2.1. Modeling
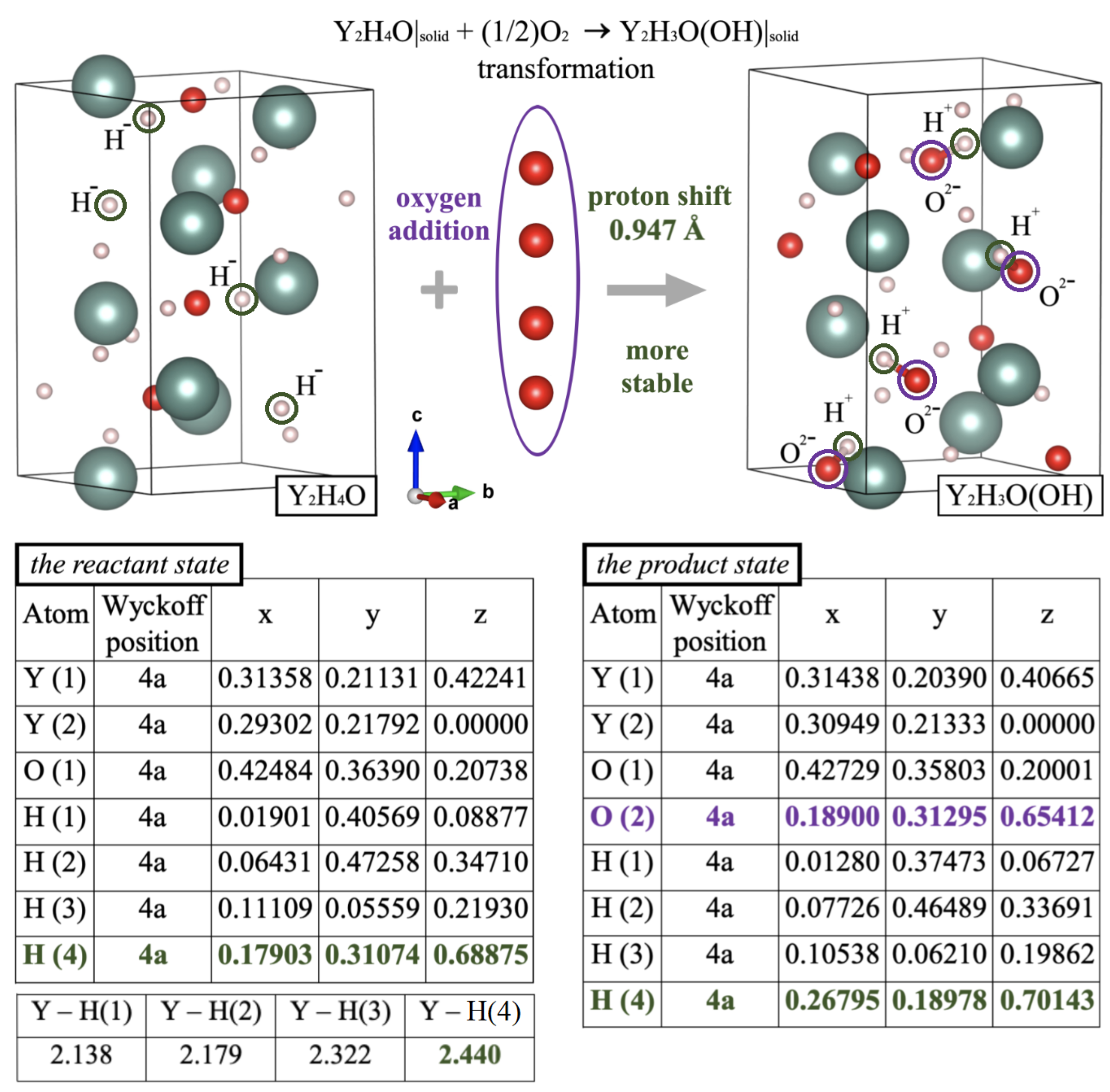
A set of procedures we used in the present study to simulate the oxidation-mediated changes of crystal structures is described in the Section “Methods”. The main purpose of our modeling was to understand how compositional, charge, and structural transformations are governed by variations in O/H stoichiometry. The first result we obtained from our computational DFT simulations is that the metastable
crystal lattice of the oxygen-poor Y
2H
4O composition may present a proper set of active sites for the accommodation of H
+ cations. In order to identify the possible number of protonic positions, we further investigated how the incorporation of additional amount of oxygen may induce the process of hydrogen exchange in the Y
2H
4O system. The model scheme we considered for the simulation of an addition reaction was based on the formal equation Y
2H
4O|solid + (1/2)O
2 -> Y
2H
3O(OH)|
solid. The main feature of the process is that the addition of oxygen (for example, upon treatment with such an oxidizing agent as hydrogen peroxide) results in converting yttrium oxyhydride into the corresponding hydroxyhydride. Evidently, the leading step here is the formation of bridging O
2- affecting the interplay of the local lattice geometry and charge states. Under such conversion, when the system combines directly with the additional oxygen atoms, it readily reorganizes the bonding geometry through generation of H
+ cations in order to keep the overall electro-neutrality for the new resultant composition. In view of chemical energetics, note that the respective enthalpy change of this transformation,
kJ/mol, indicates that, unlike the stable
and
polymorphs of Y
2H
4O [
28], its
chiral modification is highly reactive with respect to oxygen uptake. Moreover, in view of chemical kinetics of crystallization, the choice of such “early” intermediate structure of Y
2H
4O as a starting point (i.e., as a precursor) is in full compliance with Ostwald’s scenario of solid-to-solid successive transformations. That is, additional oxidation of the
chiral lattice turns out to be favorable because it leads to the formation of a more stable extended composition in which the oxygen atoms are inserted as substitutional ligands of the metal centers within the crystal structure while the
crystallographic symmetry remains unchanged.
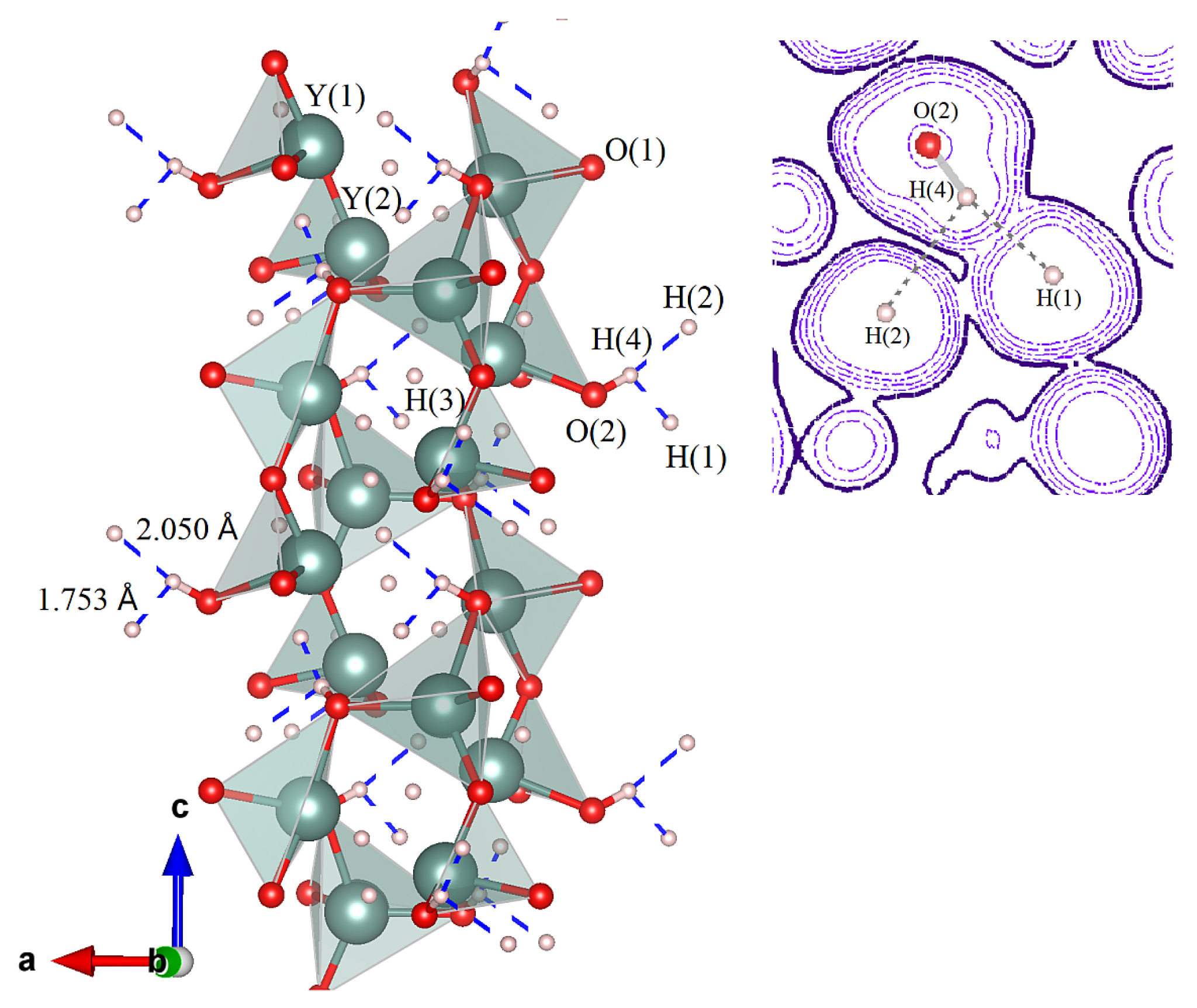
Figure 1 presents more details on electron transfer and the corresponding change of bonding/geometry configuration. Their interplay gives rise to structural patterns associated with the on-site accommodation of protons. It is also seen that a route to Y
2H
3O(OH) proceeds via distribution of the incorporated oxygen over empty interstitial voids of the
lattice. The maximum number of possible protonic sites (orbits) is equal to 1 because, in the packing motif of single-crystal architecture of the bulk Y
2H
4O, each oxygen is connected to two metal centers. This fact is well illustrated by the elongated bonding distance of
Å which Y establishes with hydridic hydrogens occupying the H(4) position. Hence, the weakness of the long Y−H(4) connections observed in Y
2H
4O positively affects the accessibility of H(4) as a potential protonic site during the oxygen incorporation. In fact, because of a stronger affinity of yttrium for oxygen, the additional oxygen plays a role as the stabilizing agent, thus providing increase in stability. Being combined with the metal center, it behaves as a radical anion that initiates the net hydrogen transfer by breaking the Y−H(4) bond, subsequently repelling a hydride ion from the yttrium site. Since this oxygen (residing in the O(2) position) becomes almost completely negatively charged, it exhibits a higher tendency to combine with the H
+ cation to form the standard OH
- ion. Note further that, as seen in
Figure 1, the displacement of the H(4) proton accompanies no noticeable reorganization of internal coordinates of the host lattice. In particular, estimating the hydrogen shift with respect to its initial position H(4) in the precursor Y
2H
4O gave a value of 0.947 Å, which is very close to the O−H bonding distance. This implies that, by establishing the O−H ionic connection, the O(2) oxygen tightly holds protonic hydrogen in at H(4), thus protecting the ionized state as the H
+ cation from reduction and elimination or merging. Moreover, from the symmetry point of view, one can suggest that crystallographic symmetry places some limitations on the possibility of H
-/H
+ coexistence in an inorganic crystalline solid. The corresponding rule could be worded as follows: the partial H
- → H
+ on-site conversion may afford subsequent displacement of the generated proton only over lattice positions matching the lowest site-symmetry. Obviously, the crystal systems described by chiral groups are the most relevant for this case, i.e., when H migration proceeds through its spontaneous reconnection from Y to O.
2.2. Crystal Structure
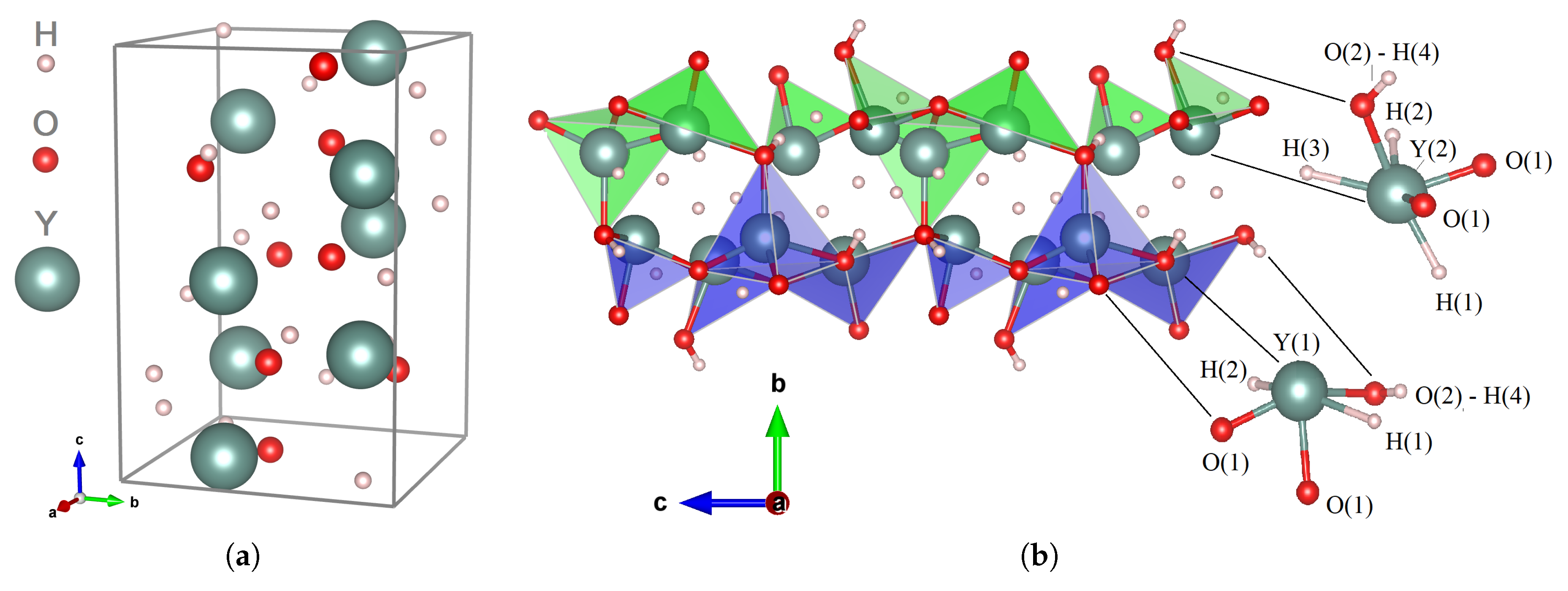
The description of the
crystal structure of Y
2H
3O(OH) is summarized in
Table 1 and
Table 2, and schematic illustrations are presented in
Figure 2. The unit cell contains eight independent atoms which occupy
orbits of the lowest point symmetry
. The arrangement of oxygen atoms matches two independent sites: O(1) surrounded tetrahedrally by four yttrium atoms and O(2) which is connected with two yttrium atoms. Y(1) coordinates with O(1) and three hydrogens, H(1)–H(3); its connection with the nearest Y(2) serves as a bridging unit in the [H
3Y
2O]
+ cation. The periodic arrangement of [H
3Y
2O]
+ underlies the 3D cationic framework in which [H
3Y
2O]
+ shares the O(2)−H(4) coupling. The Y−O bond lengths vary from
to
Å (
Table 2), where the largest values relate to the preferred coordination that O(2) chooses with yttrium. In a set of Y−O connections, O(1) forms a standard bridge between two nearest metal centers. The other oxygen, O(2), puts together the H(4) site and the metal center. Four hydrogen sites, H(1)–H(4), are filled in such a way that the hydrogen anions continue to share the “pristine" proximal ligand positions H(1)–H(3), while the new H(4) position is opened to be occupied by the released hydrogen. The interesting feature is the strong repulsive interaction between the H(1)–H(3) hydrogen anions: the lattice geometry does not allow them to approach one another closer than 2.32 Å. This implies that, according to Switendick’s criterion [
31], these negative-charged hydrogens can be considered completely separated from each other. The other interesting feature of the Y
2H
3O(OH) structure is that spatial separation of two oppositely charged hydrogen centers is accomplished by displacing the oxygen anions O(2) in order to fix the proton H
+ in the H(4) position and to form the hydroxide anion OH
- (the ion pairing effect). Note that the difference in the behavior of O(1) and O(2) atoms represents a specific example of chemical selectivity which provides the conversion of the oxyhydride host into a hydroxyhydride system.
In the crystallographic sense, the
group is minimalistic as compared to the properties of the most other space groups because it presents only a single Wyckoff set of equivalent locations, which comprises of a
point orbit. A rapid survey of crystal structures showed that inorganic materials that adopt the tetragonal
symmetry are rather scarce. Nevertheless, one can directly refer to the family of compounds A
4LiH
3(XO
4)
4 (A = K, Rb, NH
4 and X = S, Se) which crystallize in the
structure (e.g., that in Reference [
32] and references therein).
2.3. Potential of Structural and Thermal Stabilities
We investigated the structural stability of Y
2H
3O(OH) to ensure that the crystal lattice is far from dynamical instability. An analysis showed that all the necessary criteria are fulfilled: First, the stability of the tetragonal phase against the relative displacement of sublattices is provided by the positive values of the squares of zone-centered vibrational modes (See
Supplementary Materials Table S4. Second, the stiffness matrix (the elasticity tensor) is completely positive definite (
Table S8 of SI). This fact confirms the macroscopic stability of the crystalline medium in terms of the elastic energy. The knowledge of elastic constants allowed us to estimate the aggregate characteristics (
Table 3).
It is easy to verify that the structural model of Y2H3O(OH) exhibits macroscopic elastic properties which are typical for ion-covalent crystals except for one peculiarity: the elasticity tensor components, , , and , are close to those values that obey the Cauchy relations for an isotropic cubic medium: and . Moreover, the indexes and and the differences and indicate a rather low level of the elastic anisotropy. Since in a tetragonal crystal the bulk and shear moduli are directly engaged in the elastic behavior, such weak anisotropy suggests that a purely pairwise and directional character of the interatomic Coulomb forces is the distinguishing feature of the bonding situation in Y2H3O(OH). Note that a number of cubic binary and even ternary ionic compounds demonstrate a similar bonding picture. However, when the tetragonal distortion lowers the initial cubic symmetry, in the low-symmetry phase, the bonding forces acquire an angular character via indirect many-particle contributions caused by electron–ion (or electron–electron) interactions. This leads to competition between central and noncentral forces which may violate the Cauchy relations. If the effect of symmetry reduction is marginal, i.e., the lattice distortion does not produce or increase an overlap of the ionic cores, the renormalized charge distributions around the ion centers should remain localized to hold the direct/central character of the interaction between ions. To demonstrate that this particular factor underlies the weak anisotropy in Y2H3O(OH), we analyzed a topology of valence charge partitioning in terms of the electron localization function (ELF).
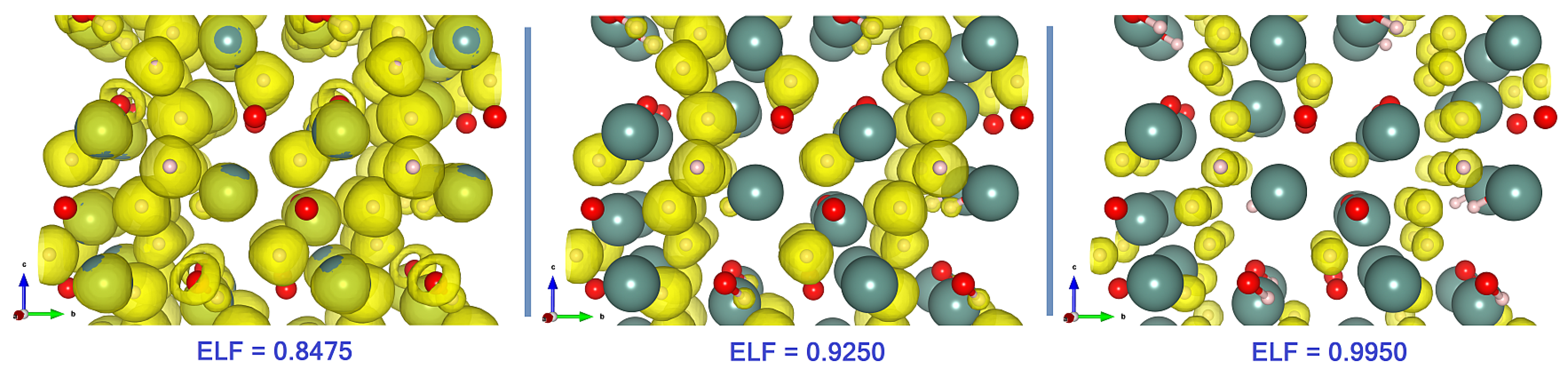
The comparison of the different ELF visualizations is presented in
Figure 3. The characteristic feature clearly seen in the limit ELF
is the global separation into electron-rich and electron-deficient spatial regions. In particular, this configuration reflects the exceptionally high level of localization of valence electron density at the H
- centers.
Utilization of the AIM protocol for the theoretical charge densities provided us with the Bader charges summarized in
Table 4. These values can be compared with the charge configuration of the nominal ionic model with a mixed-anion order: [Y
23+H
3-1O
2-(OH)
-1]. It is seen that the bonding situation is to a certain extent not complicated because the overall bonding character is determined by the interplay of ionic and covalent connectivities. Since the chiral lattice of Y
2H
3O(OH) belongs to the polar crystal class 4, we also examined how strongly the outer electron shells are distorted due to the coupling of valence electrons with dipole-active optical vibrational modes. The vibration-assisted enhancement of charge states was characterized in terms of principal values of the Born dynamical charge (
Table 4). Following the model of formal charge partitioning, one can see that the dynamical charge redistribution strengthens the yttrium donating ability, i.e., the most positive charges tend to be concentrated on the pair of yttriums while the most negative local charges are shared by the oxygen, O(1), and the three hydrogens, H(1)–H(3). In fact, we are dealing here with the cooperative effect of electron polarization which gives rise to dynamical enhancement of the valence charge localization.
The evaluation of the ground-state formation energy (shown in the last raw of
Table 1a) indicates that the bulk structure of Y
2H
3O(OH) represents an enthalpically stable condensed phase. Based on the standard dehydration scheme for a hydroxide solid, we have verified the stability of Y
2H
3O(OH) with respect to the decomposition into the mixture of oxide, hydride, and water. Since the estimates have shown that the negative enthalpy difference may be small,
eV/(f.u.), (i.e., the structure is still more stable at the zero temperature than the mixture of yttria, hydride, and water), we have further examined what may happen when the system is subjected to external heating. Accordingly, the principally important question we considered is how stable is the predicted crystal structure against the action of heat. In order to probe a destabilization role of thermal treatment and, correspondingly, to evaluate the thermal stability of the
chiral phase, we performed a series of ab initio molecular dynamics (MD) simulations.
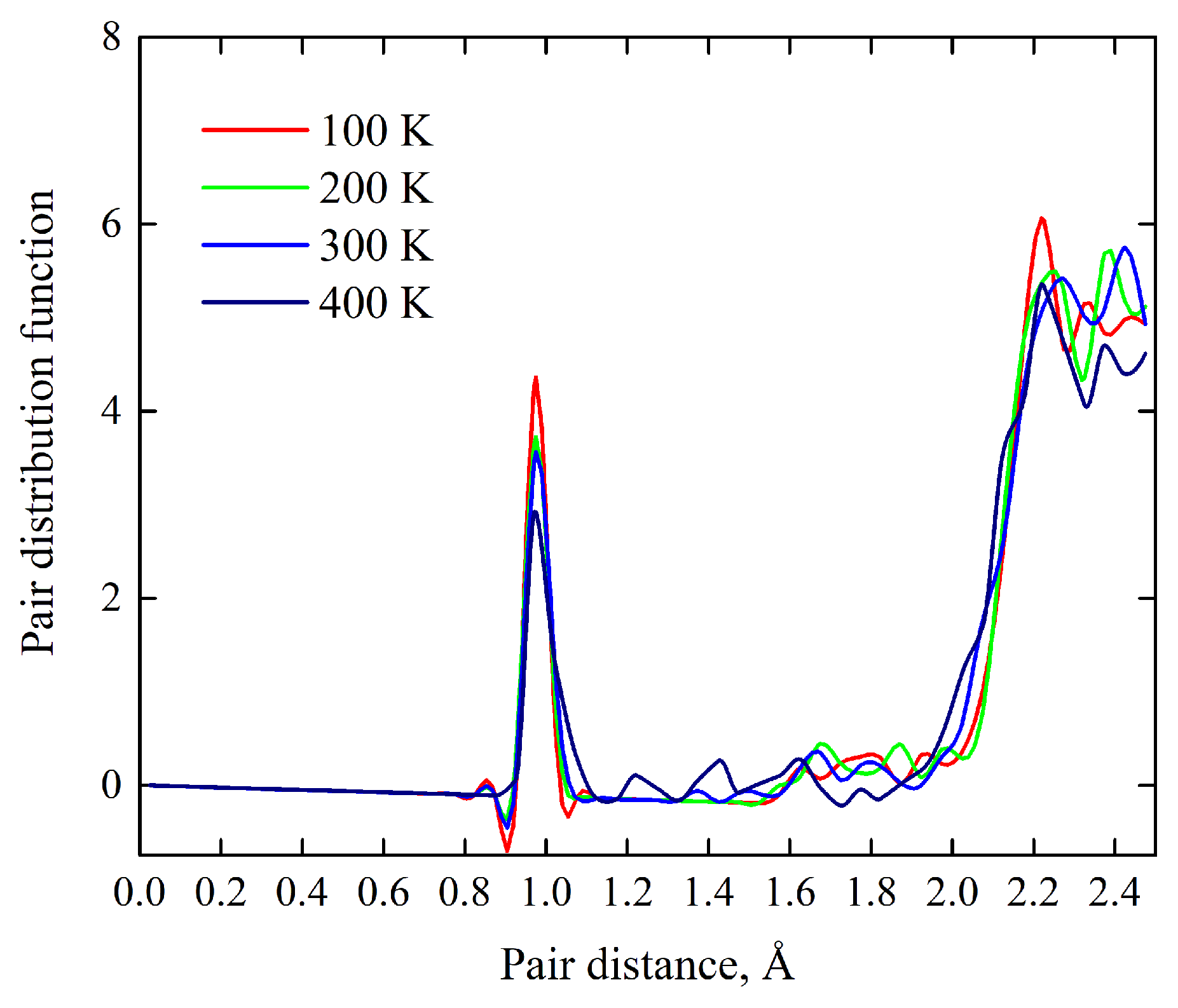
Shown in
Figure 4 are thermal motions of ions which are presented in terms of temperature evolution of the pair distribution function (PDF). It is seen that the first, second, and third peaks are intense and narrow; their high values reflect strong cation–anion interactions. The dynamical evolution of the peaks with respect to their broadening, displacements, and fluctuations shows insignificant temperature-dependent changes. Thus, these simulations indicate the smallness of the thermodynamic driving force to initiate the thermal dehydration and other thermal effects associated with the atomic disorder. Hence, the crystal lattice of Y
2H
3O(OH) remains robust against heating up to 400 K. Moreover, the theoretical estimate of the Debye temperature (a marker of the thermal behavior),
K, and the Grüneisen parameter (a marker of the thermal expansion),
made by using the elastic parameters of
Table 3 demonstrate values typical for solid hydroxides.
2.4. Dihydrogen Bonding
As it follows from the protonation chemistry of transition metal hydride complexes [
33], a dissociative pathway of metal–hydrogen bond may lead to the formation of dihydrogen bonding patterns. We have tested whether tentative signs of hydrogen bonding can be seen from vibrational analysis of Y
2H
3O(OH). In particular, we have found that the decomposition of a set of relatively low-lying frequencies (
Table S4 of SI), 1008–1393 cm
, which are associated with the stretching movements among hydrogen pairs, points to hydrogen bonding in the H(1)–H(4) and H(2)–H(4) contacts of hydridic and protonic hydrogens. On the other hand, the most high-frequency modes, 3336 and 3340 cm
, can be assigned to the decoupled stretching distortions of the OH
- anion (similarly to solid hydroxides). We have also assumed that the large value of the low-frequency shift,
ca 350 cm
, in the spectral behavior of proton donors in Y
2H
3O(OH) (calculated as the difference between high-frequency stretching vibrations in Y
2H
3O(OH) and Y(OH)
3) may serve as the other good signature of hydrogen bonding.
The structural grouping and geometry of the system of hydrogen bonds are described in
Table 5 and depicted in
Figure 5 in terms of H
H characteristic contacts. The results demonstrate that, by joining yttrium polyhedral units in the crystal lattice, the non-covalent coupling between the OH protons and hydride anions establishes a robust network of dihydrogen bonds [
33,
34,
35,
36]. On the base of hydrogen equilibrium positions (
Table 1b), the strength and directionality of the dihydrogen bonds in Y
2H
3O(OH) can be characterized as follows. Of two H
H short contacts, the smallest,
Å for the H(1)
H(4) distance, is the most representative. The other H(2)
H(4) contact elongated to 2.050 Å also belongs to a typical dihydrogen bond [
37]. Both contacts may be characterized as strong and almost strong because the distances observed are significantly smaller than the sum of van der Waals radii for hydrogen. Accordingly, the third H(4)
H(3) contact longer than 2.4 Å cannot be classified as a dihydrogen bond. We note further that strong bending of the O−H
H angle, which varies between
and
with
as an average, is a direct indication that the dihydrogen bond is realized as the O−H
interaction in full accord with the theoretical picture presented in Reference [
38]. We believe that the equilibrium distribution of dihydrogen bonds in Y
2H
3O(OH) cannot be easily destroyed because our MD simulations have clearly shown that the dihydrogen bonds are able to withstand the impact of thermal heating. The role of dihydrogen bonds is that, by linking together the [H
3Y
2O] chains via OH groups, they provide the reinforcement of the lattice structure of Y
2H
3O(OH). In other words, the dihydrogen bond represents an important structural element [
37] contributing to the stability of the chiral geometry of Y
2H
3O(OH).
One geometric characteristic of the H(1)H(4)H(2) symbolic triangle is the proximity of the H(1) and H(2) sites, which, in terms of the H(1)
H(2) contact length (
Å), is very close to the van der Waals diameter (e.g.,
Figure 5). The rationalization of this fact is interesting in the sense that the strong dihydrogen bonding tends to limit the electrostatic repulsion between the nearest, similarly charged hydridic hydrogens by placing them on the distance of closest approach. In the context of the bonding geometry, such an accommodation becomes most favorable because the proton at the H(4) position gains a possibility to simultaneously attacks the two closely located hydride centers H(1) and H(2).
2.5. Y2H3O(OH) as a Chiral Crystal
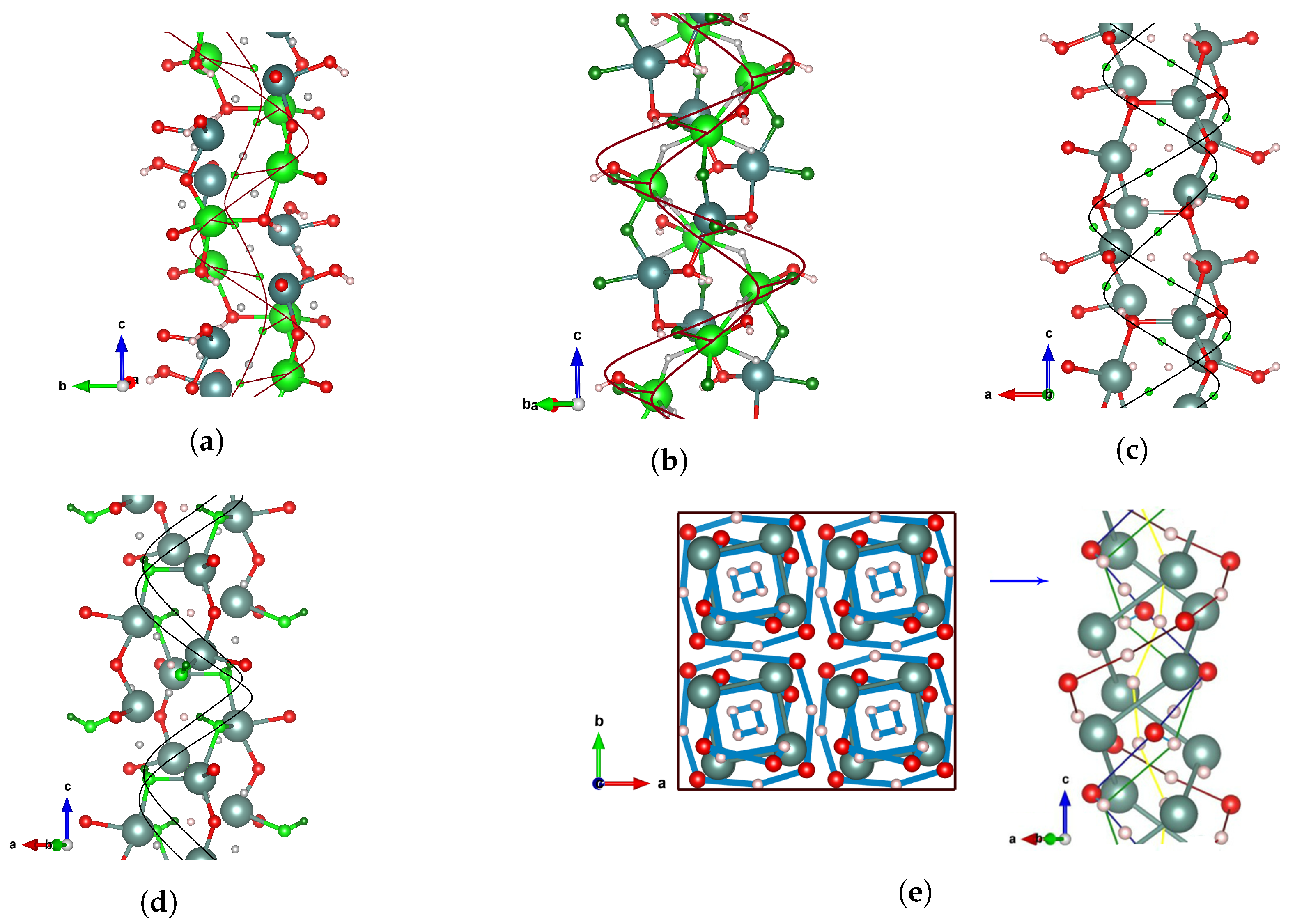
Y
2H
3O(OH) crystallizes entirely in a chiral structure in which all the atoms of the unit cell occupy the
crystallographic sites (
Table 1b). That is, the structural organization is provided by the four-atom patterns belonging to one orbit. Since the site symmetry is described by a chiral (pure rotational)
point group, a rotational atom stacking in the pattern leads to the formation of
-helices corresponding to 1d-helical chains decorated by Y(1), Y(2), O(1), O(2), H(1), H(2), H(3), and H(4) atoms. Accordingly, side-by-side spatial arrangements of all of them form a chiral framework in Y
2H
3O(OH) which consists of 8 single helices.
As shown in
Figure 6, the helical assembly comprises the right-handed helical chains around the crystallographic screw axis
, which spreads along the
z-direction of the entire structure. Interestingly, the helical organization affords a tolerable amount of freedom because some of the different
atomic positions can be grouped to compose joined helical chains (
Figure 6e). In this context, one could associate the high degree of structural orderings in the tetragonal phase with the formation of rotational stacking arrangements of the spatially separated OH
- anions. That is, in order to enforce itself via dihydrogen bonding, the lattice has to rotate relative to the oxygen fixed positions within the only possible tubular orientation around the
z axis.
2.6. Electron Band Structure
The electron structure calculations showed that the bulk Y
2H
3O(OH) is a direct-gap semiconductor with the fundamental band-gap width of
eV in the
point. This value is confirmed by the estimate,
eV, which was obtained within the
approximation (
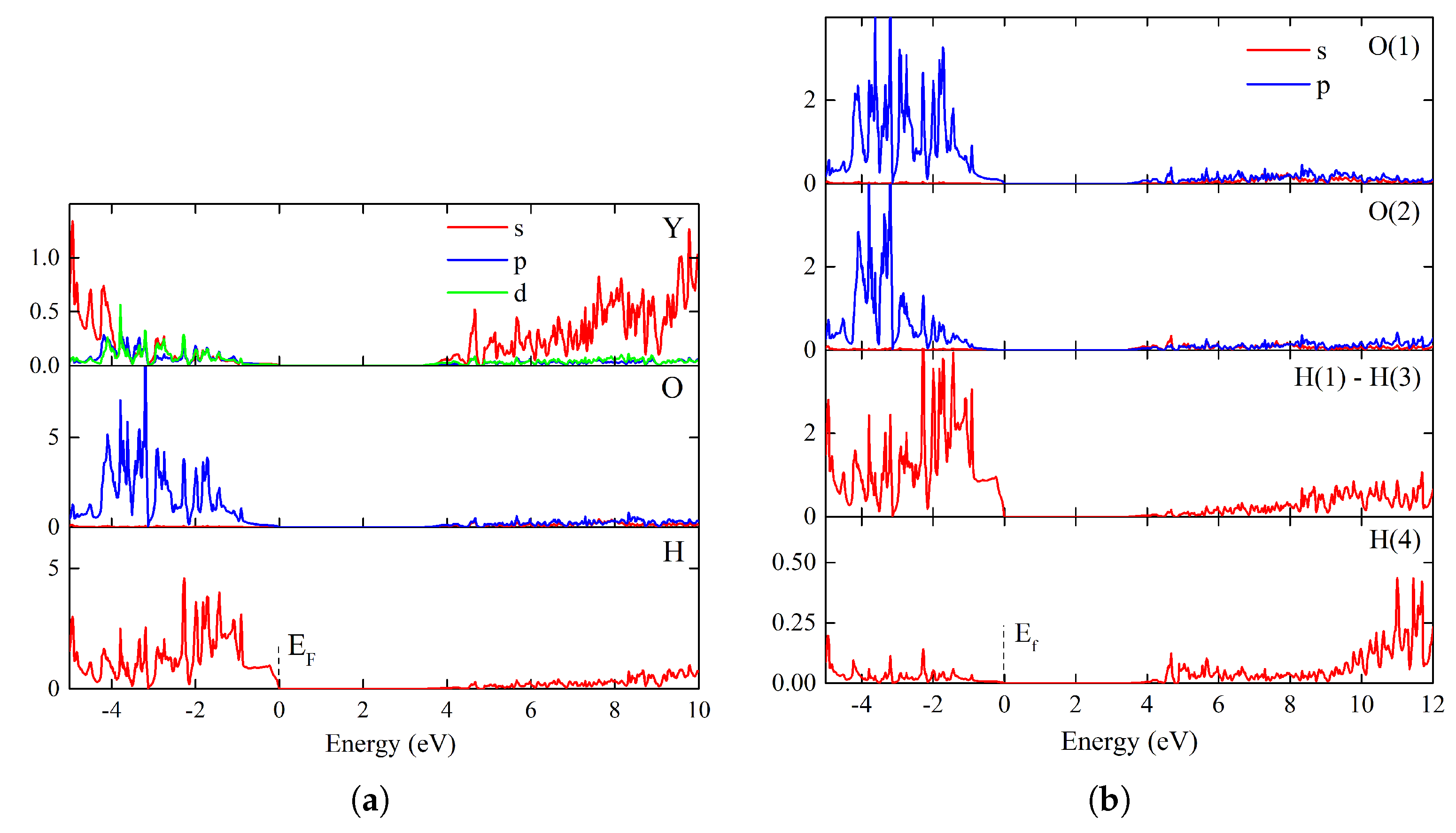
Figure S9 of SI). The density of states plots are shown in
Figure 7a,b. A comparison indicates the prevailing contribution of the hydride anion
s electron states in the valence band region near the Fermi energy. This is due to the strong localization of electrons at the hydride anion. One sees large peaks of DOS in the wide interval of the valence band, in which the behavior of
p oxygen states mimics the hydrogen
s states. However, the contribution of O(2) oxygen is narrowed and shifted by
ca 2 eV, as compared with the corresponding contribution of O(1). The other feature is that the conduction band, which is formed by contributions of the
s empty states of all the elements, is significantly broadened along the energy axis.
Figure S5 of SI indicates that the bands with s-electron character are relatively well dispersed. The calculated effective masses shown here characterize anisotropy and dispersion of the electron bonds near the
point along the
and
high-symmetry directions.
2.7. Optical Responses
Inorganic materials with the spatial chirality are of great interest for various optical applications. The structural model of Y
2H
3O(OH) corresponds to a uniaxial chiral (right-handed) material which is optically active and exhibits optical anisotropy governed by the tetragonal axis. As shown in
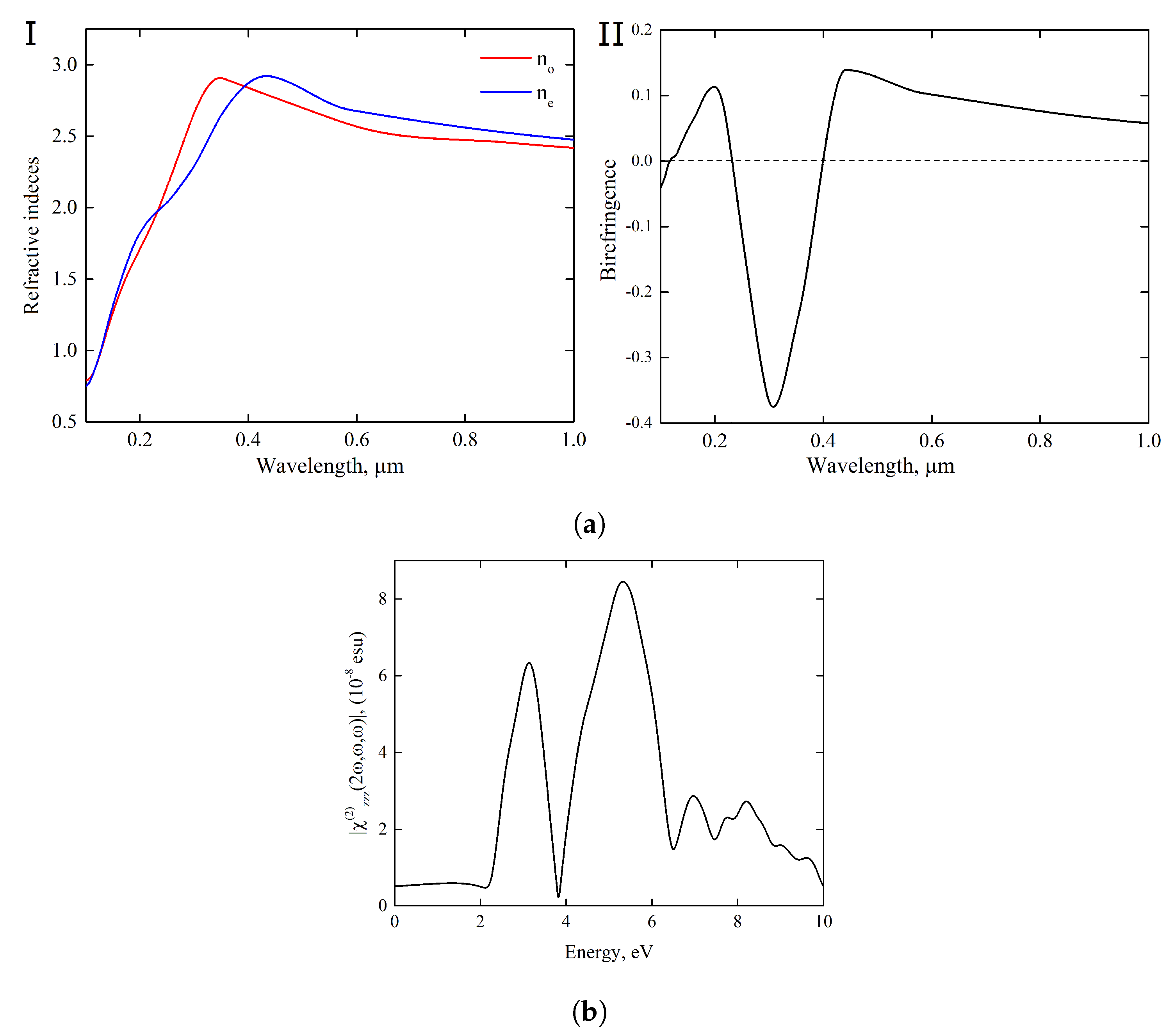
Figure 8a and in
Figures S6–S8 of SI, our calculations revealed anomalous dispersion visible at short wavelengths up to
ca 370 and 420 nm for ordinary and extraordinary rays, respectively.
The large difference in behavior of these light rays along the whole spectrum has a pronounced impact on the magnitude of . This may lead to a scenario in which the chiral material exhibits new experimentally undiscovered effect of anomalous birefringence.
Nonlinear optical (NLO) properties are more sensitive to the details of the electron band structure and to selection rules. In the crystalline medium without inversion center, the asymmetry of the electron transitions gives rise to such NLO phenomenon of the coherent nature as the second-harmonic generation (SHG) [
39,
40]. We considered a possibility of frequency doubling in terms of the second-order response of Y
2H
3O(OH) on the electric field of the incident light wave. Shown in
Figure 8b and in
Figures S10 and S11 of SI is the theoretical prediction for the
component of the SHG susceptibility tensor. According to our calculations, this component determines a dominant fraction of the bulk SHG in Y
2H
3O(OH); tensor components have been estimated in the ratio as
:
∼
: 1. The theoretical assessment for photon energies corresponding to
and 422 nm provided the evaluation of
as
and
pm/V, respectively. These values of nonlinear coefficients are close to the NLO susceptibilities typical for perovskite ferroelectrics with strong optical nonlinearities [
41].
2.8. The Role of Substitutions
Given that the chiral structure of Y2H3O(OH) is stable, one can ask whether the change of yttrium may keep crystallization in the lattice. In other words, whether the full cation exchange can afford a whole family of stoichiometric hydroxyhydrides M2H3O(OH) (where M standing on the yttrium cation site relates to a three-valence metal element) with the multianion ratio .
To resolve this issue, we have evaluated the structural stability of three systems differing in yttrium substitution by M = Sc, La, and Gd (
Tables S5–S7 of SI). The results shown in
Table 6 and in
Tables S1–S3 of SI demonstrate that the entirely substituted hydroxyhydrides crystallize in the tetragonal phase with the
chiral crystal structure. All the materials exhibit relatively similar crystallographic
ratios close to
. Calculated X-Ray diffraction patterns for predicted M
2H
3O(OH) are shown in
Figures S1–S4 of SI. The valence count shows that the compounds will behave as band insulators. Based on the smooth performance of the hydrogens which contribute to assembly of the helical structures in Y
2H
3O(OH), one can further suggest that the similar scheme of hydrogen bonding exists in M
2H
3O(OH), thus making the strong dihydrogen bonding a general feature of these chiral systems (
Table 6b). The comparison of
Table 5 and
Table 6b indicates that both the interaction strength and the proton accepting/donating abilities are roughly the same for the whole range of hydroxyhydride systems predicted in the present work.
2.9. Discussion
In summary, by modeling the oxidation process of the hydride host in terms of different structural combinations of metal–oxygen and oxygen–hydrogen interactions, we have predicted a novel class of inorganic crystalline materials—hydroxyhydrides which can be described by the chemical formula M2H3O(OH) with the cation site M occupied by the trivalent metal element. Based on the results of structure-modeling simulations, we believe that M2H3O(OH) may represent a wider range of stoichiometric compounds than those four (M = Y, Sc, La, and Gd) that have been reported in the present work. It is important to emphasize that a major common trait of these mixed-anionic systems is the absolute chirality determined by the crystal structure. In the Y2H3O(OH) case study, we have investigated a number of structural and bonding features such as the extra-high localization of valence charge densities, the strong dihydrogen bonding, and the stability of the spatial arrangement of hydridic and protonic hydrogens in the lattice. The effect of strong localization corresponds to a specific charge ordering connected with the chiral organization of the metal cations and the anions which are standing in positions that form helical curves spreading along the tetragonal axis. The effect of twisting of the H- and H+ hydrogens related to different chains causes their linking by dihydrogen bonds. In the context of structure–property relationships, valuable insights into elastic, electronic, and optical properties of the bulk Y2H3O(OH) have been gained.
In the context of application-oriented research, one can emphasize that our findings open a promising route to the development of novel relatively simple chiral materials for possible applications in optoelectronics, laser, and nonlinear optics. The development of inorganic materials with NLO properties is one of the important challenges. However, the problem is that some materials with desirable NLO properties can be formed under extreme synthesis conditions that are not so easy to reach. The organic compounds that may exhibit NLO properties under normal conditions cannot withstand external effects such as high electromagnetic fields, etc. In light of this, Y2H3O(OH) has great potential to overcome a number of current challenges and limitations.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}