TiO2 Nanotubes with Pt and Pd Nanoparticles as Catalysts for Electro-Oxidation of Formic Acid

 ,
,  ,
,
Abstract
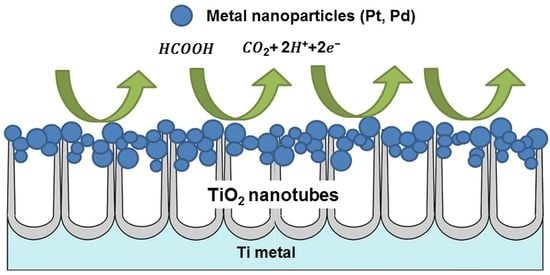
1. Introduction
2. Experimental Methods
2.1. Sample Preparation
2.2. Deposition of Metal Nanoparticles
2.3. Membrane Electrode Assemblies (MEAs) Preparation
2.4. SEM
2.5. STEM
2.6. XPS/AES
2.7. CV
2.8. Fuel Cell Tests
3. Results and Discussions
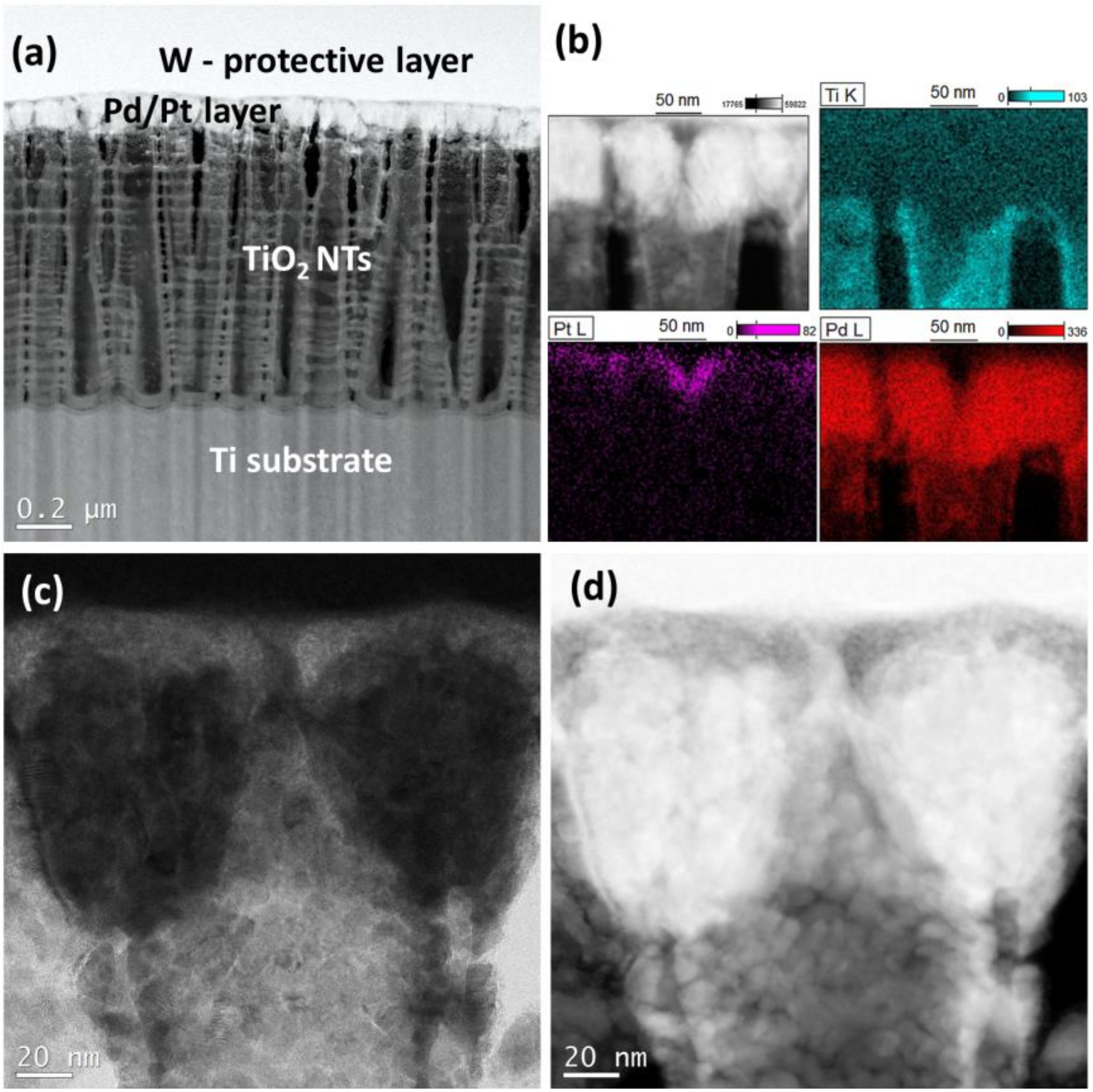
3.1. Characterization of the Catalysts
3.2. Activity of the Electrocatylysts
3.2.1. CV Tests
3.2.2. Fuel Cell Tests
3.3. Discussion
- (i)
- One of them could be metal–support interaction which may change electronic properties of metal nanoparticles. Such an interaction should manifest itself by a shift of XPS peak position. However, such evident shift was not observed in the present work. It is instructive to compare the XPS data obtained in this work (no change in the Pt 4f7/2 position 71.0 eV) with those from literature [35], where the Pt 4f7/2 maximum is shifted to 71.3 eV. The difference results from different Pt deposition methods used in both works. The magnetron sputtering used in the present work produces metal deposits of the thickness of about 150 nm, located mainly on top of TiO2 NTs, so the XPS signal comes from surface metal atoms having no direct contact with the support. In turn, metal precursor reduction method used in [35] produces more uniform and so thinner metal deposits and so the XPS signal comes from metal being in contact with the TiO2 NTs. Therefore, although such an effect was observed in [35], it cannot account for high specific activity of the TiO2 NTs supported catalyst used in the present work.
- (ii)
- It was shown that titanium dioxide is more hydrophilic than the carbon supports commonly used [46], which ensures easier formic acid access to the reaction zone, and facilitates CO2 removal. In the case of carbon-supported catalysts, CO2 bubbles block the catalyst surface, hindering access of the formic acid [29].
- (iii)
- There is a marked difference in the structures of catalyst layers for the two cases. The standard ink based catalyst layer employed for the carbon supported catalyst is about 10 μm thick and electronic conductivity between carbon support particles separated by larger particles of Nafion ionic conductor does not ensure perfect electron flow between catalyst nanoparticles and the current collector. In contrary, virtually all the Pd aglomerates, located on top of TiO2 NTs, have good electronic connection with the Ti collector and ionic (H+) contact with Nafion membrane. Also, diffusion of formic acid to the catalyst and removal of the CO2 product, are more efficient for a thin layer of metal nanoparticles on top of TiO2 NTs, than for much thicker classical catalyst layer with carbon supported catalyst.
- (iv)
- Another factor that works in favor of the TiO2 NTs supported catalyst is the difference in crystallite sizes in the two systems. From the paper on crystallite size effect on FA electrooxidation [47] it follows, that the turnover frequency (the reaction rate per 1 surface atom per second) is a complex function of the crystallite size, and, that it is the highest for an optimum crystallite size between 5 and 7 nm. One of the reasons for such a behavior is the fact, that there are two Pd neighboring atoms necessary for dissociative adsorption of FA and, therefore, small carbon supported crystallites (3 nm) having higher contribution of edges are less effective in FA electrooxidation then the larger ones (~10 nm) TiO2 NTs supported Pd nanoparticles having higher contribution of crystal planes.
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zwilling, V.; Aucouturier, M.; Darque-Ceretti, E. Anodic oxidation of titanium and TA6V alloy in chromic media. An electrochemical approach. Electrochim. Acta 1999, 45, 921–929. [Google Scholar] [CrossRef]
- Zwilling, V.; Darque-Ceretti, E.; Boutry-Forveille, A.; David, D.; Perrin, M.Y. Structure and physicochemistry of anodic oxide films on titanium and TA6V alloy. Surf. Interface Anal. 1999, 27, 629–637. [Google Scholar] [CrossRef]
- Macak, J.M.; Tsuchiya, H.; Ghicov, A.; Yasuda, K.; Hahn, R.; Bauer, S.; Schmuki, P. TiO2 nanotubes: Self-organized electrochemical formation, properties and applications. Curr. Opin. Solid State Mater. Sci. 2007, 11, 3–18. [Google Scholar] [CrossRef]
- Lee, K.; Mazare, A.; Schmuki, P. One-dimensional titanium dioxide nanomaterials: Nanotubes. Chem. Rev. 2014, 114, 9385–9454. [Google Scholar] [CrossRef]
- Roguska, A.; Pisarek, M.; Andrzejczuk, M.; Dolata, M.; Lewandowska, M.; Janik-Czachor, M. Characterization of a calcium phosphate–TiO2 nanotube composite layer for biomedical applications. Mater. Sci. Eng. C 2011, 31, 906–914. [Google Scholar] [CrossRef]
- Roguska, A.; Pisarek, M.; Belcarz, A.; Marcon, L.; Holdynski, M.; Andrzejczuk, M.; Janik-Czachor, M. Improvement of the bio-functional properties of TiO2 nanotubes. Appl. Surf. Sci. 2016, 388, 775–785. [Google Scholar] [CrossRef]
- Chen, S.; Malig, M.; Tian, M.; Chen, A.J. Electrocatalytic activity of PtAu nanoparticles deposited on TiO2 nanotubes. Phys. Chem. C 2012, 116, 3298–3304. [Google Scholar] [CrossRef]
- Rettew, R.E.; Allam, N.K.; Alamgir, F.M. Interface architecture determined electrocatalytic activity of Pt on vertically oriented TiO2 nanotubes. ACS Appl. Mater. Interfaces 2011, 3, 147–151. [Google Scholar] [CrossRef]
- Macak, J.M.; Barczuk, P.J.; Tsuchiya, H.; Nowakowska, M.Z.; Ghicov, A.; Chojak, M.; Bauer, S.; Virtanen, S.; Kulesza, P.J.; Schmuki, P. Self-organized nanotubular TiO2 matrix as support for dispersed Pt/Ru nanoparticles: Enhancement of the electrocatalytic oxidation of methanol. Electrochem. Commun. 2005, 7, 1417–1422. [Google Scholar] [CrossRef]
- Honciuc, A.; Laurin, M.; Albu, S.; Amende, M.; Sobota, M.; Lynch, R.; Schmuki, P.; Libuda, J.J. Preparation and adsorption properties of Pd nanoparticles supported on TiO2 nanotubes. Phys. Chem. C 2010, 114, 20146–20154. [Google Scholar] [CrossRef]
- Smith, Y.; Ray, R.; Carlson, K.; Sarma, B.; Misra, M. Self-ordered titanium dioxide nanotube arrays: Anodic synthesis and their photo/electro-catalytic applications. Materials 2013, 6, 2892–2957. [Google Scholar] [CrossRef] [PubMed]
- Regonini, D.; Jaroenworaluck, A.; Stevens, R.; Bowen, C.R. Effect of heat treatment on the properties and structure of TiO2 nanotubes: Phase composition and chemical composition. Surf. Interface Anal. 2010, 42, 139–144. [Google Scholar] [CrossRef]
- Pisarek, M.; Roguska, A.; Kudelski, A.; Andrzejczuk, M.; Janik-Czachor, M.; Kurzydłowski, K.J. The role of Ag particles deposited on TiO2 or Al2O3 self-organized nanoporous layers in their behavior as SERS-active and biomedical substrates. Mater. Chem. Phys. 2013, 139, 55–65. [Google Scholar] [CrossRef]
- Regonini, D.; Bowen, C.R.; Jaroenworaluck, A.; Stevens, R. A review of growth mechanism, structure and crystallinity of anodized TiO2 nanotubes. Mater. Sci. Eng. R Rep. 2013, 74, 377–406. [Google Scholar] [CrossRef]
- Yan, J.; Zhou, F.J. TiO 2 nanotubes: Structure optimization for solar cells. Mater. Chem. 2011, 21, 9406–9418. [Google Scholar] [CrossRef]
- Buchalska, M.; Kobielusz, M.; Matuszek, A.; Pacia, M.; Wojtyła, S.; Macyk, W. On oxygen activation at rutile-and anatase-TiO2. ACS Catal. 2015, 5, 7424–7431. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Huo, K.; Gao, B.; Fu, J.; Zhao, L.; Chu, P.K. Fabrication, modification, and biomedical applications of anodized TiO 2 nanotube arrays. RSC Adv. 2014, 4, 17300–17324. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Z.; Kumar, A.; Boughton, R.I.; Liu, H. Recent progress in design, synthesis, and applications of one-dimensional TiO 2 nanostructured surface heterostructures: A review. Chem. Soc. Rev. 2014, 43, 6920–6937. [Google Scholar] [CrossRef]
- Song, Y.; Wei, C.; Zhang, X.; Wei, X.; Song, X.; Sun, Z. Nanoporous Pd/TiO2 composites prepared by one-step dealloying and their electrocatalytic performance for methanol/ethanol oxidation. Mater. Chem. Phys. 2015, 161, 153–161. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, B.; Zeng, W.; Wang, Y. Plasmonic photocatalysis properties of Au nanoparticles precipitated anatase/rutile mixed TiO 2 nanotubes. Nanoscale 2013, 5, 9739–9746. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.K.; Misra, M.; Mahajan, V.K.; Raja, K.S.J. Design of a highly efficient photoelectrolytic cell for hydrogen generation by water splitting: Application of TiO2-x C x nanotubes as a photoanode and Pt/TiO2 nanotubes as a cathode. Phys. Chem. C 2007, 111, 8677–8685. [Google Scholar] [CrossRef]
- Rees, N.V.; Compton, R.G.J. Sustainable energy: A review of formic acid electrochemical fuel cells. Solid State Electrochem. 2011, 15, 2095–2100. [Google Scholar] [CrossRef]
- Jiang, J.; Wieckowski, A. Prospective direct formate fuel cell. Electrochem. Commun. 2012, 18, 41–43. [Google Scholar] [CrossRef]
- Hietala, J.; Vuori, A.; Johnsson, P.; Pollari, I.; Reutemann, W.; Kieczka, H. Formic acid. Ullmann’s Encycl. Ind. Chem. 2016, 1–23. [Google Scholar] [CrossRef]
- Assaud, L.; Brazeau, N.; Barr, M.K.S.; Hanbücken, M.; Ntais, S.; Baranova, E.A.; Santinacci, L. Atomic layer deposition of Pd nanoparticles on TiO2 nanotubes for ethanol electrooxidation: Synthesis and electrochemical properties. ACS Appl. Mater. Interfaces 2015, 7, 24533–24542. [Google Scholar] [CrossRef]
- Zhu, Y.; Khan, Z.; Masel, R.I.J. The behavior of palladium catalysts in direct formic acid fuel cells. Power Sources 2005, 139, 15–20. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G. Mechanistic study of the deactivation of carbon supported Pd during formic acid oxidation. Electrochem. Commun. 2009, 11, 2012–2014. [Google Scholar] [CrossRef]
- Mikołajczuk, A.; Borodzinski, A.; Kedzierzawski, P.; Stobinski, L.; Mierzwa, B.; Dziura, R. Direct formic acid fuel cells on Pd catalysts supported on hybrid TiO2-C materials. Appl. Surf. Sci. 2011, 257, 8211–8214. [Google Scholar] [CrossRef]
- Uhm, S.; Lee, H.J.; Lee, J. Understanding underlying processes in formic acid fuel cells. Phys. Chem. Chem. Phys. 2009, 11, 9326–9336. [Google Scholar] [CrossRef]
- Yu, X.; Pickup, P.G.J. Recent advances in direct formic acid fuel cells (DFAFC). Power Sources 2008, 182, 124–132. [Google Scholar] [CrossRef]
- Liu, Z.; Hong, L.; Tham, M.P.; Lim, T.H.; Jiang, H.J. Nanostructured Pt/C and Pd/C catalysts for direct formic acid fuel cells. Power Sources 2006, 161, 831–835. [Google Scholar] [CrossRef]
- Mikolajczuk-Zychora, A.; Borodzinski, A.; Kedzierzawski, P.; Mierzwa, B.; Mazurkiewicz-Pawlicka, M.; Stobinski, L.; Ciecierska, E.; Zimoch, A.; Opałło, M. Highly active carbon supported Pd cathode catalysts for direct formic acid fuel cells. Appl. Surf. Sci. 2016, 388, 645–652. [Google Scholar] [CrossRef]
- Chastain, J.; King, R.C., Jr. (Eds.) Handbook of X-Ray Photoelectron Spectroscopy; Physical Electronics Inc.: Eden Praire, MN, USA, 1995. [Google Scholar]
- Rasmi, K.R.; Vanithakumari, S.C.; George, R.P.; Mallika, C.; Mudali, U.K. Nanoparticles of Pt loaded on a vertically aligned TiO 2 nanotube bed: Synthesis and evaluation of electrocatalytic activity. RSC Adv. 2015, 5, 108050–108057. [Google Scholar] [CrossRef]
- Goodman, D.W. “Catalytically active Au on Titania”: Yet another example of a strong metal support interaction (SMSI)? Catal. Lett. 2005, 99, 1–4. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, Y.; Tang, Q.; Wang, L.; Zhang, H.; Quan, C.; Qi, T. Pt catalyst supported on titanium suboxide for formic acid electrooxidation reaction. Int. J. Hydrog. Energy 2014, 39, 9621–9627. [Google Scholar] [CrossRef]
- Chen, C.H.; Liou, W.J.; Lin, H.M.; Wu, S.H.; Borodzinski, A.; Stobinski, L.; Kedzierzawski, P. Palladium and palladium gold catalysts supported on MWCNTs for electrooxidation of formic acid. Fuel Cells 2010, 10, 227–233. [Google Scholar] [CrossRef]
- Nitze, F.; Mazurkiewicz, M.; Malolepszy, A.; Mikolajczuk, A.; Kędzierzawski, P.; Tai, C.-W.; Hu, G.; Kurzydłowski, K.J.; Stobinski, L.; Borodzinski, A.; et al. Synthesis of palladium nanoparticles decorated helical carbon nanofiber as highly active anodic catalyst for direct formic acid fuel cells. Electrochim. Acta 2012, 63, 323–328. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, H.-X.; Zou, S.; Cai, W.-B. Electrocatalysis of formic acid on palladium and platinum surfaces: From fundamental mechanisms to fuel cell applications. Phys. Chem. Chem. Phys. 2014, 16, 20360–20376. [Google Scholar] [CrossRef]
- Kolics, A.; Wieckowski, A.J. Adsorption of bisulfate and sulfate anions on a Pt (111) electrode. Phys. Chem. B 2001, 105, 2588–2595. [Google Scholar] [CrossRef]
- Lipkowski, J.; Wieckowski, A.J. Using thermodynamic analysis of charge density data for a solution of K2SO4 in HClO4, one can determine whether the species predominantly adsorbed at an au (111) electrode is HSO-4 or SO2-4. Electroanal. Chem. 2001, 504, 230–234. [Google Scholar] [CrossRef]
- Miki, A.; Ye, S.; Osawa, M. Surface-enhanced IR absorption on platinum nanoparticles: An application to real-time monitoring of electrocatalytic reactions. Chem. Commun. 2002, 14, 1500–1501. [Google Scholar] [CrossRef] [PubMed]
- Gralec, B.; Lewera, A. Catalytic activity of unsupported Pd-Pt nanoalloys with low Pt content towards formic acid oxidation. Appl. Catal. B Environ. 2016, 192, 304–310. [Google Scholar] [CrossRef]
- Rice, C.; Ha, S.; Masel, R.I.; Wieckowski, A.J. Catalysts for direct formic acid fuel cells. Power Sources 2003, 115, 229–235. [Google Scholar] [CrossRef]
- Matos, J.; Borodzinski, A.; Mikolajczuk-Zychora, A.; Kedzierzawski, P.; Mierzwa, B.; Juchniewicz, K.; Mazurkiewicz, M.; Hernandez-Garrido, J.C. Direct formic acid fuel cells on Pd catalysts supported on hybrid TiO2-C materials. Appl. Catal. B Environ. 2015, 163, 167–178. [Google Scholar] [CrossRef]
- Zhou, W.; Lee, J.Y.J. Particle size effects in Pd-catalyzed electrooxidation of formic acid. Phys. Chem. C 2008, 112, 3789–3793. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Specific Metal Surface Area (m2 gmetal−1) |
|---|---|
| 0.2 mg cm−2 Pt/TiO2 NT | 8.5 |
| 0.2 mg cm−2 Pd/TiO2 NT | 7.6 |
| 0.1 mg cm−2 Pd + 0.02 mg cm−2 Pt/TiO2 NT | 11.2 |
| 0.24 mg cm−2 Pd of commercial Pd/Vulcan | 65.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisarek, M.; Kędzierzawski, P.; Andrzejczuk, M.; Hołdyński, M.; Mikołajczuk-Zychora, A.; Borodziński, A.; Janik-Czachor, M. TiO2 Nanotubes with Pt and Pd Nanoparticles as Catalysts for Electro-Oxidation of Formic Acid. Materials 2020, 13, 1195. https://doi.org/10.3390/ma13051195
Pisarek M, Kędzierzawski P, Andrzejczuk M, Hołdyński M, Mikołajczuk-Zychora A, Borodziński A, Janik-Czachor M. TiO2 Nanotubes with Pt and Pd Nanoparticles as Catalysts for Electro-Oxidation of Formic Acid. Materials. 2020; 13(5):1195. https://doi.org/10.3390/ma13051195
Chicago/Turabian StylePisarek, Marcin, Piotr Kędzierzawski, Mariusz Andrzejczuk, Marcin Hołdyński, Anna Mikołajczuk-Zychora, Andrzej Borodziński, and Maria Janik-Czachor. 2020. "TiO2 Nanotubes with Pt and Pd Nanoparticles as Catalysts for Electro-Oxidation of Formic Acid" Materials 13, no. 5: 1195. https://doi.org/10.3390/ma13051195
APA StylePisarek, M., Kędzierzawski, P., Andrzejczuk, M., Hołdyński, M., Mikołajczuk-Zychora, A., Borodziński, A., & Janik-Czachor, M. (2020). TiO2 Nanotubes with Pt and Pd Nanoparticles as Catalysts for Electro-Oxidation of Formic Acid. Materials, 13(5), 1195. https://doi.org/10.3390/ma13051195