
Structural, Thermal, and Vibrational Properties of N,N-Dimethylglycine–Chloranilic Acid—A New Co-Crystal Based on an Aliphatic Amino Acid

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. X-ray Diffraction Studies
3.2. NMR Spectroscopy
3.3. Thermal Analysis (TG, DSC)
3.4. Vibrational Spectra
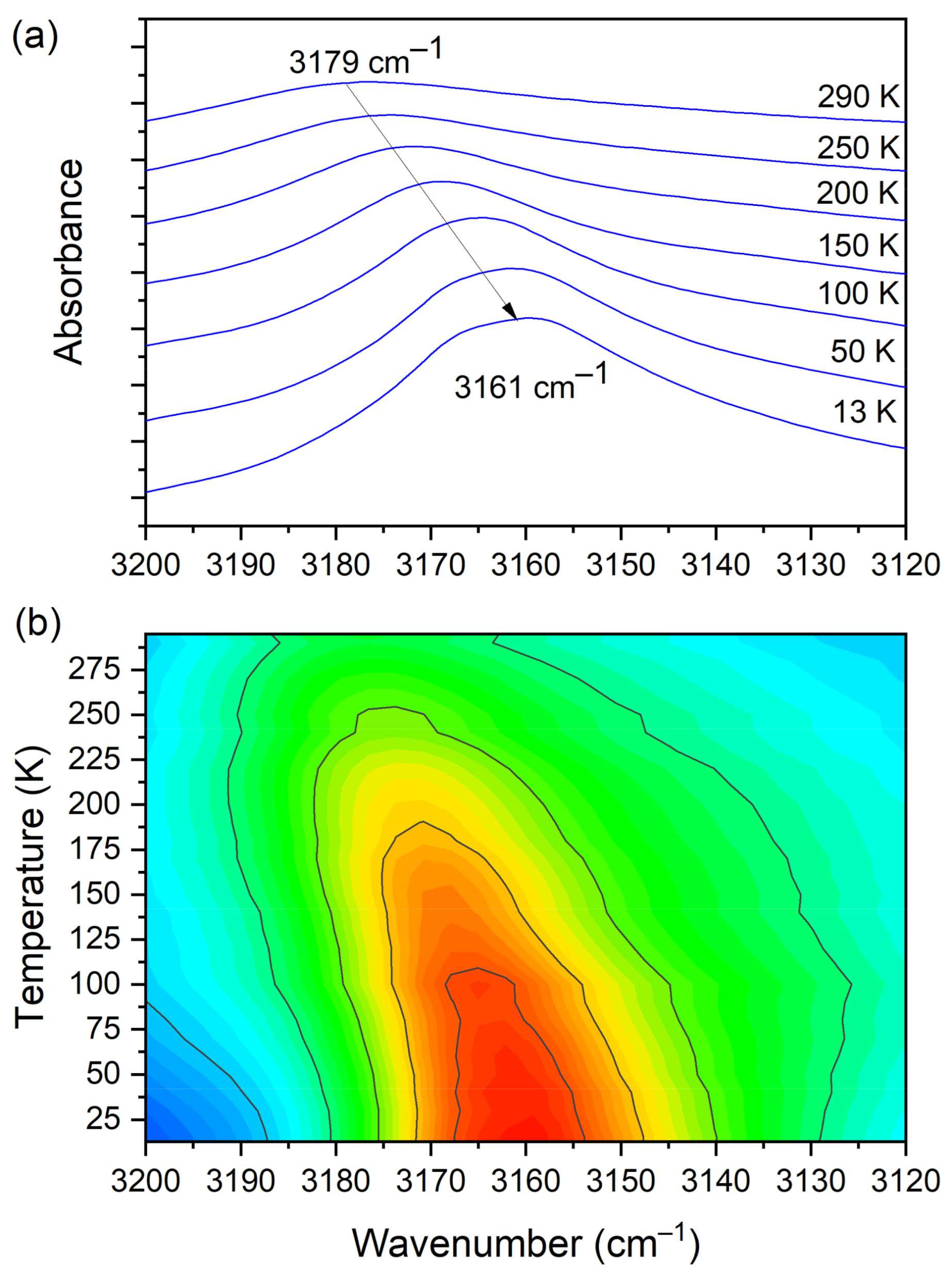
3.5. Temperature-Dependent IR Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lane, H.-Y.; Huang, C.-L.; Wu, P.-L.; Liu, Y.-C.; Chang, Y.-C.; Lin, P.-Y.; Chen, P.-W.; Tsai, G. Glycine Transporter I Inhibitor, N-Methylglycine (Sarcosine), Added to Clozapine for the Treatment of Schizophrenia. Biol. Psychiatry 2006, 60, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Anioła, M.; Dega-Szafran, Z.; Katrusiak, A.; Szafran, M. NH⋯O and OH⋯O Interactions of Glycine Derivatives with Squaric Acid. New J. Chem. 2014, 38, 3556–3568. [Google Scholar] [CrossRef]
- Hildre, A.S.; Solvang, S.-E.H.; Aarsland, D.; Midtun, Ø.; McCann, A.; Ervik, A.O.; Nygård, O.; Ueland, P.M.; Nordrehaug, J.E.; Giil, L.M. Components of the Choline Oxidation Pathway Modify the Association between the Apolipoprotein Ε4 Gene Variant and Cognitive Decline in Patients with Dementia. Brain Res. 2020, 1726, 146519. [Google Scholar] [CrossRef] [PubMed]
- Graber, C.D.; Goust, J.M.; Glassman, A.D.; Kendall, R.; Loadholt, C.B. Immunomodulating Properties of Dimethylglycine in Humans. J. Infect Dis. 1981, 143, 101–105. [Google Scholar] [CrossRef]
- Cools, A.; Maes, D.; Buyse, J.; Kalmar, I.D.; Vandermeiren, J.-A.; Janssens, G.P.J. Effect of N,N-Dimethylglycine Supplementation in Parturition Feed for Sows on Metabolism, Nutrient Digestibility and Reproductive Performance. Animal 2010, 4, 2004–2011. [Google Scholar] [CrossRef]
- Gillies, N.A.; Milan, A.M.; Chia, P.H.P.; Sharma, P.; Mitchell, S.M.; Zeng, N.; Ramzan, F.; D’Souza, R.F.; Mitchell, C.J.; Knowles, S.O.; et al. Responsiveness of One-Carbon Metabolites to a High-Protein Diet in Older Men: Results from a 10-Wk Randomized Controlled Trial. Nutrition 2021, 89, 111231. [Google Scholar] [CrossRef]
- Petrosyan, A.M.; Ghazaryan, V.V.; Giester, G.; Fleck, M.; Tylczyński, Z.; Wiesner, M. Halogenides of Dimethylglycine in Comparison with Respective Salts of Glycine, Sarcosine and Betaine. J. Mol. Struct. 2018, 1158, 106–121. [Google Scholar] [CrossRef]
- Fleck, M.; Petrosyan, A.M. Salts of Amino Acids; Springer International Publishing: Cham, Switzerland, 2014; ISBN 9783319062983. [Google Scholar]
- Schaack, G. Experimental Results on Phase Transitions in Betaine Compounds. Ferroelectrics 1990, 104, 147–158. [Google Scholar] [CrossRef]
- Abdinejad, T.; Zamanloo, M.R.; Esrafili, M.D.; Seifzadeh, D. Constructing a Dual-Mode Photochromic and Intrinsically Electrochromic Device Based on Organic Salts Prepared by Acid-Base Neutralization of Pyromellitic Diimides Bearing a Carboxyl Group with Aliphatic Amines. J. Photochem. Photobiol. A Chem. 2020, 388, 112162. [Google Scholar] [CrossRef]
- Balashova, E.V.; Krichevtsov, B.B.; Zaitseva, N.V.; Yurko, E.I.; Svinarev, F.B. Ferroelectric Thin Films of Deuterated Betaine Arsenate. Ferroelectrics 2015, 482, 60–69. [Google Scholar] [CrossRef]
- Clarke, H.T.; Gillespie, H.B.; Weisshaus, S.Z. The Action of Formaldehyde on Amines and Amino Acids1. J. Am. Chem. Soc. 1933, 55, 4571–4587. [Google Scholar] [CrossRef]
- Rodrigues, V.H.; Paixão, J.A.; Costa, M.M.; Beja, A.M. Conformation of Cationic N,N-Dimethylglycine in Dimethylglycinium Trifluoroacetate. Acta Crystallogr. C 2001, 57, 417–420. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Hetmańczyk, J.; Nowicka-Scheibe, J.; Maurin, J.K.; Schilf, W.; Rozwadowski, Z. Spectroscopic, Thermal and Structural Studies of New l-Leucine and d-Leucine Complexes with Chloranilic Acid. J. Mol. Struct. 2017, 1133, 464–471. [Google Scholar] [CrossRef]
- Hetmańczyk, J.; Nowicka-Scheibe, J.; Maurin, J.K.; Pawlukojć, A. Low Temperature Investigations of Dynamic Properties in L-Leucine—Chloranilic Acid Complex. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 200, 281–289. [Google Scholar] [CrossRef]
- Zaman, M.B.; Tomura, M.; Yamashita, Y. Crystal Engineering Using Anilic Acids and Dipyridyl Compounds through a New Supramolecular Synthon. J. Org. Chem. 2001, 66, 5987–5995. [Google Scholar] [CrossRef]
- Klinman, J.P.; Mu, D. Quinoenzymes in Biology. Annu. Rev. Biochem. 1994, 63, 299–344. [Google Scholar] [CrossRef]
- Gómez-Zavaglia, A.; Fausto, R. Low-Temperature Solid-State FTIR Study of Glycine, Sarcosine and N,N-Dimethylglycine: Observation of Neutral Forms of Simple α-Amino Acids in the Solid State. Phys. Chem. Chem. Phys. 2003, 5, 3154–3161. [Google Scholar] [CrossRef]
- Matei, A.; Drichko, N.; Gompf, B.; Dressel, M. Far-Infrared Spectra of Amino Acids. Chem. Phys. 2005, 316, 61–71. [Google Scholar] [CrossRef]
- Wolpert, M.; Hellwig, P. Infrared Spectra and Molar Absorption Coefficients of the 20 Alpha Amino Acids in Aqueous Solutions in the Spectral Range from 1800 to 500cm−1. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2006, 64, 987–1001. [Google Scholar] [CrossRef]
- Culka, A.; Jehlička, J.; Edwards, H.G.M. Acquisition of Raman Spectra of Amino Acids Using Portable Instruments: Outdoor Measurements and Comparison. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2010, 77, 978–983. [Google Scholar] [CrossRef]
- Filho, P.F.F.; Jiao, X.; Freire, P.T.; Lima, J.A., Jr.; dos Santos, A.O.; Henry, P.F.; Yokaichiya, F.; Kremner, E.; Bordallo, H.N. Structure–Property Relations in Crystalline L-Leucine Obtained from Calorimetry, X-rays, Neutron and Raman Scattering. Phys. Chem. Chem. Phys. 2011, 13, 6576–6583. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Bator, G.; Sobczyk, L.; Grech, E.; Nowicka-Scheibe, J. Inelastic Neutron Scattering, Raman, Infrared and DFT Theoretical Studies on Chloranilic Acid. J. Phys. Org. Chem. 2003, 16, 709–714. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, Agilent Technologies, Version 1.171.33.66; CrysAlis171. NET, Release 28-04-2010. Empirical Absorption Correction Using Spherical Harmonics, Implemented in SCALE3 ABSPACK Scaling Algorithm. Available online: https://www.rigakuxrayforum.com/ (accessed on 8 April 2021).
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Natkaniec, I.; Chudoba, D.; Hetmańczyk, L.; Kazimirov, V.Y.; Krawczyk, J.; Sashin, I.L.; Zalewski, S. Parameters of the NERA Spectrometer for Cold and Thermal Moderators of the IBR-2 Pulsed Reactor. J. Phys. Conf. Ser. 2014, 554, 012002. [Google Scholar] [CrossRef]
- Parker, S.F.; Lennon, D.; Albers, P.W. Vibrational Spectroscopy with Neutrons: A Review of New Directions. Appl. Spectrosc. 2011, 65, 1325–1341. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons: With Applications in Chemistry, Biology, Materials Science and Catalysis; Series on Neutron Techniques and Applications; World Scientific: Singapore, 2005; Volume 3, ISBN 9789812560131. [Google Scholar]
- Pawlukojć, A.; Sobczyk, L. Application of Inelastic Neutron Scattering (INS) in Studies on Low Frequency Molecular Vibrations. Trends Appl. Spectrosc. 2004, 5, 117–136. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Kristallogr. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational Density-Functional Perturbation Theory for Dielectrics and Lattice Dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A.; Scheffler, M. Accurate Molecular Van Der Waals Interactions from Ground-State Electron Density and Free-Atom Reference Data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Schatschneider, B.; Monaco, S.; Tkatchenko, A.; Liang, J.-J. Understanding the Structure and Electronic Properties of Molecular Crystals Under Pressure: Application of Dispersion Corrected DFT to Oligoacenes. J. Phys. Chem. A 2013, 117, 8323–8331. [Google Scholar] [CrossRef]
- Dymkowski, K.; Parker, S.F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The Modern Software for INS Interpretation. Phys. B Condens. Matter 2018, 551, 443–448. [Google Scholar] [CrossRef]
- Arnold, O.; Bilheux, J.C.; Borreguero, J.M.; Buts, A.; Campbell, S.I.; Chapon, L.; Doucet, M.; Draper, N.; Ferraz Leal, R.; Gigg, M.A.; et al. Mantid—Data Analysis and Visualization Package for Neutron Scattering and μ SR Experiments. Nucl. Instrum. Methods A 2014, 764, 156–166. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17. University of Western Australia. 2017. Available online: https://hirshfeldsurface.net (accessed on 8 April 2021).
- Pawlukojć, A.; Hetmańczyk, J.; Hetmańczyk, Ł.; Nowicka-Scheibe, J.; Maurin, J.K.; Schilf, W.; Trzybiński, D.; Woźniak, K. Evidence of Low Temperature Phase Transition in 2,6-Dimethylpyrazine—Picric Acid Cocrystal by Means of Temperature Dependent Investigations: X-Ray, DSC and IR. J. Mol. Struct. 2021, 1228, 129432. [Google Scholar] [CrossRef]
- Pawlukojć, A.; Sobczyk, L.; Prager, M.; Bator, G.; Grech, E.; Nowicka-Scheibe, J. DFT Calculations of 2,6-Dimethylpyrazine (26DMP) and Its Complex with Chloranilic Acid (CLA): Comparison to INS, IR and Raman Vibration Spectra. J. Mol. Struct. 2008, 892, 261–267. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 8 April 2021).
- Sawka-Dobrowolska, W.; Bator, G.; Sobczyk, L.; Grech, E.; Nowicka-Scheibe, J.; Pawlukojć, A. Structure and Vibrational Spectra of 1:1 Chloranilic Acid (CLA)—Tetramethylpyrazine (TMP) Complex. Struct. Chem. 2005, 16, 281–286. [Google Scholar] [CrossRef]
- Bator, G.; Sawka-Dobrowolska, W.; Sobczyk, L.; Owczarek, M.; Pawlukojć, A.; Grech, E.; Nowicka-Scheibe, J. Hydrogen Bonded NHO Chains Formed by Chloranilic Acid (CLA) with 4,4′-Di-t-Butyl-2,2′-Bipyridyl (DtBBP) in the Solid State. Chem. Phys. 2012, 392, 114–121. [Google Scholar] [CrossRef]
- Carabatos-Nédelec, C.; Becker, P. Order–Disorder and Structural Phase Transitions in Solid-State Materials by Raman Scattering Analysis. J. Raman Spectrosc. 1997, 28, 663–671. [Google Scholar] [CrossRef]
- da R. Andrade, P.; Rao, A.D.P.; Katiyar, R.S.; Porto, S.P.S. Analysis of the Relationship between Temperature Dependence of the Libration Mode and Dielectric Relaxation in NaNO2. Solid State Commun. 1973, 12, 847–851. [Google Scholar] [CrossRef]
- da R. Andrade, P.; Porto, S.P.S. On Linewidth of Phonons Associated to a Disorder Mechanism. Solid State Commun. 1973, 13, 1249–1254. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C10H11Cl2NO6 |
|---|---|
| Formula weight | 312.10 |
| Temperature | 293(2) K |
| Wavelength | 1.54184 Å |
| Crystal system | Triclinic |
| Space group | P-1 |
| Unit cell dimensions | a = 9.6199(4) Å, α = 97.749(4)°. |
| b = 12.0092(5) Å, β = 110.173(4)°. | |
| c = 12.8763(6) Å, γ = 111.415(4)°. | |
| Volume | 1240.68(10) Å3 |
| Z | 4 |
| Density (calculated) | 1.671 Mg/m3 |
| Absorption coefficient | 4.960 mm−1 |
| F(000) | 640 |
| Crystal size | 0.2 × 0.2 × 0.03 mm3 |
| Theta range for data collection | 5.16 to 71.16°. |
| Index ranges | −11 ≤ h ≤ 10, −14 ≤ k ≤ 14, −15 ≤ l ≤ 15 |
| Reflections collected | 15,069 |
| Independent reflections | 4409 (R(int) = 0.1036) |
| Completeness to theta = 71.16° | 91.9% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4409/0/361 |
| Goodness-of-fit on F2 | 0.980 |
| Final R indices (I > 2σ(I)) | R1 = 0.0471, wR2 = 0.1233 |
| R indices (all data) | R1 = 0.0553, wR2 = 0.1283 |
| Largest diff. peak and hole | 0.546 and −0.303 e·Å−3 |
| Coordinates | Experimental X-ray | Calculated CASTEP/PBE + vdW | Coordinates | Experimental X-ray | Calculated CASTEP/PBE + vdW |
|---|---|---|---|---|---|
| Bond lengths (Å) | N1A–C4A–H4A1 | 109.5(1) | 107.8 | ||
| C1A–C2A | 1.512(3) | 1.521 | N1A–C4A–H4A3 | 109.5(1) | 109.2 |
| C1C–C2C | 1.466(4) | 1.452 | N1B–C3B–H3B2 | 109.5(1) | 108.6 |
| C1D–C2D | 1.458(4) | 1.449 | O1A–C1A–O2A | 121.8(2) | 121.8 |
| C2A–H2A1 | 0.971 | 1.097 | O1B–C1B–C2B | 122.2(2) | 122.6 |
| C2A–H2A2 | 0.970 | 1.097 | O1D–C1D–C6D | 118.8(2) | 118.2 |
| C2C–C3C | 1.332(4) | 1.363 | O2A–C1A–C2A | 116.1(2) | 115.9 |
| C3A–H3A1 | 0.960 | 1.091 | O3D–C3D–C2D | 122.5(2) | 123.0 |
| C3C–C4C | 1.512(4) | 1.513 | O4D–C4D–C3D | 115.6(2) | 115.0 |
| C4A–H4A1 | 0.960 | 1.092 | O6D–C6D–C5D | 126.6(2) | 126.1 |
| C4C–C5C | 1.421(4) | 1.422 | Torsion angles (°) | ||
| C5C–C6C | 1.371(4) | 1.395 | H2A–O2A–C1A–O1A | −180(3) | 179.1 |
| Cl2C–C2C | 1.723(3) | 1.731 | H2A–O2A–C1A–C2A | 1(3) | −0.2 |
| Cl5D–C5D | 1.731(3) | 1.737 | H1A–N1A–C2A–H2A2 | 76 | 73 |
| N1A–H1A | 0.860(4) | 1.061 | H1A–N1A–C3A–H3A1 | −62 | −57 |
| N1A–C3A | 1.490(4) | 1.505 | H1A–N1A–C3A–H3A2 | 178 | −176.1 |
| N1A–C4A | 1.484(3) | 1.504 | H1A–N1A–C3A–H3A3 | 58 | 63.7 |
| N1A–C2A | 1.483(2) | 1.498 | C2A–N1A–C4A–H4A1 | −179.7 | 171.9 |
| O1A–C1A | 1.208(4) | 1.225 | C2A–N1A–C4A–H4A2 | 60.3 | 52.5 |
| O2A–H2A | 0.820(4) | 1.032 | C3A–N1A–C4A–H4A2 | −174.7 | 177.4 |
| O2A–C1A | 1.303(2) | 1.320 | C3A–N1A–C4A–H4A3 | 65.4 | 57.6 |
| O2B–H2B | 0.730(4) | 1.030 | O2A–C1A–C2A–N1A | −173.9(2) | −172.6 |
| O3D–C3D | 1.325(3) | 1.319 | O2A–C1A–C2A–H2A1 | 52.8 | −50.8 |
| O4C–C4C | 1.227(3) | 1.243 | H2B–O2B–C1B–C2B | −3(3) | −0.8 |
| O3D–H3D | 0.840(4) | 1.002 | C3B–N1B–C2B–C1B | −162.1(2) | −161.2 |
| Bond angles (°) | C3B–N1B–C2B–H2B1 | 76.7 | 75.5 | ||
| C1A–C2A–H2A1 | 109.2(1) | 111.1 | C4B–N1B–C2B–H2B1 | 179.6(2) | 177.7 |
| C1B–C2B–H2B2 | 109.1(1) | 109.0 | C2B–N1B–C3B–H3B1 | 180 | 173.4 |
| C1C–C2C–C3C | 120.7(2) | 120.2 | C2B–N1B–C3B–H3B2 | 59.8 | 53.9 |
| C1C–C6C–C5C | 117.8(2) | 118.4 | H1B–N1B–C4B–H4B2 | 179 | −175.3 |
| C2A–N1A–C3A | 112.4(2) | 112.2 | C2B–N1B–C4B–H4B3 | −59.8 | −55.6 |
| C2A–N1A–C4A | 111.0(2) | 111.1 | O1B–C1B–C2B–N1B | 4.3(4) | 4.3 |
| C2C–C1C–C6C | 118.2(2) | 118.3 | O2B–C1B–C2B–N1B | −176.2(2) | −176.4 |
| C2B–N1B–C4B | 110.4(2) | 110.1 | H3C–O3C–C3C–C4C | 0(3) | −3.7 |
| C3C–C4C–C5C | 117.2(2) | 117.8 | C6C–C1C–C2C–Cl2C | 176.7(2) | 177.1 |
| C4C–C5C–C6C | 123.7(2) | 122.4 | O1C–C1C–C6C–C5C | −176.2(2) | −175.5 |
| Cl2C–C2C–C1C | 117.5(2) | 118.6 | C1C–C2C–C3C–O3C | −179.0(2) | −178.6 |
| Cl2C–C2C–C3C | 121.7(2) | 121.0 | O3C–C3C–C4C–O4C | 4.1(3) | 4.2 |
| H1A–N1A–C2A | 107.0(3) | 108.0 | O4C–C4C–C5C–Cl5C | −4.7(4) | −5.3 |
| H1A–N1A–C4A | 108.0(3) | 106.2 | C3C–C4C–C5C–C6C | −6.1(4) | −5.7 |
| H2A–O2A–C1A | 110.0(3) | 114.5 | O1D–C1D–C2D–Cl2D | 0.7(4) | 0.2 |
| H2B–O2B–C1B | 118.0(3) | 113.9 | O1D–C1D–C2D–C3D | −178.3(3) | −178.3 |
| H2B1–C2B–H2B2 | 107.8(1) | 107.2 | O1D–C1D–C6D–C5D | −179.9(2) | −179.5 |
| H3A1–C3A–H3A2 | 109.4(1) | 109.4 | Cl2D–C2D–C3D–C4D | −179.7(2) | −179.9 |
| H3C–O3C–C3C | 107.0(3) | 108.0 | C1D–C2D–C3D–C4D | −0.7(4) | −1.6 |
| H4A1–C4A–H4A3 | 109.5(1) | 111.0 | C2D–C3D–C4D–C5D | −2.0(4) | −0.5 |
| H4B2–C4B–H4B3 | 109.5(1) | 110.1 | C3D–C4D–C5D–C6D | 4.0(4) | 2.9 |
| N1A–C2A–H2A1 | 109.2(1) | 108.8 | C4D–C5D–C6D–O6D | 176.4(2) | 176.9 |
| N1A–C3A–H3A1 | 109.5(1) | 108.8 | C4D–C5D–C6D–C1D | −3.0(4) | −3.0 |
| D–H···A | d(D–H) | d(H···A) | d(D···A) |
|---|---|---|---|
| O(2A)–H(2A)···O(6C) | 0.82(3) | 1.76(3) | 2.555(2) |
| N(1A)–H(1A)···O(1D)#1 | 0.86(3) | 2.39(3) | 2.933(2) |
| N(1A)-H(1A)···O(6D)#1 | 0.86(3) | 1.99(3) | 2.813(2) |
| O(2B)-H(2B)···O(6D)#2 | 0.73(3) | 1.85(4) | 2.578(2) |
| N(1B)-H(1B)···O(1C) | 0.83(3) | 2.43(3) | 2.971(2) |
| N(1B)-H(1B)···O(6C) | 0.83(3) | 2.03(3) | 2.826(2) |
| O(3C)-H(3C)···O(1B)#3 | 0.87(4) | 2.11(4) | 2.810(2) |
| O(3D)-H(3D)···O(1A)#4 | 0.84(4) | 2.15(4) | 2.765(2) |
| Reagents Used and Measurement Conditions | C=O | C–OH | C–O | C–Cl | CH3 | CH2 | COOH | N |
|---|---|---|---|---|---|---|---|---|
| CLA in DMSO | 165.9 | 110.01 | ||||||
| DMG in DMSO | 43.8 2.59 | 60.8 3.24 | 167.2 | −345.2 | ||||
| CLA solid state CPMAS | 176.0 # | 154.9 # | 110.8 114.3 | |||||
| DMG solid state CPMAS | 41.9, 43.3, 44.1, 44.5 | 58.7, 59.3 | 169.4, 170.97 | −342.4 | ||||
| CLA + DMG Complex DMSO | 167.9 ν 1/2 = 88 Hz | 105.1 | 43.75 2.80 | 56.97 4.04 | 168.0 ν 1/2 = 1 Hz | −344.1 | ||
| CLA + DMG Complex CPMAS | 178.7 ” 170.1 *” | 154.7 ” | 164.5 ” | 110.0 106.3 | 45.7 | 60.1 | 170.1 * | −342.2 |
| CLA + TMG 1:1 in DMSO | 167.95 | 105.3 | ||||||
| CLA + double excess of TMG in DMSO | 172.4 | 105.4 | ||||||
| Dimethylglycine In DMSO | 43.8 2.59 | 60.8 3.24 | 167.2 | −345.2 | ||||
| Dimethylglycine + TMG in DMSO | 45.76 2.10 | 65.25 2.56 | 173.0 | −357.2 | ||||
| N-dimethylglycine D2O | 43.45 2.70 | 59.82 3.58 | 170.25 | −342.1 | ||||
| N-dimethylglycine in D2O + HClO4 | 43.73 2.84 | 57.33 3.93 | 168.13 | −344.8 | ||||
| UCG | Lattice Modes | Internal Modes | Selection Rules | ||||
|---|---|---|---|---|---|---|---|
| Ci | Ac. | Trans. | Rot. | DMG+ | CLA− | IR | Raman |
| Ag | 12 | 12 | 90 | 66 | x2, y2, z2, xy, xz, yz | ||
| Au | 3 | 9 | 12 | 90 | 66 | x, y, z | |
| Experimental | Calculated | Approximate Assignments | ||
|---|---|---|---|---|
| IR (290 K) | Raman (290 K) | INS | CASTEP/PBE + vdW | |
| 3122(IR) 3126(R) | CH3 str. asym. (DMG+) | |||
| 3120(IR) 3120(R) | CH3 str. asym. (DMG+) | |||
| 3119(IR) 3119(R) | CH3 str. asym. (DMG+), O–H str. (CLA−) | |||
| 3118(IR) 3118(R) | CH3 str. asym. (DMG+) | |||
| 3179 | 3113(IR) 3112(R) | O–H str. (CLA−) | ||
| 3102(IR) 3102(R) | CH3 str. asym. (DMG+) | |||
| 3050 | 3051 | 3052(IR) 3052(R) | CH2 str. asym. (DMG+) | |
| 3032 | 3044(IR) 3044(R) | CH2 str. asym. (DMG+) | ||
| 3018(IR) 3017(R) | CH3 str. sym. (DMG+) | |||
| 3013(IR) 3013(R) | CH3 str. sym., CH2 str. asym. (DMG+) | |||
| 3006(IR) 3006(R) | CH3 str. sym. (DMG+) | |||
| 3006 2996 | 3000(IR) 3000(R) | CH2 str. sym. (DMG+) | ||
| 2974 2947 | 2976 | 2995(IR) 2995(R) | CH2 str. sym. (DMG+) | |
| 2911 2887 | 2857(IR) 2855(R) | N–H str. (DMG+) | ||
| 2863 2840 | 2874 | 2846(IR) 2846(R) | N–H str. (DMG+) | |
| 2788 | N–H str. (DMG+) | |||
| 2722 2698 | 2720 | 2595(IR) 2592(R) | O–H str. (DMG+) | |
| 2654 2626 | 2562(IR) 2558(R) | O–H str. (DMG+) | ||
| 2510 2468 | O–H str. (DMG+) | |||
| 2441 | O–H str. (DMG+) | |||
| 1994 | C–H2 bend. overtone | |||
| 1874 | CH2 bend. + CH2 wagg. summation | |||
| 1734 | 1682(IR) 1684(R) | C=O str., NH+ bend., COH bend. (DMG+) | ||
| 1727 | 1726 | 1671(IR) 1670(R) | C=O str., NH+ bend., COH bend. (DMG+, CLA−) | |
| 1629(IR) 1629(R) | C=O str., C−C str., COH bend. (CLA−) | |||
| 1627(IR) 1628(R) | C=O str., C−C str., COH bend. (CLA−) | |||
| 1676 | 1679 | 1606(IR) | C=O str., C−C str., COH bend. (CLA−), NH+ bend. (DMG+) | |
| 1655 | 1640 | 1575(IR) 1581(R) | C=O str., C−C str., COH bend. (CLA−) | |
| 1523 | 1541 1582 | 1515(IR) 1529(R) | NH+ bend. (DMG+), C−C str., COH bend. (CLA−, DMG+) | |
| 1480 | 1483 | 1470(IR) 1470(R) | CH3 bend., NH+ bend. (DMG+) | |
| 1464 | 1453(IR) 1453(R) | CH3 bend., NH+ bend. (DMG+) | ||
| 1435(IR) 1435(R) | CH3 bend., NH+ bend. (DMG+) | |||
| 1447 | 1455 | 1433(IR) 1433(R) | CH3 bend., NH+ bend. (DMG+) | |
| 1431(IR) 1432(R) | CH3 bend., NH+ bend. (DMG+) | |||
| 1399(IR) 1399(R) | CH3 bend., NH+ bend., CH2 bend. (DMG+) | |||
| 1416 | 1396(IR) 1396(R) | CH3 bend., NH+ bend., CH2 bend. (DMG+) | ||
| 1407 | 1403 | 1371(IR) 1372(R) | CH3 bend., NH+ bend., CH2 wagg., COH bend. (DMG+) | |
| 1390 | 1351(IR) 1352(R) | CH2 wagg., COH bend. (DMG+) | ||
| 1391 | 1346(IR) 1347(R) | CH2 wagg., COH bend. (DMG+, CLA−), C−C str. (CLA−) | ||
| 1372 | 1324(IR) 1324(R) | C=O str., C−C str., COH bend. (CLA−) | ||
| 1363 | 1345 1365 | 1315(IR) 1318(R) | C=O str., C−C str., COH bend. (CLA−) | |
| 1328 | 1277(IR) 1276(R) | CH2 twist. (DMG+), COH bend., CH3 rock.(CLA−) | ||
| 1279 | 1294 | 1262(IR) 1262(R) | C−C str., COH bend. (CLA−), CH2 wagg. (DMG+) | |
| 1254(IR) 1251(R) | C−C str., C−O str. (CLA−) | |||
| 1248(IR) 1247(R) | C−C str., C−O str. (CLA−), CH2 wagg., C−O str. (DMG+) | |||
| 1255 | 1259 | 1243(IR) 1241(R) | C−C str., COH bend. (CLA−), CH2 rock., CH3 rock. (DMG+) | |
| 1214(IR) 1215(R) | CH3 rock. (DMG+) | |||
| 1160(IR) 1161(R) | CH3 rock. (DMG+), COH bend. (CLA−) | |||
| 1177 | 1175 | 1154(IR) 1152(R) | CH3 rock. (DMG+), COH bend. (CLA−) | |
| 1140 | 1142 | 1118(IR) 1117(R) | CH3 rock. (DMG+) | |
| 1114 | 1121 | 1075(IR) 1071(R) | OH bend., CH3 rock. (DMG+) | |
| 1062(IR) 1057(R) | OH bend., CH3 rock. (DMG+) | |||
| 1054 | 1055 | 1025(IR) 1025(R) | NH+ rock., CH3 rock. (DMG+) | |
| 1017 | 1020 | 990(IR) 989(R) | C−N str., C−C str., CH3 rock. (DMG+) | |
| 976(IR) 976(R) | C−N str., CH3 rock., CH2 rock. (DMG+) | |||
| 1001 | 1002 | 973(IR) 974(R) | C−N str., CH3 rock., CH2 rock. (DMG+) | |
| 962(IR) 961(R) | C−C str., COH bend. (CLA−) | |||
| 980 | 983 | 958(IR) 958(R) | C−C str., COH bend. (CLA−) | |
| 957 | 961 | 937(IR) 936(R) | C−N str., CH3 rock., CH2 rock. (DMG+) | |
| 904 886 | 910 889 | 891(IR) 888(R) | C−C str., C−N str. (DMG+), OH tors. (CLA−) | |
| 870(IR) 868(R) | OH tors. (CLA−) | |||
| 862 | 863 | 859(IR) 856(R) | OH tors. (CLA−) | |
| 839 | 837 | 844(IR) 844(R) | C−N str. (DMG+) | |
| 771 | 790 | 798 781 | 817(IR) 816(R) | C−C str., C−Cl str. (CLA−) |
| 747 | 752 | 755 | 762(IR) 762(R) | Ring def. (CLA−) |
| 675 | 677 | 679 | 663(IR) 663(R) | COOH bend. (DMG+) |
| 616 | 608 | 621 | 567(IR) 561(R) | Ring def. (CLA−), COOH rock. (DMG+) |
| 571 | 573 | 570 | 555(IR) 556(R) | CH2 rock., COOH rock. (DMG+) |
| 535 | 536 | 539 | 530(IR) 530(R) | Ring def. (CLA−) |
| 504 | 505 | 509 | 504(IR) 505(R) | Ring def. (CLA−) |
| 461 | 466 | 469 | 486(IR) 487(R) | C−C−N bend., C−N−C bend., N−CH3 bend. (DMG+) |
| 434 | 436 | 431(IR) 430(R) | Ring def. (CLA−), C−C−N bend., N−CH3 bend. (DMG+) | |
| 405 | 408 | 417 | 401(IR) 400(R) | Ring def. (CLA−) |
| 361(IR) 361(R) | Ring def. (CLA−), C−C−N bend. (DMG+) | |||
| 372 | 369 352 | 380 364 | 358(IR) 356(R) | C=O bend., C−O bend. (CLA−), CH2 rock. (DMG+) |
| 340(IR) 340(R) | C−N−C bend., N−CH3 bend. (DMG+), COH bend. (CLA−) | |||
| 307 | 300(IR) 300(R) | Ring def. (CLA−) | ||
| 286 | 287 | 284 | 281(IR) 280(R) | COH bend. (CLA−), C−N−C bend., CH3 tors. (DMG+) |
| 266(IR) 266(R) | CH3 tors., CH2 rock. (DMG+) | |||
| 232(IR) 232(R) | CH3 tors., CH2 rock. (DMG+) | |||
| 236 | 236 | 235 | 227(IR) 229(R) | CH3 tors. (DMG+) |
| 213(IR) 212(R) | CH3 tors. (DMG+) | |||
| 209(IR) 208(R) | CH3 tors. (DMG+) | |||
| 220 | 214 | 223 | 205(IR) 203(R) | Ring tors. C=O wagg. (CLA−) |
| 208 | 212 | 200(IR) 199(R) | Ring tors. C=O wagg. (CLA−) | |
| 175 | 178(IR) 178(R) | C−C tors. (DMG+) | ||
| 156 | 163 | 163 | 168(IR) 168(R) | C−C tors. (DMG+) |
| 141 | 152 | 149(IR) 151(R) | ||
| 137 | 135 | 146 | 138(IR) 140(R) | Ring tors. (CLA−), C−N tors. (DMG+) |
| 122 | 127 120 | 25–135 | Lattice vibrations | |
| 101 92 | 92 | 94 | ||
| 65 | 79 | |||
| 54 | 59 | 64 | ||
| Parameter | Value |
|---|---|
| a (cm−1) | 6.53 ± 0.07 |
| b (cm−1·K) | 2.65 × 10−3 ± 8.6 × 10−5 |
| c (cm−1) | 44.00 ± 9.18 |
| Ea (kJ·mol−1) | 6.7 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hetmańczyk, J.; Hetmańczyk, Ł.; Nowicka-Scheibe, J.; Pawlukojć, A.; Maurin, J.K.; Schilf, W. Structural, Thermal, and Vibrational Properties of N,N-Dimethylglycine–Chloranilic Acid—A New Co-Crystal Based on an Aliphatic Amino Acid. Materials 2021, 14, 3292. https://doi.org/10.3390/ma14123292
Hetmańczyk J, Hetmańczyk Ł, Nowicka-Scheibe J, Pawlukojć A, Maurin JK, Schilf W. Structural, Thermal, and Vibrational Properties of N,N-Dimethylglycine–Chloranilic Acid—A New Co-Crystal Based on an Aliphatic Amino Acid. Materials. 2021; 14(12):3292. https://doi.org/10.3390/ma14123292
Chicago/Turabian StyleHetmańczyk, Joanna, Łukasz Hetmańczyk, Joanna Nowicka-Scheibe, Andrzej Pawlukojć, Jan K. Maurin, and Wojciech Schilf. 2021. "Structural, Thermal, and Vibrational Properties of N,N-Dimethylglycine–Chloranilic Acid—A New Co-Crystal Based on an Aliphatic Amino Acid" Materials 14, no. 12: 3292. https://doi.org/10.3390/ma14123292
APA StyleHetmańczyk, J., Hetmańczyk, Ł., Nowicka-Scheibe, J., Pawlukojć, A., Maurin, J. K., & Schilf, W. (2021). Structural, Thermal, and Vibrational Properties of N,N-Dimethylglycine–Chloranilic Acid—A New Co-Crystal Based on an Aliphatic Amino Acid. Materials, 14(12), 3292. https://doi.org/10.3390/ma14123292