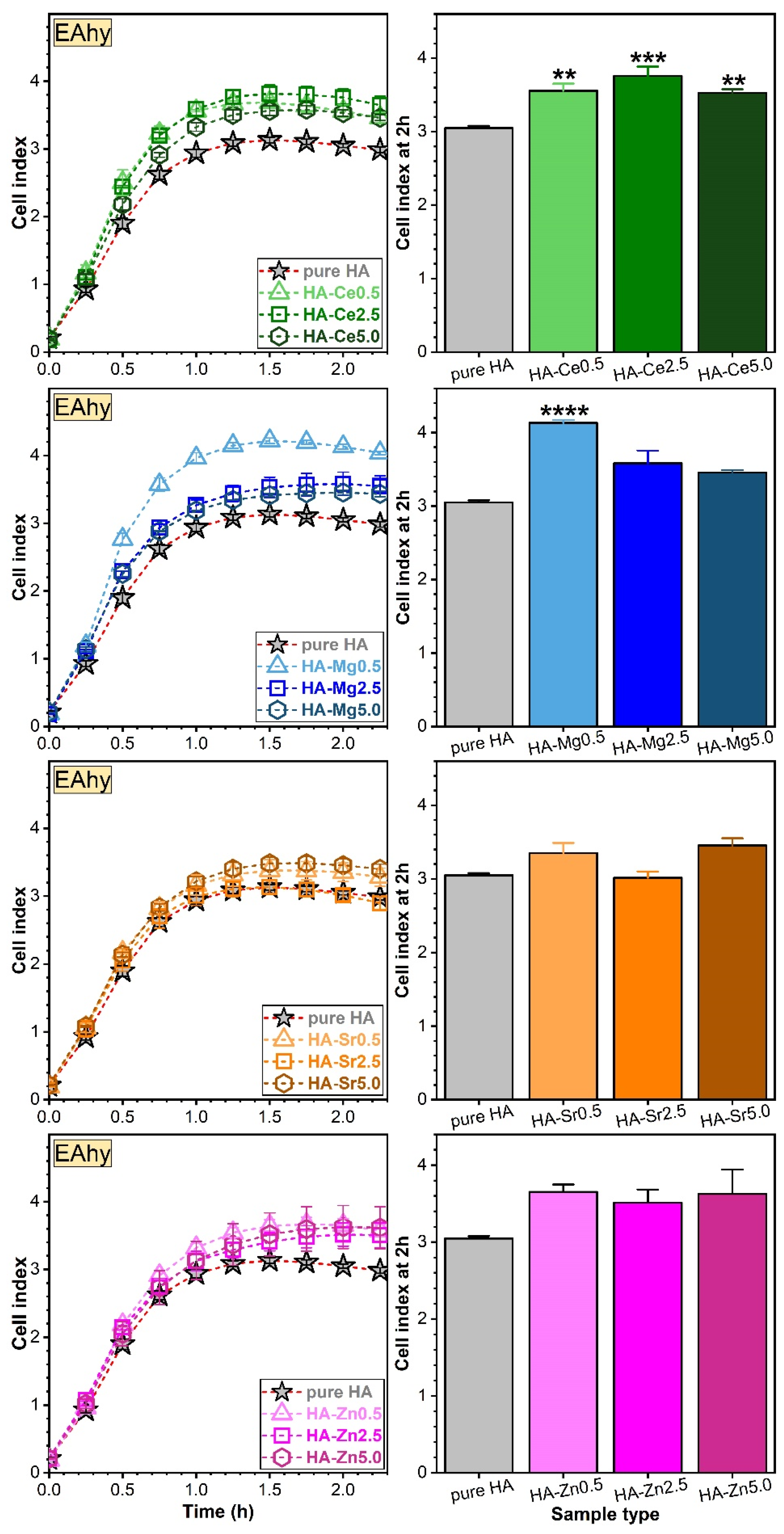
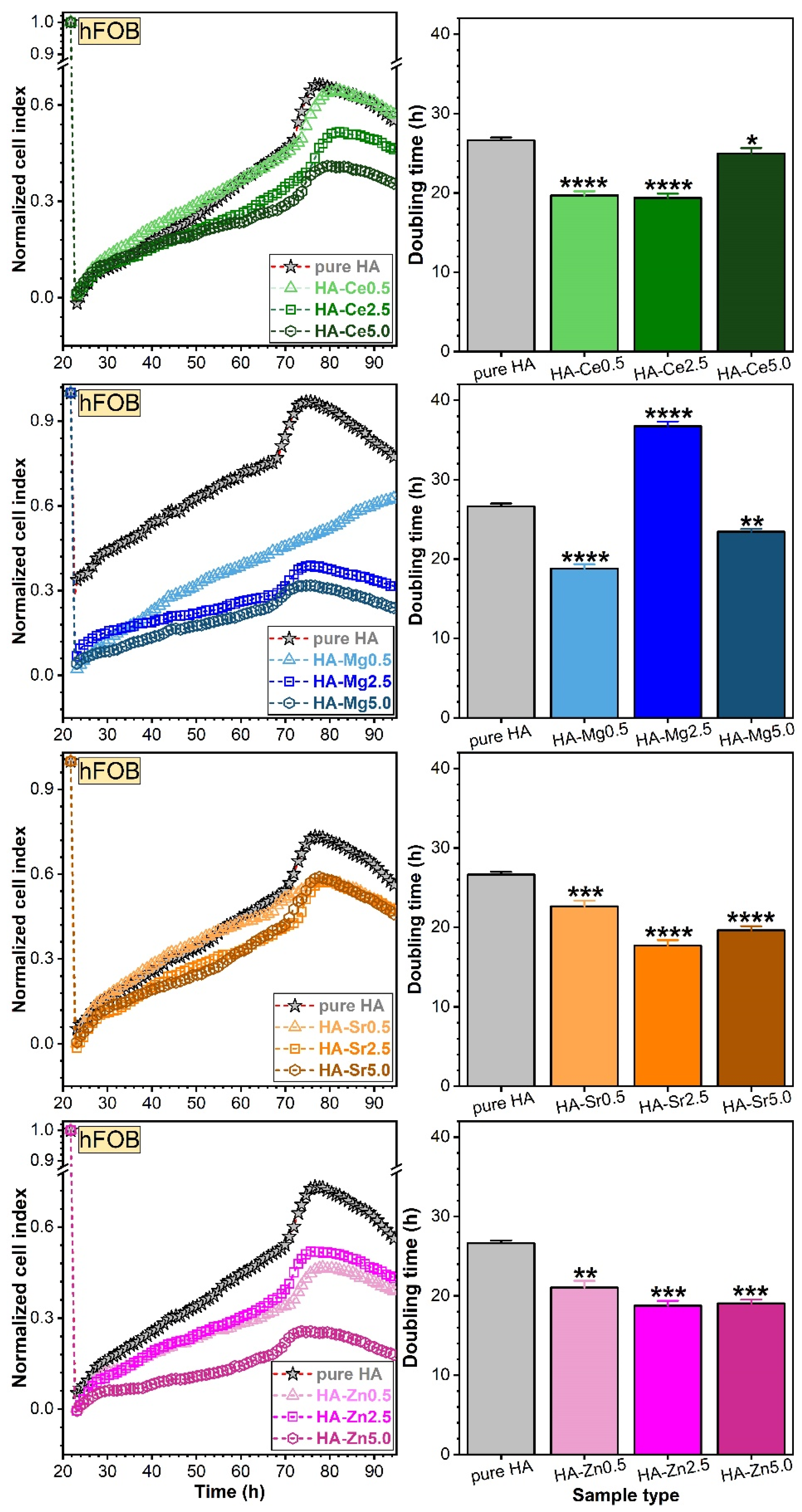
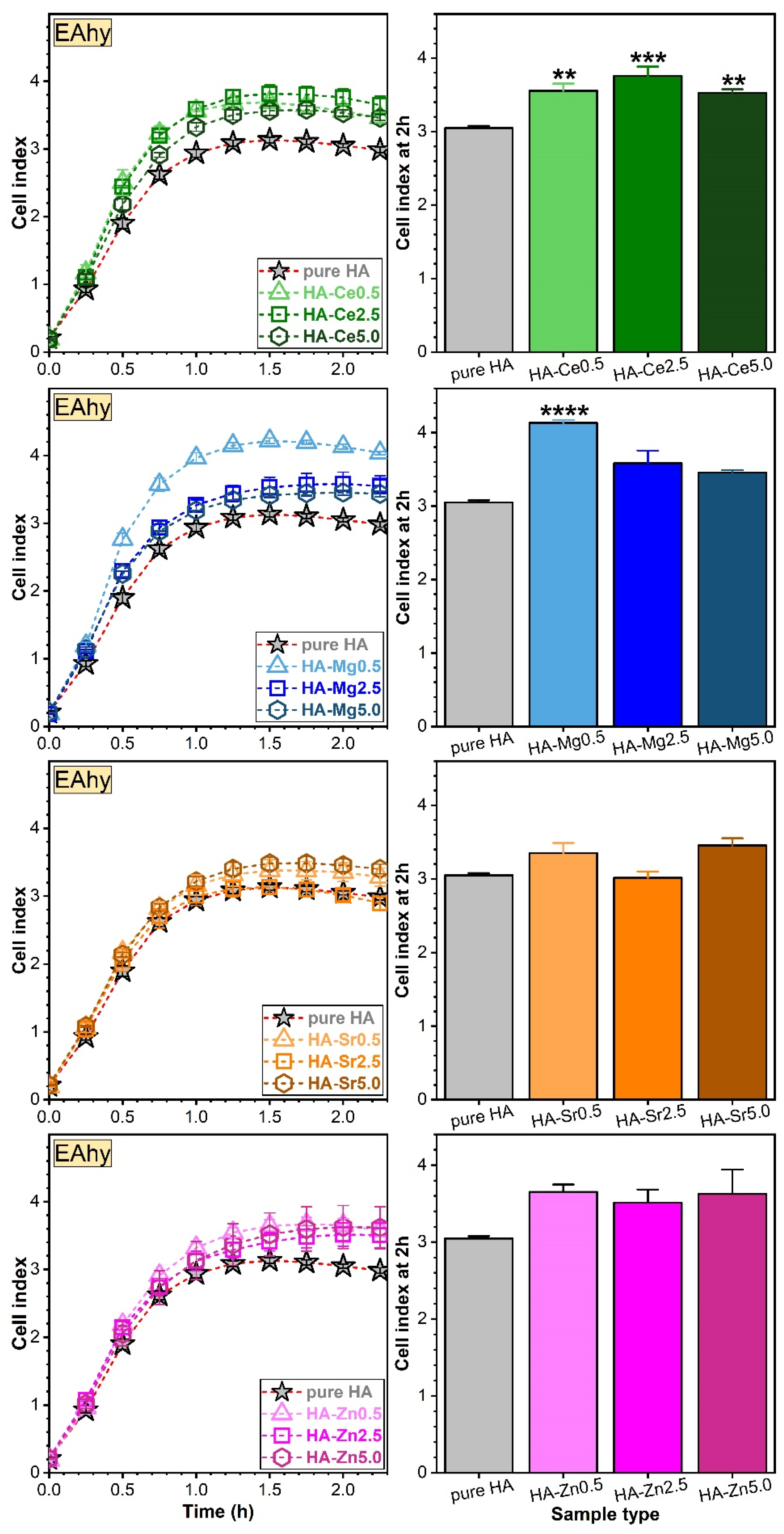
Assessment of Osteoblast (hFOB) and Endothelial (EAhy) Cell Adhesion via RTCA
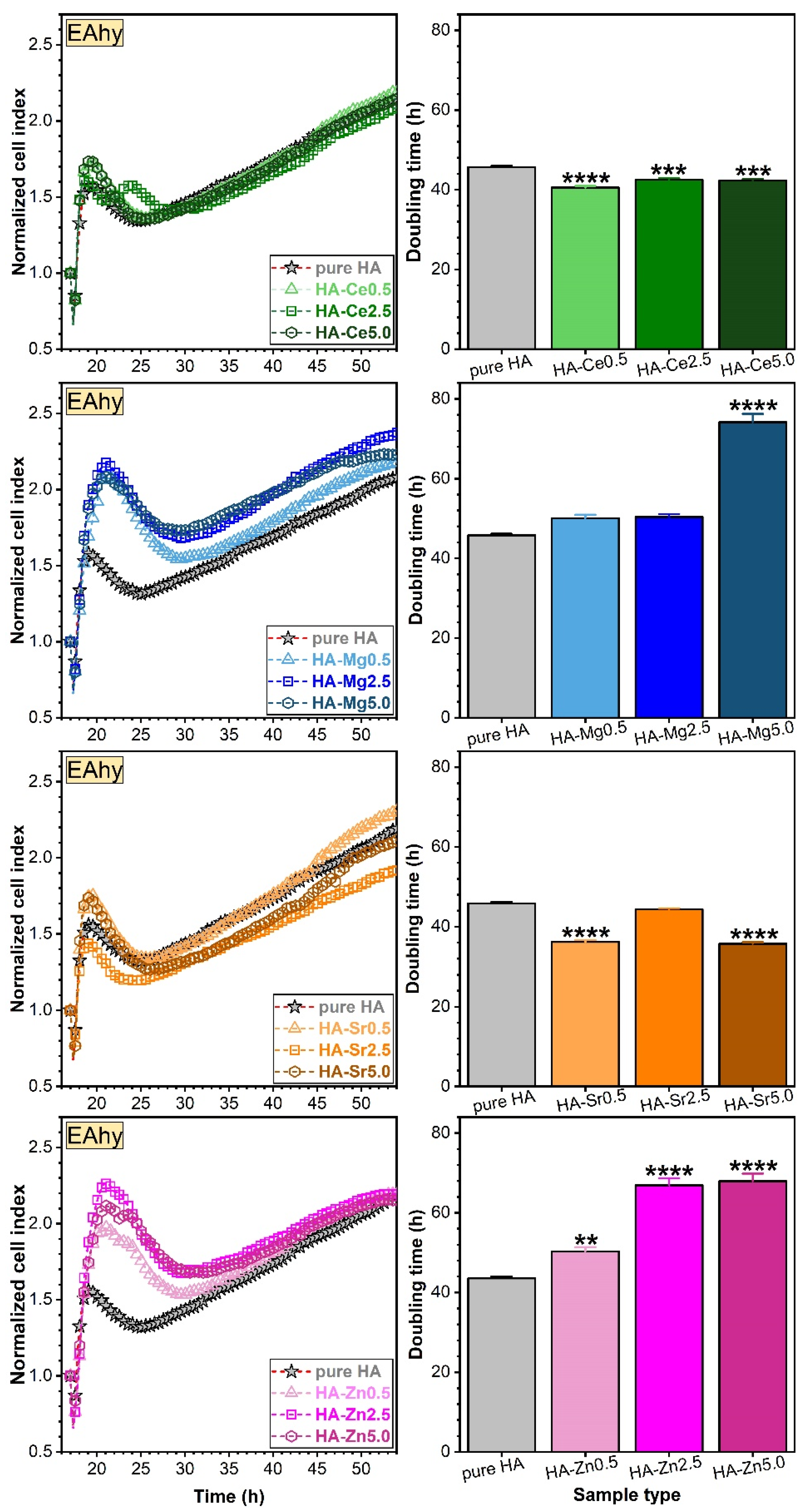
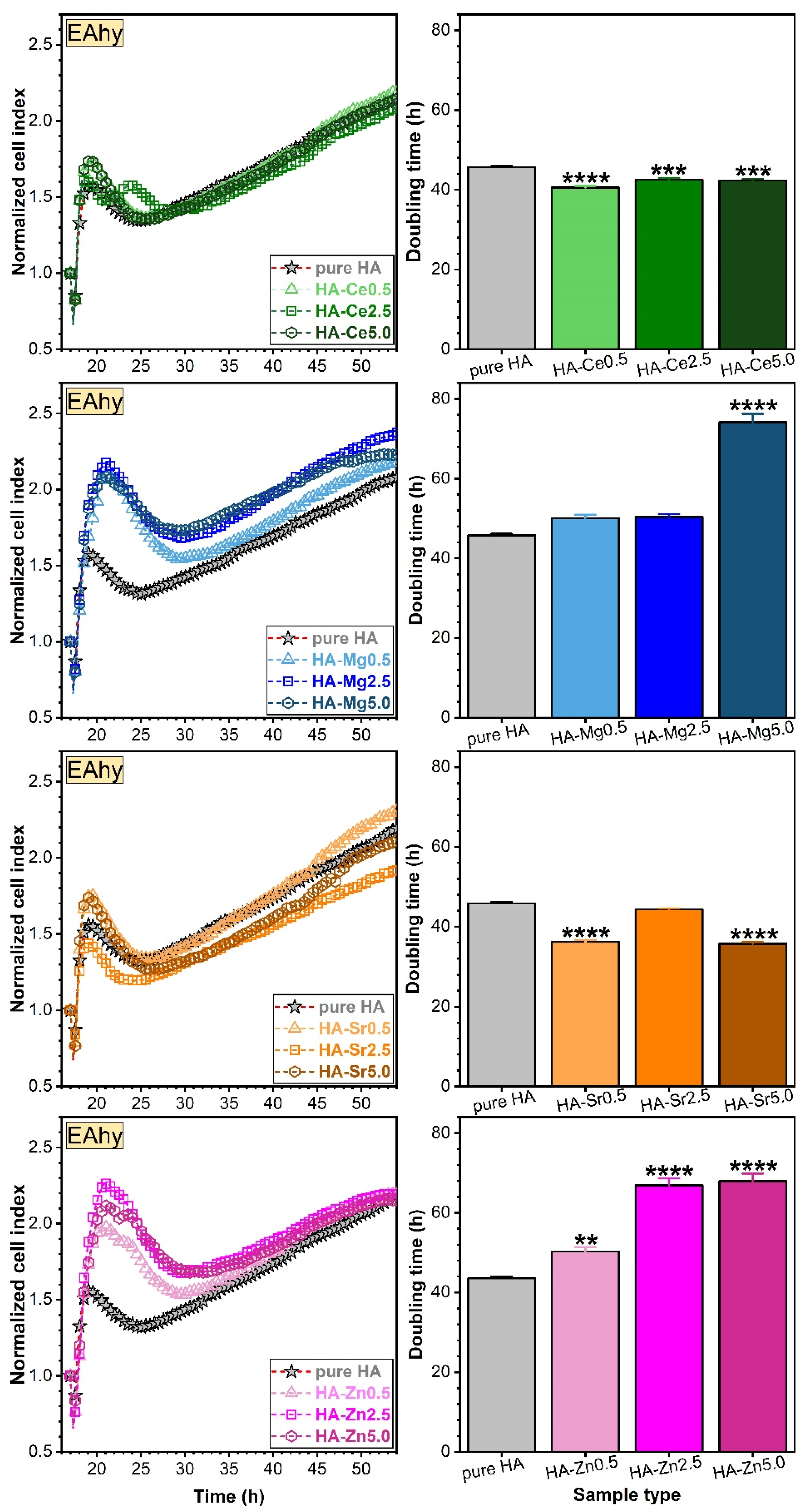
Assessment of Osteoblast (hFOB) and Endothelial (EAhy) Cell Proliferations via RTCA
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antoniac, I.V. (Ed.) Handbook of Bioceramics and Biocomposites; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-12459-9. [Google Scholar]
- Hassan, M.N.; Mahmoud, M.M.; El-Fattah, A.A.; Kandil, S. Microwave-assisted preparation of Nano-hydroxyapatite for bone substitutes. Ceram. Int. 2016, 42, 3725–3744. [Google Scholar] [CrossRef]
- Pajor, K.; Pajchel, L.; Kolmas, J. Hydroxyapatite and fluorapatite in conservative dentistry and oral implantology—A review. Materials 2019, 12, 2683. [Google Scholar] [CrossRef] [Green Version]
- Kattimani, V.S.; Kondaka, S.; Lingamaneni, K.P. Hydroxyapatite–-Past, Present, and Future in Bone Regeneration. Bone Tissue Regen. Insights 2016, 7, BTRI.S36138. [Google Scholar] [CrossRef] [Green Version]
- Tite, T.; Popa, A.-C.; Balescu, L.; Bogdan, I.; Pasuk, I.; Ferreira, J.; Stan, G. Cationic Substitutions in Hydroxyapatite: Current Status of the Derived Biofunctional Effects and Their In Vitro Interrogation Methods. Materials 2018, 11, 2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šupová, M. Substituted hydroxyapatites for biomedical applications: A review. Ceram. Int. 2015, 41, 9203–9231. [Google Scholar] [CrossRef]
- Sammons, R. Biological responses to hydroxyapatite. In Hydroxyapatite (Hap) for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2015; pp. 53–83. [Google Scholar]
- Bose, S.; Tarafder, S.; Bandyopadhyay, A. Hydroxyapatite coatings for metallic implants. In Hydroxyapatite (Hap) for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2015; pp. 143–157. [Google Scholar]
- Zakharov, N.A.; Polunina, I.A.; Polunin, K.E.; Rakitina, N.M.; Kochetkova, E.I.; Sokolova, N.P.; Kalinnikov, V.T. Calcium Hydroxyapatite for Medical Applications. Inorg. Mater. 2004, 40, 641–648. [Google Scholar] [CrossRef]
- Palazzo, B.; Sidoti, M.C.; Roveri, N.; Tampieri, A.; Sandri, M.; Bertolazzi, L.; Galbusera, F.; Dubini, G.; Vena, P.; Contro, R. Controlled drug delivery from porous hydroxyapatite grafts: An experimental and theoretical approach. Mater. Sci. Eng. C 2005, 25, 207–213. [Google Scholar] [CrossRef]
- Ghiasi, B.; Sefidbakht, Y.; Rezaei, M. Hydroxyapatite for Biomedicine and Drug Delivery. In Nanomaterials for Advanced Biological Applications; Springer: Cham, Switzerland, 2019; pp. 85–120. [Google Scholar]
- LeGeros, R.Z. Calcium Phosphate-Based Osteoinductive Materials. Chem. Rev. 2008, 108, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Dorozhkin, S. Calcium Orthophosphates in Nature, Biology and Medicine. Materials 2009, 2, 399–498. [Google Scholar] [CrossRef] [Green Version]
- Uskoković, V. Ion-doped hydroxyapatite: An impasse or the road to follow? Ceram. Int. 2020, 46, 11443–11465. [Google Scholar] [CrossRef]
- Royer, A.; Viguie, J.C.; Heughebaert, M.; Heughebaert, J.C. Stoichiometry of hydroxyapatite: Influence on the flexural strength. J. Mater. Sci. Mater. Med. 1993, 4, 76–82. [Google Scholar] [CrossRef]
- Miculescu, F.; Stan, G.E.; Ciocan, L.T.; Miculescu, M.; Berbecaru, A.; Antoniac, I. Cortical bone as resource for producing biomimetic materials for clinical use. Dig. J. Nanomater. Biostructures 2012, 7, 1667–1677. [Google Scholar]
- Zyman, Z.; Tkachenko, M.; Epple, M.; Polyakov, M.; Naboka, M. Magnesium-substituted hydroxyapatite ceramics. Materwiss. Werksttech. 2006, 37, 474–477. [Google Scholar] [CrossRef]
- Duta, L.; Oktar, F.N.; Stan, G.E.; Popescu-Pelin, G.; Serban, N.; Luculescu, C.; Mihailescu, I.N. Novel doped hydroxyapatite thin films obtained by pulsed laser deposition. Appl. Surf. Sci. 2013, 265, 41–49. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, X.; Gu, Z.; Qin, H.; Li, L.; Liu, J.; Yu, X. In vitro study on the degradation of lithium-doped hydroxyapatite for bone tissue engineering scaffold. Mater. Sci. Eng. C 2016, 66, 185–192. [Google Scholar] [CrossRef]
- Sang Cho, J.; Um, S.H.; Su Yoo, D.; Chung, Y.C.; Hye Chung, S.; Lee, J.C.; Rhee, S.H. Enhanced osteoconductivity of sodium-substituted hydroxyapatite by system instability. J. Biomed. Mater. Res. Part B Appl. Biomater. 2014, 102, 1046–1062. [Google Scholar] [CrossRef]
- Li, H.; Zhao, X.; Cao, S.; Li, K.; Chen, M.; Xu, Z.; Lu, J.; Zhang, L. Na-doped hydroxyapatite coating on carbon/carbon composites: Preparation, in vitro bioactivity and biocompatibility. Appl. Surf. Sci. 2012, 263, 163–173. [Google Scholar] [CrossRef]
- Dubnika, A.; Loca, D.; Rudovica, V.; Parekh, M.B.; Berzina-Cimdina, L. Functionalized silver doped hydroxyapatite scaffolds for controlled simultaneous silver ion and drug delivery. Ceram. Int. 2017, 43, 3698–3705. [Google Scholar] [CrossRef]
- Fielding, G.A.; Roy, M.; Bandyopadhyay, A.; Bose, S. Antibacterial and biological characteristics of silver containing and strontium doped plasma sprayed hydroxyapatite coatings. Acta Biomater. 2012, 8, 3144–3152. [Google Scholar] [CrossRef] [Green Version]
- AL-Haddad, A.; Che Ab Aziz, Z.A. Bioceramic-Based Root Canal Sealers: A Review. Int. J. Biomater. 2016, 2016, 9753210. [Google Scholar] [CrossRef] [Green Version]
- Kaygili, O.; Keser, S.; Dorozhkin, S.V.; Yakuphanoglu, F.; Al-Ghamdi, A.A.; Kirbag, S.; Sertkaya, D.; Ates, T.; Gursoy, N.C. Structural and Dielectrical Properties of Ag- and Ba-Substituted Hydroxyapatites. J. Inorg. Organomet. Polym. Mater. 2014, 24, 1001–1008. [Google Scholar] [CrossRef]
- Landi, E.; Logroscino, G.; Proietti, L.; Tampieri, A.; Sandri, M.; Sprio, S. Biomimetic Mg-substituted hydroxyapatite: From synthesis to in vivo behaviour. J. Mater. Sci. Mater. Med. 2008, 19, 239–247. [Google Scholar] [CrossRef]
- Andrés, N.C.; D’Elía, N.L.; Ruso, J.M.; Campelo, A.E.; Massheimer, V.L.; Messina, P.V. Manipulation of Mg 2+ –Ca 2+ Switch on the Development of Bone Mimetic Hydroxyapatite. ACS Appl. Mater. Interfaces 2017, 9, 15698–15710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, J.; Jiang, P.; Sun, G.; Ma, Z.; Hu, J.; Shen, X.; Tong, H. Comparisons among Mg, Zn, Sr, and Si doped nano-hydroxyapatite/chitosan composites for load-bearing bone tissue engineering applications. Mater. Chem. Front. 2017, 1, 900–910. [Google Scholar] [CrossRef]
- Aina, V.; Bergandi, L.; Lusvardi, G.; Malavasi, G.; Imrie, F.E.; Gibson, I.R.; Cerrato, G.; Ghigo, D. Sr-containing hydroxyapatite: Morphologies of HA crystals and bioactivity on osteoblast cells. Mater. Sci. Eng. C 2013, 33, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Hosick, H.L.; Bandyopadhyay, A.; Bose, S.; Ding, C.; Luk, K.D.K.; Cheung, K.M.C.; Lu, W.W. Preparation and cell–materials interactions of plasma sprayed strontium-containing hydroxyapatite coating. Surf. Coat. Technol. 2007, 201, 4685–4693. [Google Scholar] [CrossRef]
- Shanmugam, S.; Gopal, B. Copper substituted hydroxyapatite and fluorapatite: Synthesis, characterization and antimicrobial properties. Ceram. Int. 2014, 40, 15655–15662. [Google Scholar] [CrossRef]
- Stanić, V.; Dimitrijević, S.; Antić-Stanković, J.; Mitrić, M.; Jokić, B.; Plećaš, I.B.; Raičević, S. Synthesis, characterization and antimicrobial activity of copper and zinc-doped hydroxyapatite nanopowders. Appl. Surf. Sci. 2010, 256, 6083–6089. [Google Scholar] [CrossRef]
- Cox, S.C.; Jamshidi, P.; Grover, L.M.; Mallick, K.K. Preparation and characterisation of nanophase Sr, Mg, and Zn substituted hydroxyapatite by aqueous precipitation. Mater. Sci. Eng. C 2014, 35, 106–114. [Google Scholar] [CrossRef]
- Webster, T.J.; Massa-Schlueter, E.A.; Smith, J.L.; Slamovich, E.B. Osteoblast response to hydroxyapatite doped with divalent and trivalent cations. Biomaterials 2004, 25, 2111–2121. [Google Scholar] [CrossRef]
- Begam, H.; Kundu, B.; Chanda, A.; Nandi, S.K. MG63 osteoblast cell response on Zn doped hydroxyapatite (HAp) with various surface features. Ceram. Int. 2017, 43, 3752–3760. [Google Scholar] [CrossRef]
- Kolekar, T.V.; Thorat, N.D.; Yadav, H.M.; Magalad, V.T.; Shinde, M.A.; Bandgar, S.S.; Kim, J.H.; Agawane, G.L. Nanocrystalline hydroxyapatite doped with aluminium: A potential carrier for biomedical applications. Ceram. Int. 2016, 42, 5304–5311. [Google Scholar] [CrossRef]
- Ciobanu, G.; Bargan, A.M.; Luca, C. New cerium(IV)-substituted hydroxyapatite nanoparticles: Preparation and characterization. Ceram. Int. 2015, 41, 12192–12201. [Google Scholar] [CrossRef]
- Sundarabharathi, L.; Chinnaswamy, M.; Ponnamma, D.; Parangusan, H.; Al-Maadeed, M.A.A. Investigation of antimicrobial properties and in-vitro bioactivity of Ce 3+ -Sr 2+ dual-substituted nano hydroxyapatites. J. Am. Ceram. Soc. 2019, 102, 144–157. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, P.; Udduttula, A.; Uthirapathy, V. Development and characterisation of novel Ce-doped hydroxyapatite–Fe 3 O 4 nanocomposites and their in vitro biological evaluations for biomedical applications. IET Nanobiotechnol. 2018, 12, 138–146. [Google Scholar] [CrossRef]
- Mellier, C.; Fayon, F.; Boukhechba, F.; Verron, E.; LeFerrec, M.; Montavon, G.; Lesoeur, J.; Schnitzler, V.; Massiot, D.; Janvier, P.; et al. Design and properties of novel gallium-doped injectable apatitic cements. Acta Biomater. 2015, 24, 322–332. [Google Scholar] [CrossRef]
- Vladescu, A.; Padmanabhan, S.C.; Ak Azem, F.; Braic, M.; Titorencu, I.; Birlik, I.; Morris, M.A.; Braic, V. Mechanical properties and biocompatibility of the sputtered Ti doped hydroxyapatite. J. Mech. Behav. Biomed. Mater. 2016, 63, 314–325. [Google Scholar] [CrossRef]
- Nakazawa, M.; Yamada, M.; Wakamura, M.; Egusa, H.; Sakurai, K. Activation of Osteoblastic Function on Titanium Surface with Titanium-Doped Hydroxyapatite Nanoparticle Coating: An In Vitro Study. Int. J. Oral Maxillofac. Implant. 2017, 32, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Stipniece, L.; Stepanova, V.; Narkevica, I.; Salma-Ancane, K.; Boyd, A.R. Comparative study of surface properties of Mg-substituted hydroxyapatite bioceramic microspheres. J. Eur. Ceram. Soc. 2018, 38, 761–768. [Google Scholar] [CrossRef]
- Mehrjoo, M.; Javadpour, J.; Shokrgozar, M.A.; Farokhi, M.; Javadian, S.; Bonakdar, S. Effect of magnesium substitution on structural and biological properties of synthetic hydroxyapatite powder. Mater. Express 2015, 5, 41–48. [Google Scholar] [CrossRef]
- Popa, C.L.; Ciobanu, C.S. Synthesis and characterization of fluorescent hydroxyapatite. Rom. Rep. Phys. 2016, 68, 1170–1177. [Google Scholar]
- Xu, C.; Qu, X. Cerium oxide nanoparticle: A remarkably versatile rare earth nanomaterial for biological applications. NPG Asia Mater. 2014, 6, e90. [Google Scholar] [CrossRef]
- Pu’ad, N.M.; Koshy, P.; Abdullah, H.Z.; Idris, M.I.; Lee, T.C. Syntheses of hydroxyapatite from natural sources. Heliyon 2019, 5, e01588. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Basu, B. Doped biphasic calcium phosphate: Synthesis and structure. J. Asian Ceram. Soc. 2019, 7, 265–283. [Google Scholar] [CrossRef] [Green Version]
- Mucalo, M. Special Issue: Novel Advances and Approaches in Biomedical Materials Based on Calcium Phosphates. Materials 2019, 12, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qadir, M.; Li, Y.; Wen, C. Ion-substituted calcium phosphate coatings by physical vapor deposition magnetron sputtering for biomedical applications: A review. Acta Biomater. 2019, 89, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Arcos, D.; Vallet-Regí, M. Substituted hydroxyapatite coatings of bone implants. J. Mater. Chem. B 2020, 8, 1781–1800. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, M.; Negrini, N.C.; Scialla, S.; Marelli, B.; Farè, S.; Danti, S.; Buehler, M.J. Additive Manufacturing Approaches for Hydroxyapatite-Reinforced Composites. Adv. Funct. Mater. 2019, 29, 1903055. [Google Scholar] [CrossRef] [Green Version]
- Ofudje, E.A.; Adeogun, A.I.; Idowu, M.A.; Kareem, S.O. Synthesis and characterization of Zn-Doped hydroxyapatite: Scaffold application, antibacterial and bioactivity studies. Heliyon 2019, 5, e01716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Caetano, G.; Vyas, C.; Blaker, J.; Diver, C.; Bártolo, P. Polymer-Ceramic Composite Scaffolds: The Effect of Hydroxyapatite and β-tri-Calcium Phosphate. Materials 2018, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Pierantozzi, D.; Scalzone, A.; Jindal, S.; Stīpniece, L.; Šalma-Ancāne, K.; Dalgarno, K.; Gentile, P.; Mancuso, E. 3D printed Sr-containing composite scaffolds: Effect of structural design and material formulation towards new strategies for bone tissue engineering. Compos. Sci. Technol. 2020, 191, 108069. [Google Scholar] [CrossRef]
- Paredes, C.; Martínez-Vázquez, F.J.; Pajares, A.; Miranda, P. Development by robocasting and mechanical characterization of hybrid HA/PCL coaxial scaffolds for biomedical applications. J. Eur. Ceram. Soc. 2019, 39, 4375–4383. [Google Scholar] [CrossRef]
- Patterson, A.L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Burroughs, P.; Hamnett, A.; Orchard, A.F.; Thornton, G. Satellite structure in the X-ray photoelectron spectra of some binary and mixed oxides of lanthanum and cerium. J. Chem. Soc. Dalt. Trans. 1976, 1686–1698. [Google Scholar] [CrossRef]
- Popa, C.L.; Deniaud, A.; Michaud-Soret, I.; Guégan, R.; Motelica-Heino, M.; Predoi, D. Structural and Biological Assessment of Zinc Doped Hydroxyapatite Nanoparticles. J. Nanomater. 2016, 2016, 1062878. [Google Scholar] [CrossRef] [Green Version]
- Ciobanu, C.; Popa, C.; Predoi, D. Cerium doped hydroxyapatite nanoparticles synthesized by coprecipitation method. J. Serb. Chem. Soc. 2016, 81, 433–446. [Google Scholar] [CrossRef]
- Zhu, H.; Guo, D.; Sun, L.; Li, H.; Hanaor, D.A.H.; Schmidt, F.; Xu, K. Nanostructural insights into the dissolution behavior of Sr-doped hydroxyapatite. J. Eur. Ceram. Soc. 2018, 38, 5554–5562. [Google Scholar] [CrossRef] [Green Version]
- Markovic, M.; Fowler, B.O.; Tung, M.S. Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 553–568. [Google Scholar] [CrossRef]
- Stan, G.E. Adherent functional graded hydroxylapatite coatings produced by sputtering deposition techniques. J. Optoelectron. Adv. Mater. 2009, 11, 1132–1135. [Google Scholar]
- Popescu, A.C.; Florian, P.E.; Stan, G.E.; Popescu-Pelin, G.; Zgura, I.; Enculescu, M.; Oktar, F.N.; Trusca, R.; Sima, L.E.; Roseanu, A.; et al. Physical-chemical characterization and biological assessment of simple and lithium-doped biological-derived hydroxyapatite thin films for a new generation of metallic implants. Appl. Surf. Sci. 2018, 439, 724–735. [Google Scholar] [CrossRef]
- Madupalli, H.; Pavan, B.; Tecklenburg, M.M.J. Carbonate substitution in the mineral component of bone: Discriminating the structural changes, simultaneously imposed by carbonate in A and B sites of apatite. J. Solid State Chem. 2017, 255, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Dutreilh-Colas, M.; Hjezi, Z.; Cornette, J.; El Felss, N.; Champion, E.; Damia, C. Functionalization of Hydroxyapatite Ceramics: Raman Mapping Investigation of Silanization. Ceramics 2019, 2, 372–384. [Google Scholar] [CrossRef] [Green Version]
- Aina, V.; Lusvardi, G.; Annaz, B.; Gibson, I.R.; Imrie, F.E.; Malavasi, G.; Menabue, L.; Cerrato, G.; Martra, G. Magnesium- and strontium-co-substituted hydroxyapatite: The effects of doped-ions on the structure and chemico-physical properties. J. Mater. Sci. Mater. Med. 2012, 23, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef]
- Duta, L.; Mihailescu, N.; Popescu, A.C.; Luculescu, C.R.; Mihailescu, I.N.; Çetin, G.; Gunduz, O.; Oktar, F.N.; Popa, A.C.; Kuncser, A.; et al. Comparative physical, chemical and biological assessment of simple and titanium-doped ovine dentine-derived hydroxyapatite coatings fabricated by pulsed laser deposition. Appl. Surf. Sci. 2017, 413, 129–139. [Google Scholar] [CrossRef]
- Sa, Y.; Guo, Y.; Feng, X.; Wang, M.; Li, P.; Gao, Y.; Yang, X.; Jiang, T. Are different crystallinity-index-calculating methods of hydroxyapatite efficient and consistent? New J. Chem. 2017, 41, 5723–5731. [Google Scholar] [CrossRef]
- Tang, Y.; Chappell, H.F.; Dove, M.T.; Reeder, R.J.; Lee, Y.J. Zinc incorporation into hydroxylapatite. Biomaterials 2009, 30, 2864–2872. [Google Scholar] [CrossRef]
- Ren, F.; Leng, Y.; Xin, R.; Ge, X. Synthesis, characterization and ab initio simulation of magnesium-substituted hydroxyapatite. Acta Biomater. 2010, 6, 2787–2796. [Google Scholar] [CrossRef]
- Rincón-López, J.; Hermann-Muñoz, J.; Giraldo-Betancur, A.; De Vizcaya-Ruiz, A.; Alvarado-Orozco, J.; Muñoz-Saldaña, J. Synthesis, Characterization and In Vitro Study of Synthetic and Bovine-Derived Hydroxyapatite Ceramics: A Comparison. Materials 2018, 11, 333. [Google Scholar] [CrossRef] [Green Version]
- Cawthray, J.F.; Creagh, A.L.; Haynes, C.A.; Orvig, C. Ion Exchange in Hydroxyapatite with Lanthanides. Inorg. Chem. 2015, 54, 1440–1445. [Google Scholar] [CrossRef]
- Dean, J.A. Lange’s Handbook of Chemistry, 15th ed.; McGraw-Hill: New York, NY, USA, 1999; ISBN 0-07-016384-7. [Google Scholar]
- Nouri-Felekori, M.; Khakbiz, M.; Nezafati, N. Synthesis and characterization of Mg, Zn and Sr-incorporated hydroxyapatite whiskers by hydrothermal method. Mater. Lett. 2019, 243, 120–124. [Google Scholar] [CrossRef]
- Sigma Life Science—Cell Culture. 2014. Available online: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/Brochure/1/cell-culture-manual.pdf (accessed on 5 July 2021).
- Kumari, M.; Singh, S.P.; Chinde, S.; Rahman, M.F.; Mahboob, M.; Grover, P. Toxicity study of cerium oxide nanoparticles in human neuroblastoma cells. Int. J. Toxicol. 2014, 33, 86–97. [Google Scholar] [CrossRef]
- Kho, D.; MacDonald, C.; Johnson, R.; Unsworth, C.; O’Carroll, S.; Mez, E.; Angel, C.; Graham, E. Application of xCELLigence RTCA Biosensor Technology for Revealing the Profile and Window of Drug Responsiveness in Real Time. Biosensors 2015, 5, 199–222. [Google Scholar] [CrossRef] [Green Version]
- Bertuzzi, A.; Gandolfi, A.; Sinisgalli, C.; Starace, G.; Ubezio, P. Cell loss and the concept of potential doubling time. Cytometry 1997, 29, 34–40. [Google Scholar] [CrossRef]
- Oraiopoulou, M.E.; Tzamali, E.; Tzedakis, G.; Vakis, A.; Papamatheakis, J.; Sakkalis, V. In Vitro/In silico study on the role of doubling time heterogeneity among primary glioblastoma cell lines. BioMed Res. Int. 2017, 2017, 8569328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yeung, K.W.K. Bone grafts and biomaterials substitutes for bone defect repair: A review. Bioact. Mater. 2017, 2, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Stipniece, L.; Wilson, S.; Curran, J.M.; Chen, R.; Salma-Ancane, K.; Sharma, P.K.; Meenan, B.J.; Boyd, A.R. Strontium substituted hydroxyapatite promotes direct primary human osteoblast maturation. Ceram. Int. 2021, 47, 3368–3379. [Google Scholar] [CrossRef]
- Pan, H.B.; Li, Z.Y.; Wang, T.; Lam, W.M.; Wong, C.T.; Darvell, B.W.; Luk, K.D.K.; Hu, Y.; Lu, W.W. Nucleation of strontium-substituted apatite. Cryst. Growth Des. 2009, 9, 3342–3345. [Google Scholar] [CrossRef]
- Suganthi, R.V.; Elayaraja, K.; Joshy, M.I.A.; Chandra, V.S.; Girija, E.K.; Kalkura, S.N. Fibrous growth of strontium substituted hydroxyapatite and its drug release. Mater. Sci. Eng. C 2011, 31, 593–599. [Google Scholar] [CrossRef]
- O’Donnell, M.D.; Fredholm, Y.; de Rouffignac, A.; Hill, R.G. Structural analysis of a series of strontium-substituted apatites. Acta Biomater. 2008, 4, 1455–1464. [Google Scholar] [CrossRef]
- Blumenthal, N.C.; Posner, A.S. Hydroxyapatite: Mechanism of formation and properties. Calcif. Tissue Res. 1973, 13, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Loong, C.K.; Rey, C.; Kuhn, L.T.; Combes, C.; Wu, Y.; Chen, S.H.; Glimcher, M.J. Evidence of hydroxyl-ion deficiency in bone apatites: An inelastic neutron-scattering study. Bone 2000, 26, 599–602. [Google Scholar] [CrossRef]
- Pasteris, J.D.; Wopenka, B.; Freeman, J.J.; Rogers, K.; Valsami-Jones, E.; Van Der Houwen, J.A.M.; Silva, M.J. Lack of OH in nanocrystalline apatite as a function of degree of atomic order: Implications for bone and biomaterials. Biomaterials 2004, 25, 229–238. [Google Scholar] [CrossRef]
- Khalid, H.; Chaudhry, A.A. Basics of hydroxyapatite—structure, synthesis, properties, and clinical applications. In Handbook of Ionic Substituted Hydroxyapatites; Woodhead Publishing: Cambridge, UK, 2020. [Google Scholar]
- Terra, J.; Dourado, E.R.; Eon, J.-G.; Ellis, D.E.; Gonzalez, G.; Rossi, A.M. The structure of strontium-doped hydroxyapatite: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2009, 11, 568–577. [Google Scholar] [CrossRef]
- Gomes, S.; Nedelec, J.-M.; Jallot, E.; Sheptyakov, D.; Renaudin, G. Unexpected Mechanism of Zn 2+ Insertion in Calcium Phosphate Bioceramics. Chem. Mater. 2011, 23, 3072–3085. [Google Scholar] [CrossRef]
- Baino, F.; Hamzehlou, S.; Kargozar, S. Bioactive glasses: Where are we and where are we going? J. Funct. Biomater. 2018, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Bhugra, C.; Pikal, M.J. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. J. Pharm. Sci. 2008, 97, 1329–1349. [Google Scholar] [CrossRef]
- Chen, S.; Shi, Y.; Zhang, X.; Ma, J. Biomimetic synthesis of Mg-substituted hydroxyapatite nanocomposites and three-dimensional printing of composite scaffolds for bone regeneration. J. Biomed. Mater. Res. Part A 2019, 107, 2512–2521. [Google Scholar] [CrossRef]
- Reger, N.C.; Kundu, B.; Balla, V.K.; Bhargava, A.K. In vitro cytotoxicity and ion release of multi-ion doped hydroxyapatite. Int. J. Appl. Ceram. Technol. 2019, 16, 503–516. [Google Scholar] [CrossRef]
- Teng, Z.; Kuang, X.; Wang, J.; Zhang, X. Real-time cell analysis—A new method for dynamic, quantitative measurement of infectious viruses and antiserum neutralizing activity. J. Virol. Methods 2013, 193, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Şener, L.T.; Albeniz, G.; Dinç, B.; Albeniz, I. iCELLigence real-time cell analysis system for examining the cytotoxicity of drugs to cancer cell lines. Exp. Ther. Med. 2017, 14, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz-Hajduk, J.; Ochocka, J.R. Real-time cell analysis system in cytotoxicity applications: Usefulness and comparison with tetrazolium salt assays. Toxicol. Rep. 2020, 7, 335–344. [Google Scholar] [CrossRef]
- Moniri, M.R.; Young, A.; Reinheimer, K.; Rayat, J.; Dai, L.-J.; Warnock, G.L. Dynamic assessment of cell viability, proliferation and migration using real time cell analyzer system (RTCA). Cytotechnology 2015, 67, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvinova, L.S.; Shupletsova, V.V.; Khaziakhmatova, O.G.; Yurova, K.A.; Malashchenko, V.V.; Melashchenko, E.S.; Todosenko, N.M.; Khlusova, M.Y.; Sharkeev, Y.P.; Komarova, E.G.; et al. Behavioral Changes of Multipotent Mesenchymal Stromal Cells in Contact with Synthetic Calcium Phosphates in vitro. Cell Tissue Biol. 2018, 12, 112–119. [Google Scholar] [CrossRef]
- Pirosa, A.; Gottardi, R.; Alexander, P.G.; Tuan, R.S. Engineering in-vitro stem cell-based vascularized bone models for drug screening and predictive toxicology. Stem Cell Res. Ther. 2018, 9, 112. [Google Scholar] [CrossRef] [Green Version]
- Zreiqat, H.; Howlett, C.R.; Zannettino, A.; Evans, P.; Schulze-Tanzil, G.; Knabe, C.; Shakibaei, M. Mechanisms of magnesium-stimulated adhesion of osteoblastic cells to commonly used orthopaedic implants. J. Biomed. Mater. Res. 2002, 62, 175–184. [Google Scholar] [CrossRef]
- Sader, M.S.; LeGeros, R.Z.; Soares, G.A. Human osteoblasts adhesion and proliferation on magnesium-substituted tricalcium phosphate dense tablets. J. Mater. Sci. Mater. Med. 2009, 20, 521–527. [Google Scholar] [CrossRef]
- Kazimierczak, P.; Kolmas, J.; Przekora, A. Biological Response to Macroporous Chitosan-Agarose Bone Scaffolds Comprising Mg- and Zn-Doped Nano-Hydroxyapatite. Int. J. Mol. Sci. 2019, 20, 3835. [Google Scholar] [CrossRef] [Green Version]
- Lange, T.S.; Bielinsky, A.K.; Kirchberg, K.; Bank, I.; Herrmann, K.; Krieg, T.; Scharffetter-Kochanek, K. Mg2+ and Ca2+ Differentially Regulate β1 Integrin-Mediated Adhesion of Dermal Fibroblasts and Keratinocytes to Various Extracellular Matrix Proteins. Exp. Cell Res. 1994, 214, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Inomata, K.; Honda, M. Co-Culture of Osteoblasts and Endothelial Cells on a Microfiber Scaffold to Construct Bone-Like Tissue with Vascular Networks. Materials 2019, 12, 2869. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chirică, I.M.; Enciu, A.-M.; Tite, T.; Dudău, M.; Albulescu, L.; Iconaru, S.L.; Predoi, D.; Pasuk, I.; Enculescu, M.; Radu, C.; et al. The Physico-Chemical Properties and Exploratory Real-Time Cell Analysis of Hydroxyapatite Nanopowders Substituted with Ce, Mg, Sr, and Zn (0.5–5 at.%). Materials 2021, 14, 3808. https://doi.org/10.3390/ma14143808
Chirică IM, Enciu A-M, Tite T, Dudău M, Albulescu L, Iconaru SL, Predoi D, Pasuk I, Enculescu M, Radu C, et al. The Physico-Chemical Properties and Exploratory Real-Time Cell Analysis of Hydroxyapatite Nanopowders Substituted with Ce, Mg, Sr, and Zn (0.5–5 at.%). Materials. 2021; 14(14):3808. https://doi.org/10.3390/ma14143808
Chicago/Turabian StyleChirică, Iuliana Maria, Ana-Maria Enciu, Teddy Tite, Maria Dudău, Lucian Albulescu, Simona Liliana Iconaru, Daniela Predoi, Iuliana Pasuk, Monica Enculescu, Cristian Radu, and et al. 2021. "The Physico-Chemical Properties and Exploratory Real-Time Cell Analysis of Hydroxyapatite Nanopowders Substituted with Ce, Mg, Sr, and Zn (0.5–5 at.%)" Materials 14, no. 14: 3808. https://doi.org/10.3390/ma14143808
APA StyleChirică, I. M., Enciu, A.-M., Tite, T., Dudău, M., Albulescu, L., Iconaru, S. L., Predoi, D., Pasuk, I., Enculescu, M., Radu, C., Mihalcea, C. G., Popa, A.-C., Rusu, N., Niţă, S., Tănase, C., & Stan, G. E. (2021). The Physico-Chemical Properties and Exploratory Real-Time Cell Analysis of Hydroxyapatite Nanopowders Substituted with Ce, Mg, Sr, and Zn (0.5–5 at.%). Materials, 14(14), 3808. https://doi.org/10.3390/ma14143808