Vanadium-Substituted Phosphomolybdic Acids for the Aerobic Cleavage of Lignin Models—Mechanistic Aspect and Extension to Lignin


Abstract
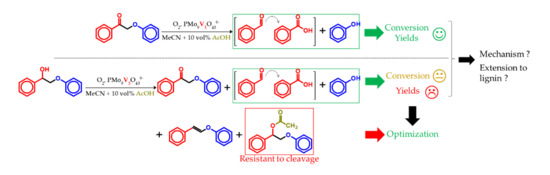
1. Introduction
2. Materials and Methods
2.1. Materials Synthesis
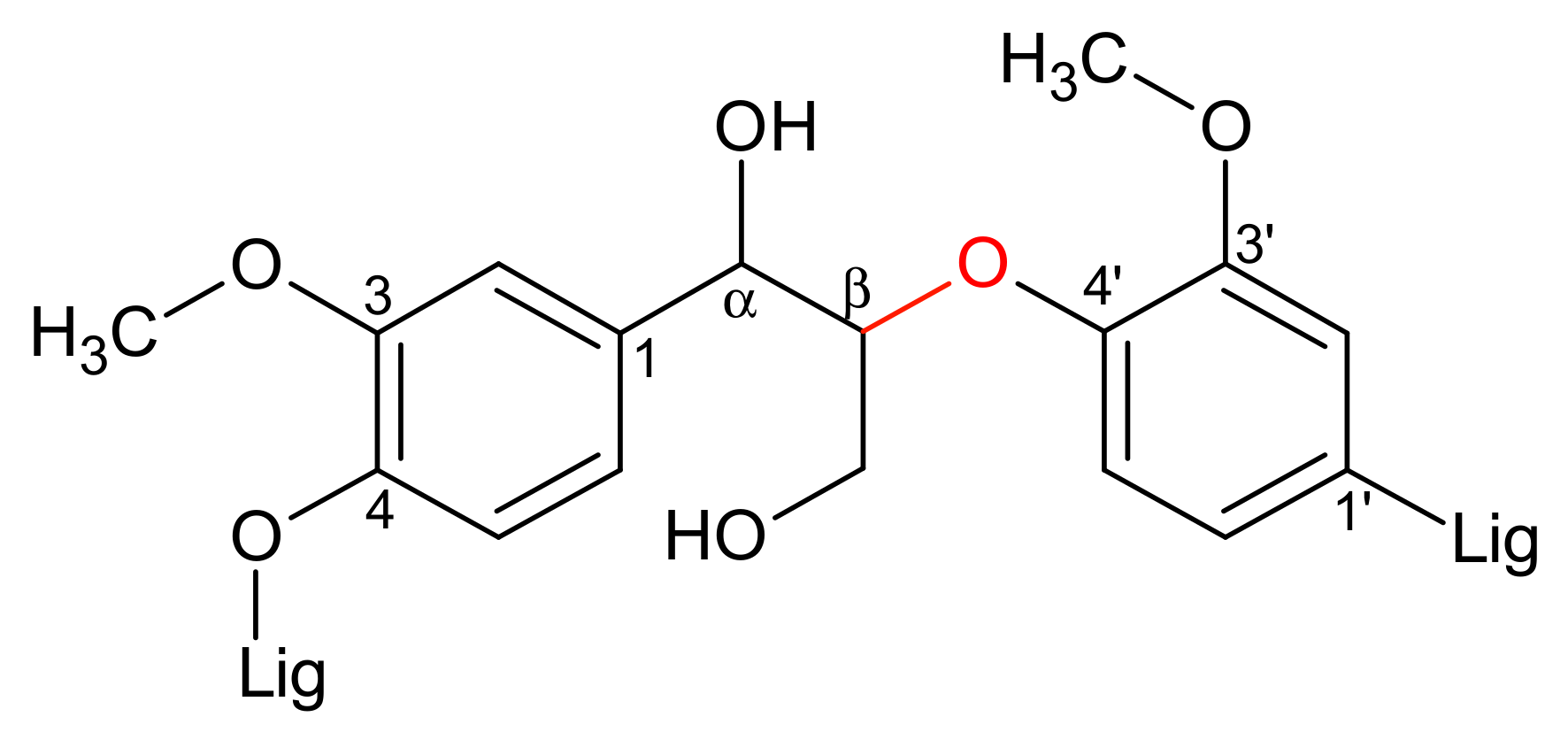
2.2. H6PV3 Characterization
2.3. Catalytic Tests
2.4. Monitoring of the Tests
2.5. Preliminary Tests on an Organosolv Lignin
3. Results
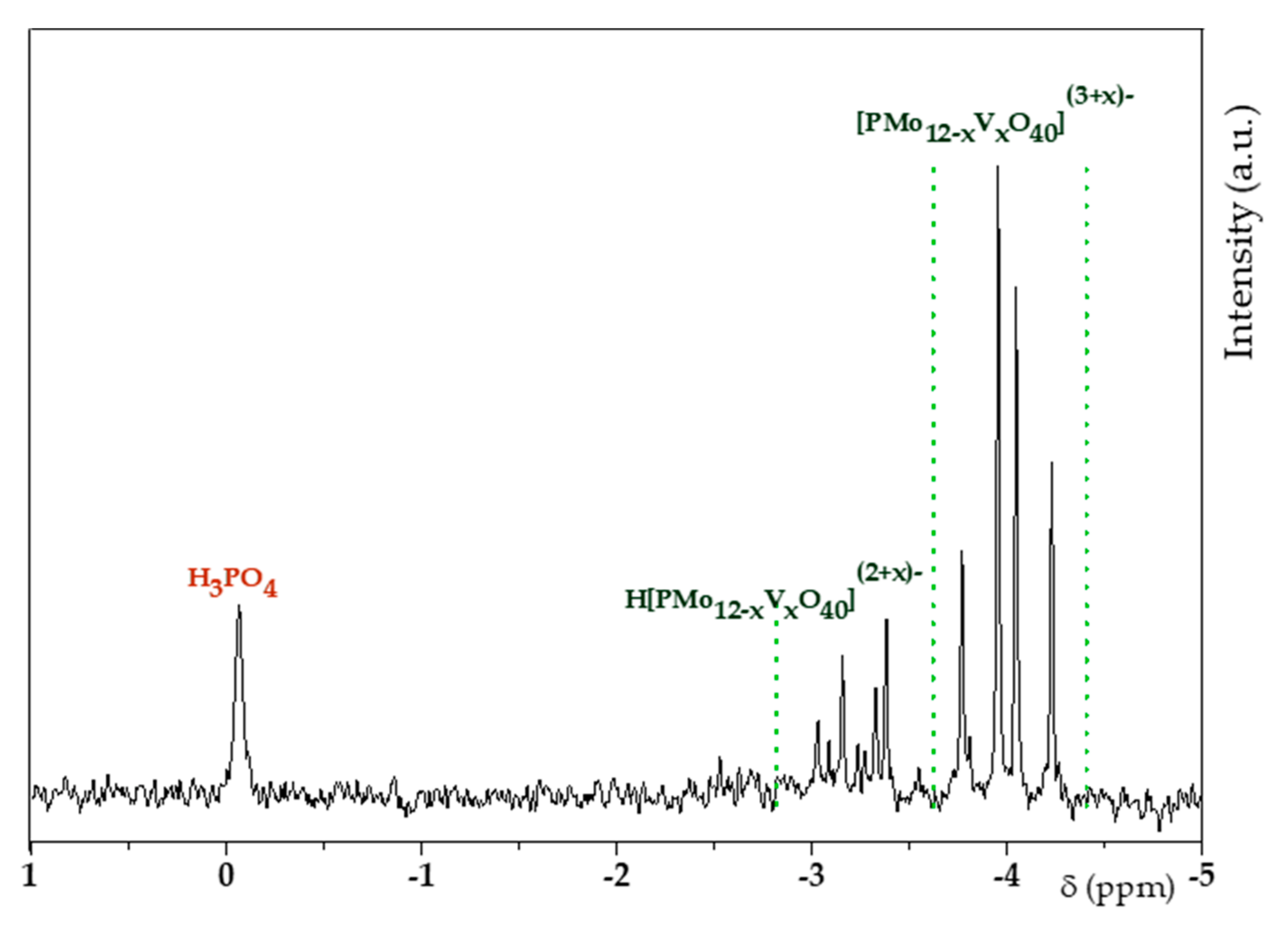
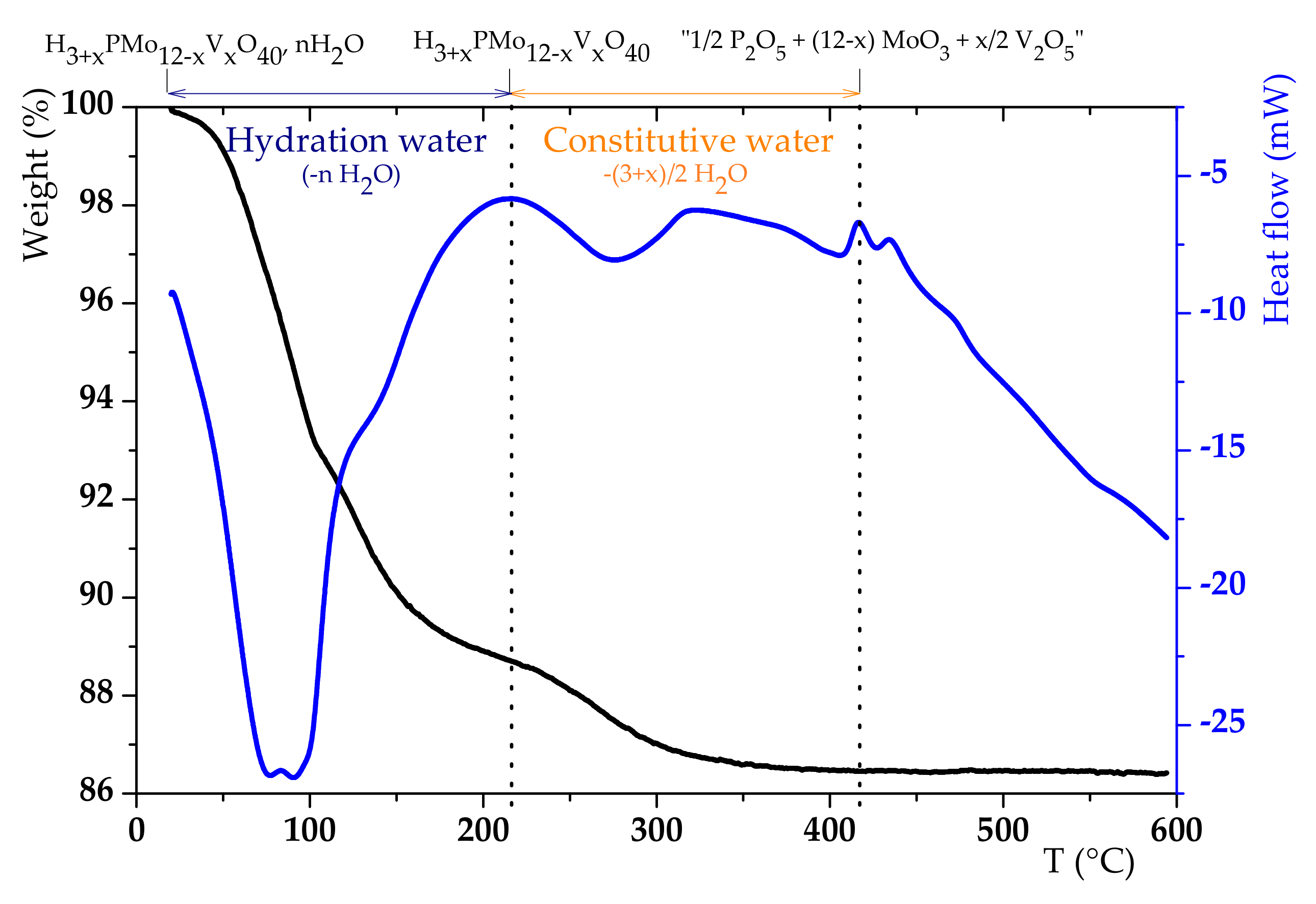
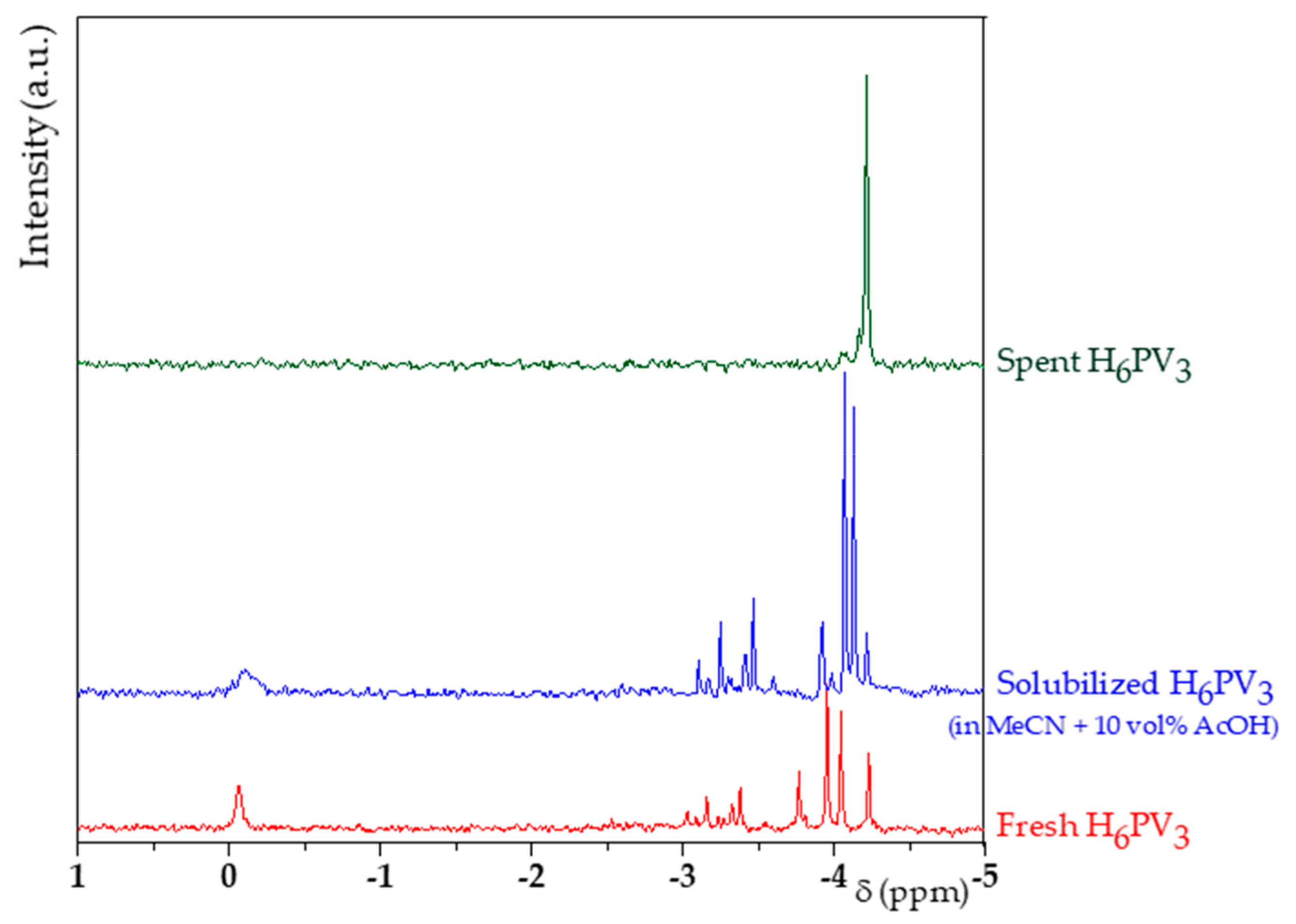
3.1. H6PV3 Synthetic Procedure
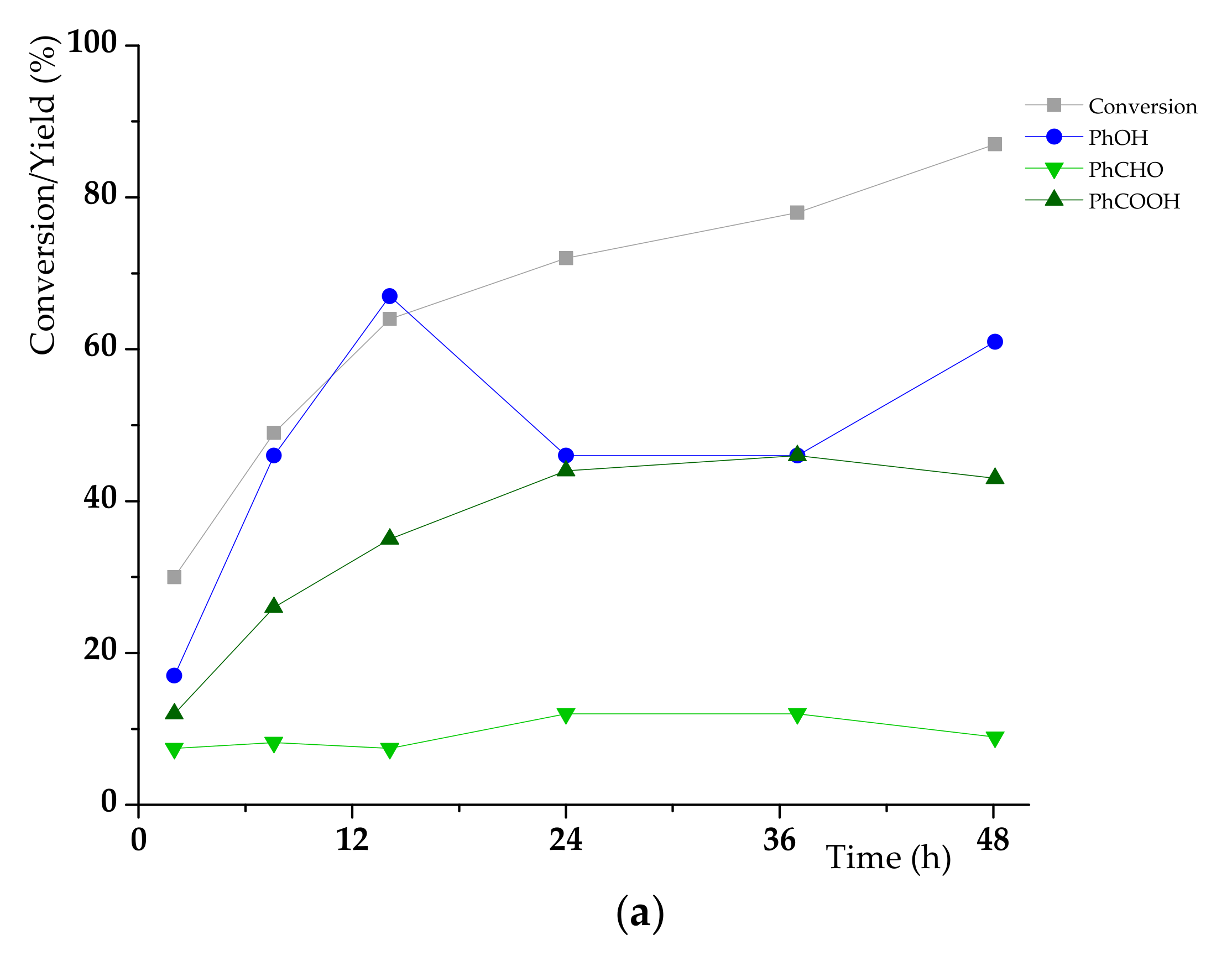
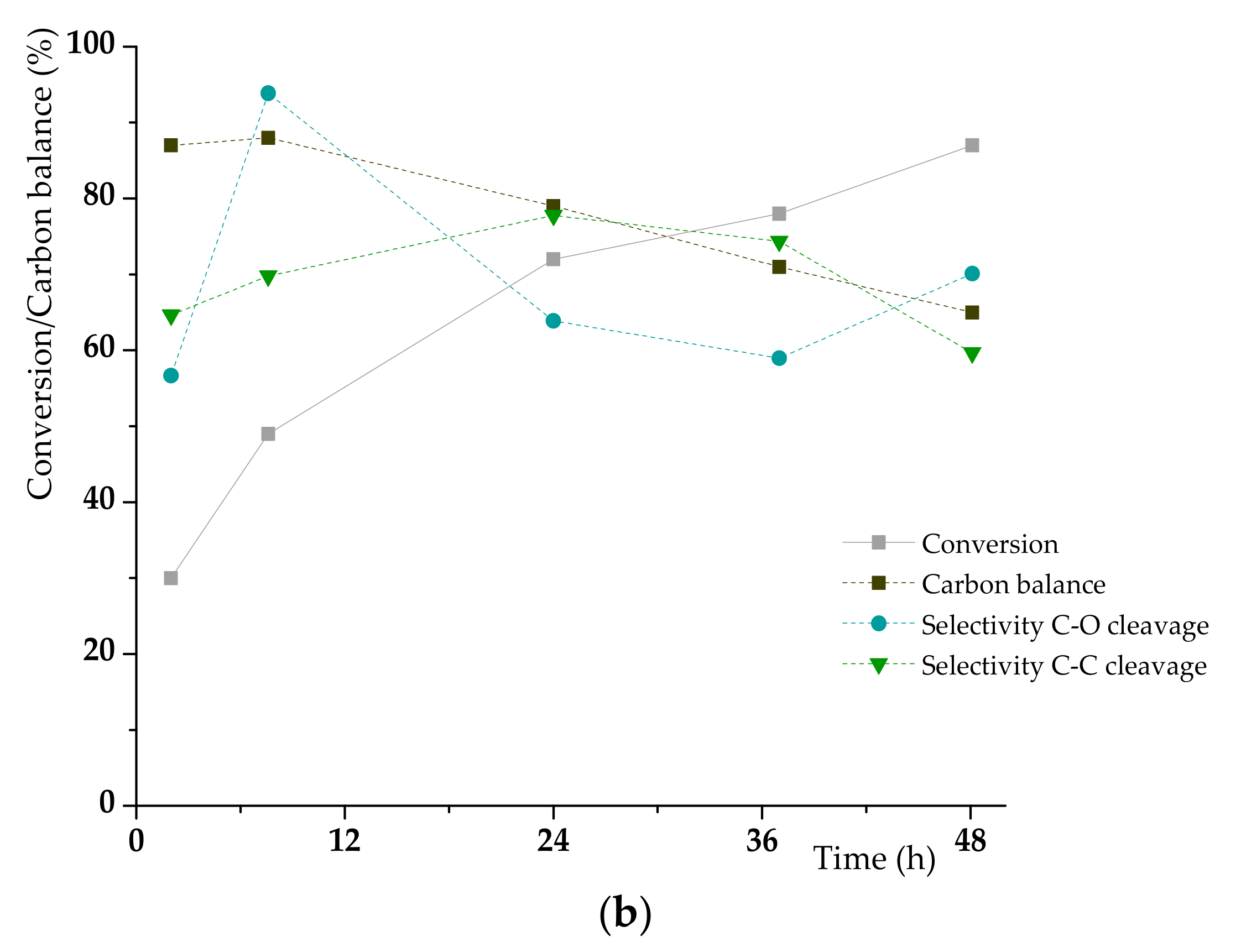
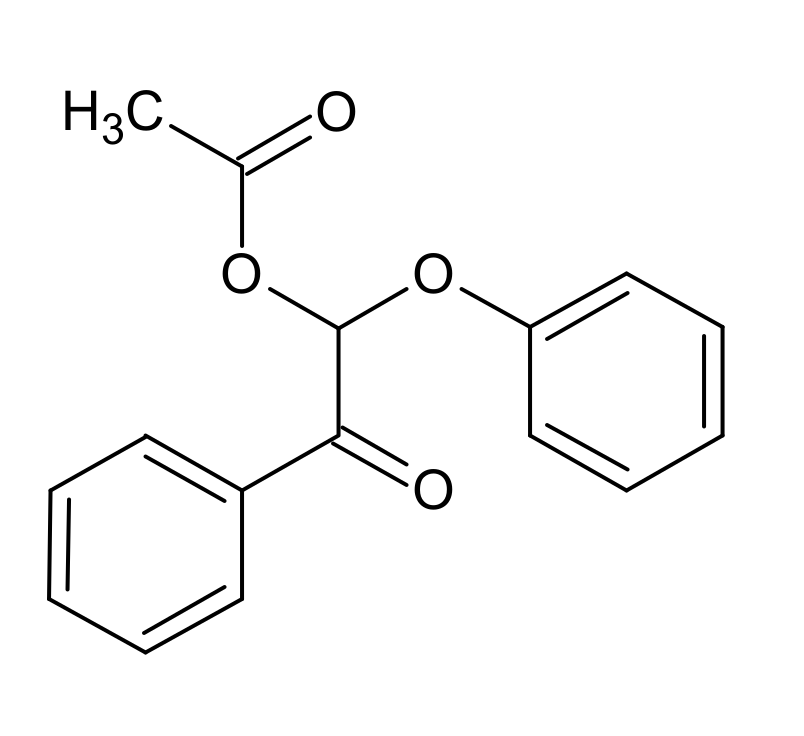
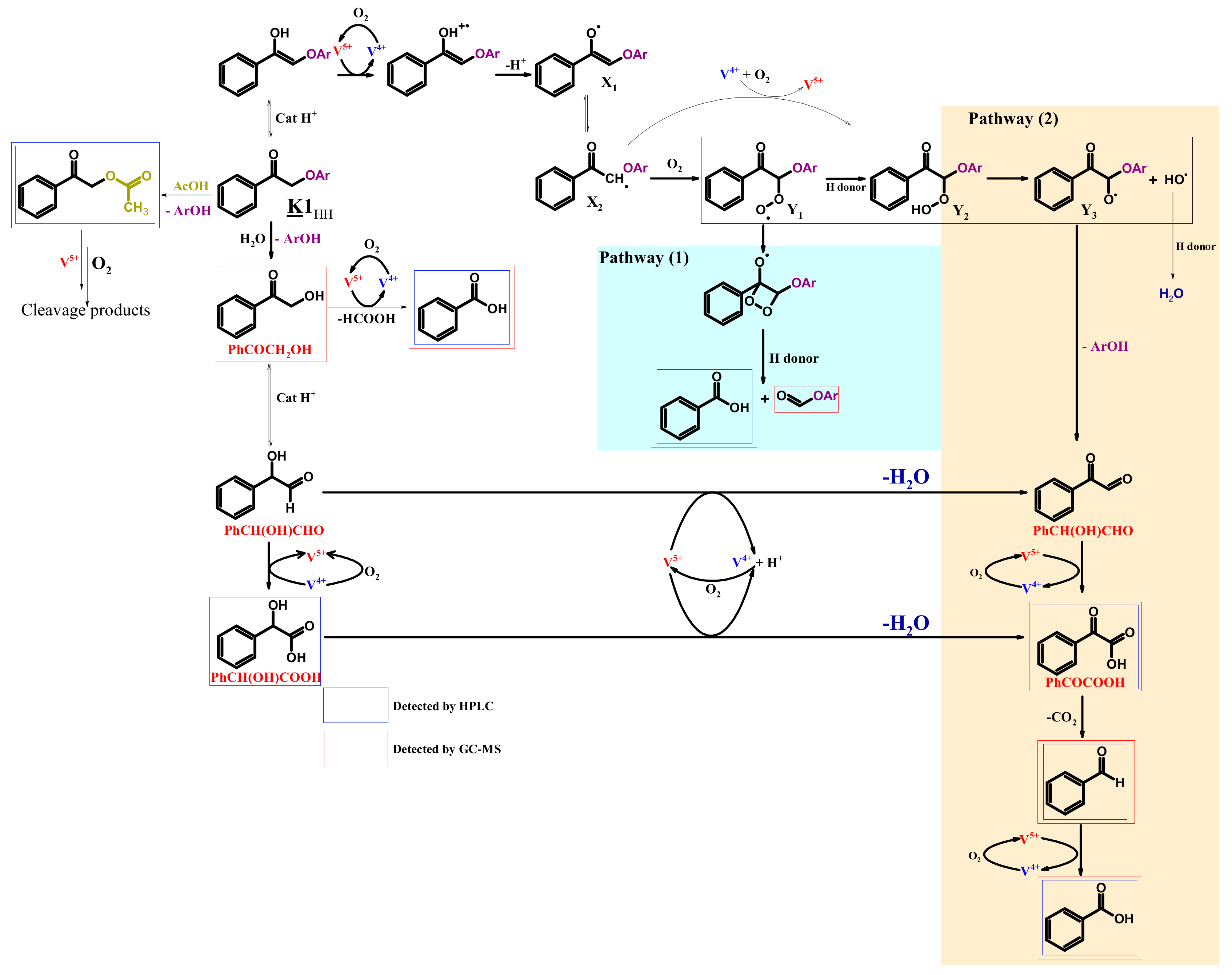
3.2. K1HH aerobic cleavage
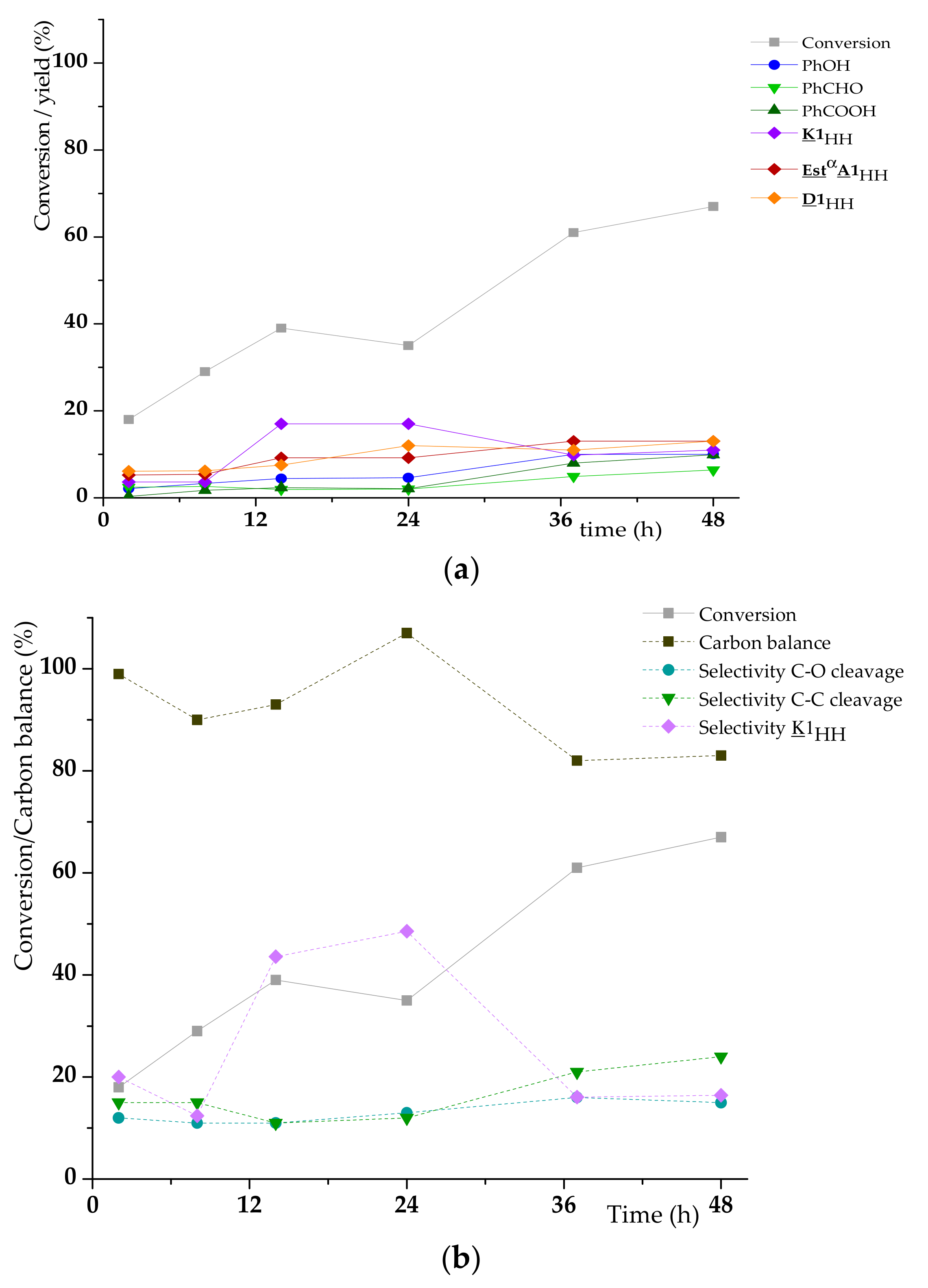
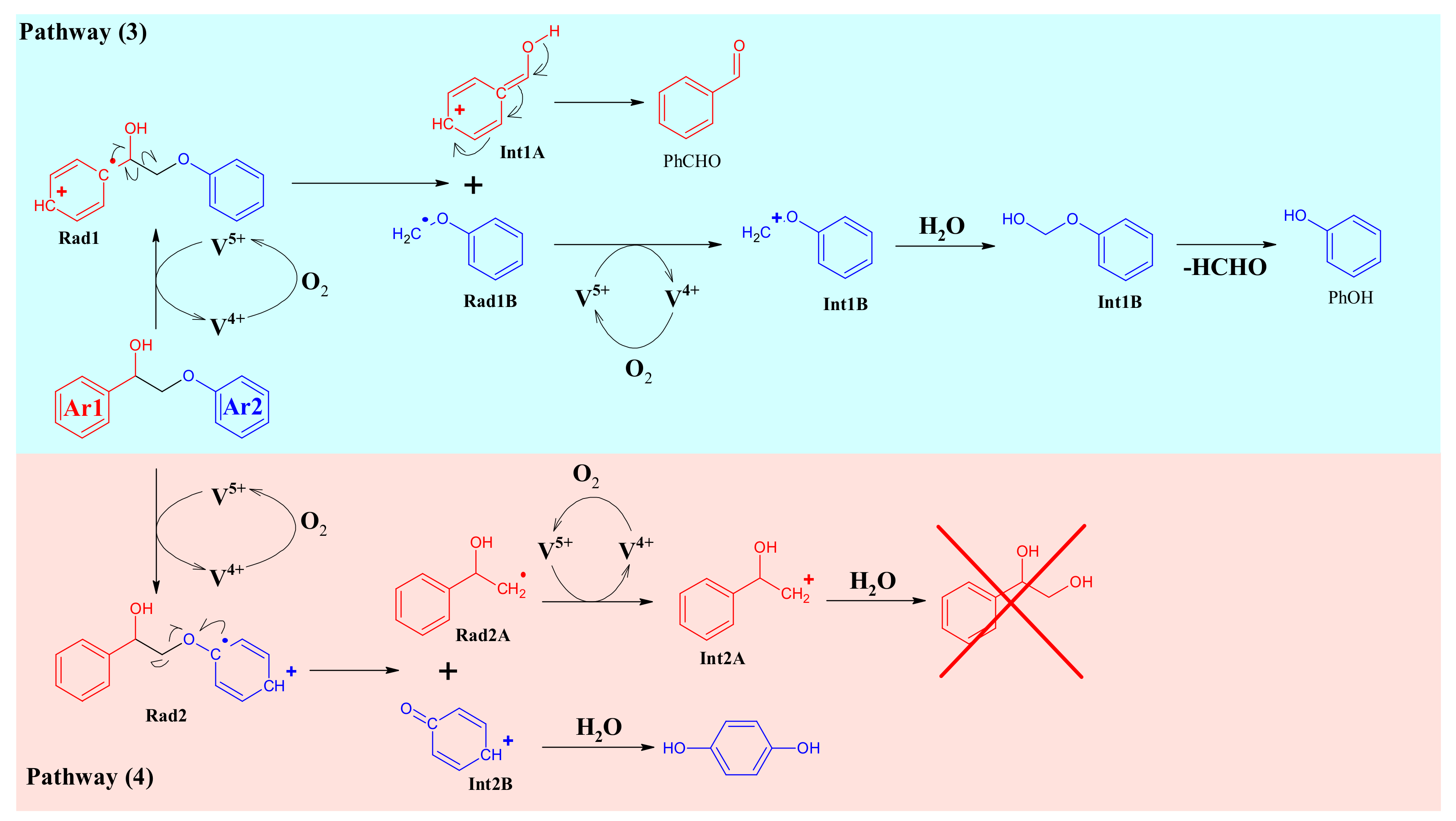
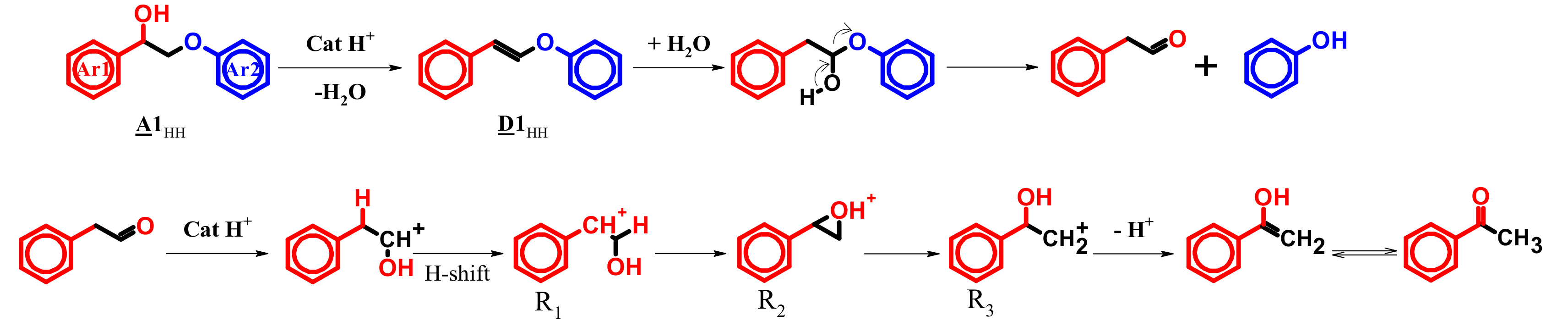
3.3. A1HH aerobic Cleavage
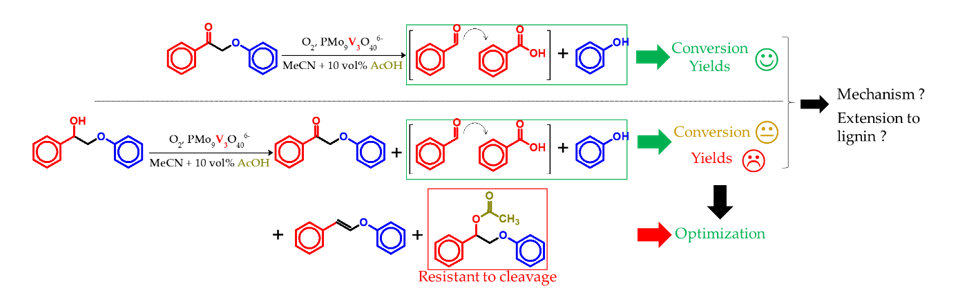
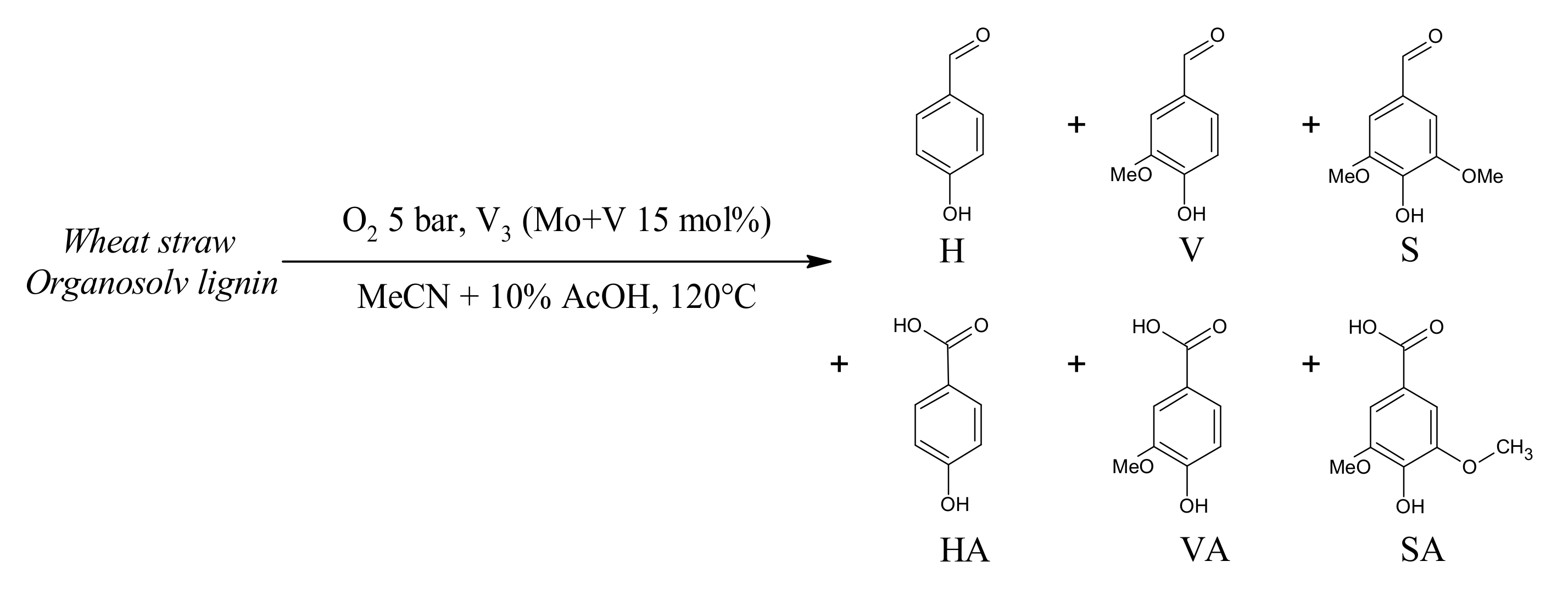
3.4. Lignin Aerobic Cleavage: Preliminary Tests
4. Discussion
4.1. H6PV3 Synthesis
4.2. Evolution of the Catalyst
4.3. K1HH and A1HH Cleavage
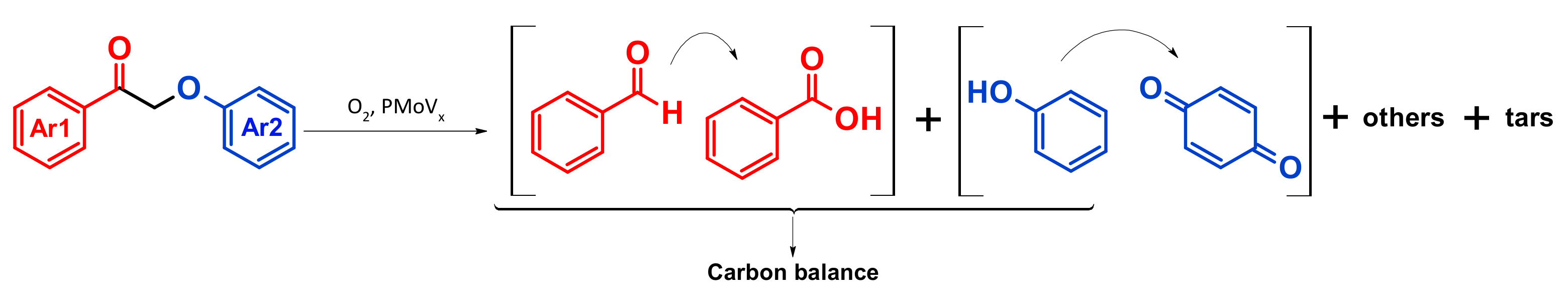
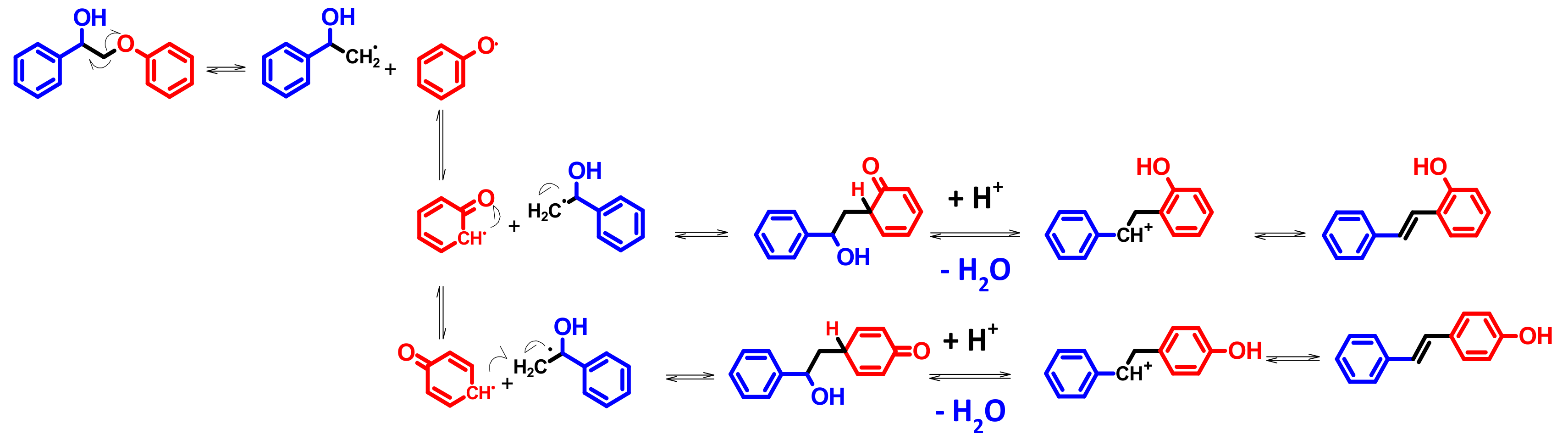
4.4. Mechanism Proposal
4.5. Extension to Lignin
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Behling, R.; Valange, S.; Chatel, G. Heterogeneous catalytic oxidation for lignin valorization into valuable chemicals: what results? What limitations? What trends? Green Chem. 2016, 18, 1839–1854. [Google Scholar] [CrossRef]
- Hocking, M.B. Vanillin: Synthetic Flavoring from Spent Sulfite Liquor. J. Chem. Educ. 1997, 74, 1055. [Google Scholar] [CrossRef]
- Lange, H.; Decina, S.; Crestini, C. Oxidative upgrade of lignin—Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar] [CrossRef]
- Ma, R.; Guo, M.; Zhang, X. Recent advances in oxidative valorization of lignin. Catal. Today 2018, 302, 50–60. [Google Scholar] [CrossRef]
- Mottweiller, J.; Puche, M.; Räuber, C.; Schmidt, T.; Concepción, P.; Corma, A.; Bolm, C. Copper- and Vanadium-Catalyzed Oxidative Cleavage of Lignin using Molecular oxygen. ChemSusChem 2015, 8, 2106–2113. [Google Scholar] [CrossRef]
- Díaz-Urrutia, C.; Hurisso, B.B.; Gauthier, P.M.; Sedai, B.; Singer, R.D.; Baker, R.T. Catalytic aerobic oxidation of lignin-derived bio-oils using oxovanadium and copper complex catalysts and ionic liquids. J. Mol. Catal. A Chem. 2016, 423, 414–422. [Google Scholar] [CrossRef]
- Amadio, E.; Di Lorenzo, R.; Zonta, C.; Licini, G.M. Vanadium catalyzed aerobic carbon–carbon cleavage. Co-ord. Chem. Rev. 2015, 301, 147–162. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, Q.; Pan, T.; Zuo, Y.; Fu, Y.; Guo, Q.-X. Depolymerization of lignin by catalytic oxidation with aqueous polyoxometalates. Appl. Catal. A Gen. 2013, 467, 504–508. [Google Scholar] [CrossRef]
- De Gregorio, G.F.; Prado, R.; Vriamont, C.; Erdocia, X.; Labidi, J.; Hallett, J.P.; Welton, T. Oxidative Depolymerization of Lignin Using a Novel Polyoxometalate-Protic Ionic Liquid System. ACS Sustain. Chem. Eng. 2016, 4, 6031–6036. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, H.; Rogers, R.D. Oxygen Enhances Polyoxometalate-based Catalytic Dissolution and Delignification of Woody Biomass in Ionic Liquids. ACS Sustain. Chem. Eng. 2014, 2, 2859–2865. [Google Scholar] [CrossRef]
- Weinstock, I.A. Delignification of Wood Pulp by Vanadium-Substituted Polyoxometalates. US Patent 5302248, 12 April 1994. [Google Scholar]
- Gaspar, A.R.; Gamelas, J.A.F.; Evtuguin, D.V.; Neto, C.P. Alternatives for lignocellulosic pulp delignification using polyoxometalates and oxygen: a review. Green Chem. 2007, 9, 717. [Google Scholar] [CrossRef]
- Weinstock, I.A.; Schreiber, R.E.; Neumann, R. Molecular oxygen in polyoxometalate mediated reactions. Chem. Rev. 2018, 118, 2680–2717. [Google Scholar] [CrossRef] [PubMed]
- El Aakel, L.; Launay, F.; Atlamsani, A.; Brégeault, J.-M. Efficient and selective catalytic oxidative cleavage of α-hydroxy ketones using vanadium-based HPA and dioxygen. Chem. Commun. 2001, 21, 2218. [Google Scholar] [CrossRef] [PubMed]
- El Ali, B.; El-Ghanam, A.M.; Fettouhi, M. H3+nPMo12−nVnO40-catalyzed selective oxidation of benzoins to benzils or aldehydes and esters by molecular oxygen. J. Mol. Catal. A 2001, 165, 283–290. [Google Scholar] [CrossRef]
- Ballarini, N.; Casagrandi, L.; Cavani, F.; D’Alessandro, T.; Frattini, A.; Accorinti, P.; Alini, S.; Babini, P. The liquid-phase oxidation of cyclohexanone with oxygen, catalysed by Keggin-type polyoxometalates. A cleaner alternative to the current industrial process for adipic acid synthesis. DGMK Tagungsbericht 2008, 225–232. [Google Scholar]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef]
- Sedai, B.; Díaz-Urrutia, C.; Baker, R.T.; Wu, R.; Silks, L.P.; Hanson, S.K. Comparison of Copper and Vanadium Homogeneous Catalysts for Aerobic Oxidation of Lignin Models. ACS Catal. 2011, 1, 794–804. [Google Scholar] [CrossRef]
- Hanson, S.K.; Baker, R.T.; Gordon, J.C.; Scott, B.L.; Thorn, D.L. Aerobic Oxidation of Lignin Models Using a Base Metal Vanadium Catalyst. Inorg. Chem. 2010, 49, 5611–5618. [Google Scholar] [CrossRef]
- Son, S.; Toste, F.D. Non-Oxidative Vanadium-Catalyzed C-O Bond Cleavage: Application to Degradation of Lignin Model Compounds. Angew. Chem. Int. Ed. 2010, 49, 3791–3794. [Google Scholar] [CrossRef]
- Zhang, G.; Scott, B.L.; Wu, R.; Silks, L.P.; Hanson, S.K. Aerobic Oxidation Reactions Catalyzed by Vanadium Complexes of Bis(Phenolate) Ligands. Inorg. Chem. 2012, 51, 7354–7361. [Google Scholar] [CrossRef]
- Hanson, S.K.; Wu, R.; Silks, L.P. C-C or C-O Bond Cleavage in a Phenolic Lignin Model Compound: Selectivity Depends on Vanadium Catalyst. Angew. Chem. Int. Ed. 2012, 51, 3410–3413. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Du, Z.; Liu, J.; Xia, F.; Xu, J. Selective oxidative C–C bond cleavage of a lignin model compound in the presence of acetic acid with a vanadium catalyst. Green Chem. 2015, 17, 4968–4973. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Yan, L.; Yu, H.-Z.; Zhang, Q.; Fu, Y. Mechanism of Vanadium-Catalyzed Selective C–O and C–C Cleavage of Lignin Model Compound. ACS Catal. 2016, 6, 4399–4410. [Google Scholar] [CrossRef]
- Parker, H.J.; Chuck, C.J.; Woodman, T.; Jones, M.D. Degradation of β-O-4 model lignin species by vanadium Schiff-base catalysts: Influence of catalyst structure and reaction conditions on activity and selectivity. Catal. Today 2016, 269, 40–47. [Google Scholar] [CrossRef]
- Gazi, S.; Đokić, M.; Moeljadi, A.M.P.; Ganguly, R.; Hirao, H.; Soo, H.S. Kinetics and DFT Studies of Photoredox Carbon–Carbon Bond Cleavage Reactions by Molecular Vanadium Catalysts under Ambient Conditions. ACS Catal. 2017, 7, 4682–4691. [Google Scholar] [CrossRef]
- Evtuguin, D.V.; Daniel, A.I.; Silvestre, A.J.; Amado, F.M.; Neto, C.P. Lignin aerobic oxidation promoted by molybdovanadophosphate polyanion [PMo7V5O40]8−. Study on the oxidative cleavage of β-O-4 aryl ether structures using model compounds. J. Mol. Catal. A Chem. 2000, 154, 217–224. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef]
- Bujanovic, B.; Ralph, S.; Reiner, R.; Hirth, K.; Atalla, R. Polyoxometalates in Oxidative Delignification of Chemical Pulps: Effect on Lignin. Materials 2010, 3, 1888–1903. [Google Scholar] [CrossRef]
- Odyakov, V.F.; Zhizhina, E.G.; Rodikova, Y.A.; Gogin, L.L. Mo-V-Phosphoric Heteropoly Acids and Their Salts: Aqueous Solution Preparation—Challenges and Perspectives. Eur. J. Inorg. Chem. 2015, 2015, 3618–3631. [Google Scholar] [CrossRef]
- Kern, F.; Ruf, S.; Emig, G. Vapour-phase trimerization of formaldehyde to trioxane catalysed by 1-vanado-11-molybdophosphoric acid. Appl. Catal. A Gen. 1997, 150, 143–151. [Google Scholar] [CrossRef]
- Tsigdinos, G.A.; Hallada, C.J. Molybdovanadophosphoric acids and their salts. I. Investigation of methods of preparation and characterization. Inorg. Chem. 1968, 7, 437–441. [Google Scholar] [CrossRef]
- Derwent, R.; Jenkin, M.; Passant, N.; Pilling, M. Reactivity-based strategies for photochemical ozone control in Europe. Environ. Sci. Policy 2007, 10, 445–453. [Google Scholar] [CrossRef]
- Atlamsani, A.; Ziyad, M.; Brégeault, J. Les hétéropolyacides comme catalyseurs bifonctionnels pour la coupure oxydante de cyclanones. J. Chim. Phys. 1995, 92, 1344–1364. [Google Scholar] [CrossRef]
- Odyakov, V.F.; Zhizhina, E.G. New process for preparing aqueous solutions of Mo-V-phosphoric heteropoly acids. Russ. J. Inorg. Chem. 2009, 54, 361–367. [Google Scholar] [CrossRef]
- Grate, J.H.; Hamm, D.R.; Mahajan, S. Palladium and phosphomolybdovanadate catalyzed olefin oxidation to carbonyls. Mol. Eng. 1993, 3, 205–229. [Google Scholar] [CrossRef]
- Delmas, M.; Benjelloun-Mlayah, B. Procédé de Prétraitement d’une Matière Végétale Lignocellulosique en vue de la Production de Bioethanol; Pat. Fr 2 926 824; INPI: Paris, France, 21 January 2008. [Google Scholar]
- Mbotchak, L.; Le Morvan, C.; Duong, K.L.; Rousseau, B.; Tessier, M.; Fradet, A. Purification, Structural Characterization, and Modification of Organosolv Wheat Straw Lignin. J. Agric. Food Chem. 2015, 63, 5178–5188. [Google Scholar] [CrossRef]
- Marchal-Roch, C.; Bayer, R.; Moisan, J.F.; Teze, A.; Hervé, G. Oxidative dehydrogenation of isobutyric acid: Characterization and modeling of vanadium containing polyoxometalate catalysts. Top. Catal. 1996, 3, 407–419. [Google Scholar] [CrossRef]
- Pettersson, L.; Andersson, I.; Selling, A.; Grate, J.H. Multicomponent polyanions. 46. Characterization of the isomeric Keggin decamolybdodivanadophosphate ions in aqueous solution by 31P and 51V NMR. Inorg. Chem. 1994, 33, 982–993. [Google Scholar] [CrossRef]
- Barats-Damatov, D.; Shimon, L.J.W.; Feldman, Y.; Bendikov, T.; Neumann, R. Solid-State Crystal-to-Crystal Phase Transitions and Reversible Structure–Temperature Behavior of Phosphovanadomolybdic Acid, H5PV2Mo10O40. Inorg. Chem. 2015, 54, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Villabrille, P.; Romanelli, G.; Gassa, L.; Vazquez, P.; Caceres, C. Synthesis and characterization of Fe- and Cu-doped molybdovanadophosphoric acids and their application in catalytic oxidation. Appl. Catal. A Gen. 2007, 324, 69–76. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wei, L.; Hu, H.; Huang, Z.; Yang, M.; Huang, A.; Wu, J.; Feng, Z. Esterification mechanism of lignin with different catalysts based on lignin model compounds by mechanical activation-assisted solid-phase synthesis. RSC Adv. 2017, 7, 52382–52390. [Google Scholar] [CrossRef]
- Sato, T.; Hamada, Y.; Sumikawa, M.; Araki, S.; Yamamoto, H. Solubility of Oxygen in Organic Solvents and Calculation of the Hansen Solubility Parameters of Oxygen. Ind. Eng. Chem. Res. 2014, 53, 19331–19337. [Google Scholar] [CrossRef]
- Quaranta, M.; Murkovic, M.; Klimant, I. A new method to measure oxygen solubility in organic solvents through optical oxygen sensing. Analyst 2013, 138, 6243. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Deng, Z.; Yan, J.; Zhang, Z.; Zhang, F.; Zhang, Z. Experimental Investigation on the Solubility of Oxygen in Toluene and Acetic Acid. Ind. Eng. Chem. Res. 2014, 53, 9932–9937. [Google Scholar] [CrossRef]
- Khenkin, A.M.; Neumann, R. Oxidative C−C Bond Cleavage of Primary Alcohols and Vicinal Diols Catalyzed by H5PV2Mo10O40 by an Electron Transfer and Oxygen Transfer Reaction Mechanism. J. Am. Chem. Soc. 2008, 130, 14474–14476. [Google Scholar] [CrossRef]
- Stadler, R.H.; Welti, D.H.; Stämpfli, A.A.; Fay, L.B. Thermal Decomposition of Caffeic Acid in Model Systems: Identification of Novel Tetraoxygenated Phenylindan Isomers and Their Stability in Aqueous Solution. J. Agric. Food Chem. 1996, 44, 898–905. [Google Scholar] [CrossRef]
- Neumann, R.; Khenkin, A.M. Molecular oxygen and oxidation catalysis by phosphovanadomolybdates. Chem. Commun. 2006, 24, 2529. [Google Scholar] [CrossRef]
- Brégeault, J.-M. Transition-metal complexes for liquid-phase catalytic oxidation: some aspects of industrial reactions and of emerging technologies. Dalton Trans. 2003, 17, 3289–3302. [Google Scholar] [CrossRef]
- El Amrani, I.; Atlamsani, A.; Dakkach, M.; Rodríguez, M.; Romero, I.; Amthiou, S. Efficient and selective oxidation of aldehydes with dioxygen catalysed by vanadium-containing heteropolyanions. Comptes Rendus Chim. 2017, 20, 888–895. [Google Scholar] [CrossRef]
- Cavani, F.; Ferroni, L.; Frattini, A.; Lucarelli, C.; Mazzini, A.; Raabova, K.; Alini, S.; Accorinti, P.; Babini, P. Evidence for the presence of alternative mechanisms in the oxidation of cyclohexanone to adipic acid with oxygen, catalysed by Keggin polyoxometalates. Appl. Catal. A Gen. 2011, 391, 118–124. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Skone, J.H.; Ning, M.; Wasada-Tsutsui, Y. Toward the accurate calculation of pKa values in water and acetonitrile. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1827, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, G.S.; Cady, G.H. Inorganic Chemistry in Trifluoroacetic Acid1. J. Am. Chem. Soc. 1957, 79, 2451–2454. [Google Scholar] [CrossRef]
- Bailey, P.S.; Chang, Y.-G. Rearrangements of Certain α-Alkoxy-α-hydroperoxyacophenones. Alcoholysis of Acetic Benzoic Anhydride. J. Org. Chem. 1962, 27, 1192–1197. [Google Scholar] [CrossRef]
- Efremenko, I.; Neumann, R. Computational Insight into the Initial Steps of the Mars–van Krevelen Mechanism: Electron Transfer and Surface Defects in the Reduction of Polyoxometalates. J. Am. Chem. Soc. 2012, 134, 20669–20680. [Google Scholar] [CrossRef] [PubMed]
- Geletii, Y.V.; Hill, C.L.; Atalla, R.H.; Weinstock, I.A. Reduction of O2 to Superoxide Anion (O2•−) in Water by Heteropolytungstate Cluster-Anions. J. Am. Chem. Soc. 2006, 128, 17033–17042. [Google Scholar] [CrossRef] [PubMed]
- De Gregorio, G.F.; Weber, C.C.; Gräsvik, J.; Welton, T.; Brandt, A.; Hallett, J.P. Mechanistic insights into lignin depolymerisation in acidic ionic liquids. Green Chem. 2016, 18, 5456–5465. [Google Scholar] [CrossRef]
- Tian, Y.; Jürgens, E.; Mill, K.; Jordan, R.; Maulbetsch, T.; Kunz, D. Nucleophilic Isomerization of Epoxides by Pincer-Rhodium Catalysts: Activity Increase and Mechanistic Insights. ChemCatChem 2019, 11, 4028–4035. [Google Scholar] [CrossRef]
- Kuhn, W. Method for stabilizing phenylacetaldehyde. US Patent 6 624 330, 23 September 2003. [Google Scholar]
- Schutyser, W.; Renders, T.; Bosch, S.V.D.; Koelewijn, S.-F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef]
- Freudenberg, K. Biosynthesis and constitution of lignin. Nature 1959, 183, 1152–1155. [Google Scholar] [CrossRef]
- Akim, L.G.; Colodette, J.L.; Argyropoulos, D.S. Factors limiting oxygen delignification of kraft pulp. Can. J. Chem. 2001, 79, 201–210. [Google Scholar] [CrossRef]
- Yu, X.; Wei, Z.; Lu, Z.; Pei, H.; Wang, H. Activation of lignin by selective oxidation: An emerging strategy for boosting lignin depolymerization to aromatics. Bioresour. Technol. 2019, 291, 121885. [Google Scholar] [CrossRef] [PubMed]
- Magallanes, G.; Kärkäs, M.D.; Bosque, I.; Lee, S.; Maldonado, S.; Stephenson, C.R.J. Selective C–O Bond Cleavage of Lignin Systems and Polymers Enabled by Sequential Palladium-Catalyzed Aerobic Oxidation and Visible-Light Photoredox Catalysis. ACS Catal. 2019, 9, 2252–2260. [Google Scholar] [CrossRef]
- Liu, C.; Wu, S.; Zhang, H.; Xiao, R. Catalytic oxidation of lignin to valuable biomass-based platform chemicals: A review. Fuel Process. Technol. 2019, 191, 181–201. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; He, J.; Zhang, Y. Highly effective C–C bond cleavage of lignin model compounds. Green Chem. 2017, 19, 3135–3141. [Google Scholar] [CrossRef]
- Wang, M.; Lu, J.; Zhang, X.; Li, L.; Li, H.; Luo, N.; Wang, F. Two-Step, Catalytic C–C Bond Oxidative Cleavage Process Converts Lignin Models and Extracts to Aromatic Acids. ACS Catal. 2016, 6, 6086–6090. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | RP (%) | Chi2 (%) |
|---|---|---|---|---|---|---|---|---|
| V3 | 14.219 | 14.410 | 13.616 | 112.54 | 110.13 | 60.13 | 15.1 | 13.1 |
| H3PMo12 (JCPDS 00-043-0317) | 14.100 | 14.130 | 13.550 | 112.10 | 109.80 | 60.70 | - | - |
| Entry | z (vol%) | Mo + V Loading (mol%) | Conv. (%) | Yield (%) | O2 sol. (%) a | CB (%) | O2 Consumption b | ||
|---|---|---|---|---|---|---|---|---|---|
| PhOH | PhCHO | PhCOOH | |||||||
| 1c | 0 | 15 | 14 | 8.1 | 0 | 4 | 2.1 d | 91 | n. i. |
| 2 | 0 | 15 | 48 | 32 | 2.5 | 19 | 2.3 d | 76 | 0.9 |
| 3 | 100 | 15 | 100 | 80 | 0 | 80 | 7.8 d | 74 | 0 |
| 4 | 2.5 | 15 | 59 | 34 | 17 | 28 | 2.36 | 78 | 2.0 |
| 5 | 5 | 15 | 68 | 47 | 20 | 38 | 2.42 | 81 | 2.9e |
| 6 | 10 | 15 | 72 | 55 | 12 | 44 | 2.56 | 80 | 1.5 |
| 7f | 10 | 15 | 44 | 24 | 5.6 | 24 | 2.56 | 81 | 1.5 |
| 8 | 20 | 15 | 80 | 67 | 0 | 47 | 2.85 | 72 | 1.4 |
| 9g | 10 | 0 | 39 | 35 | 11 | 0.9 | 2.56 | 82 | 1.3 |
| 10 | 10 | 7 | 43 | 36 | 8 | 24 | 2.56 | 88 | 1.4 |
| 11 | 10 | 36 | 88 | 47 | 2.2 | 50 | 2.56 | 58 | 1.1 |
| Entry | T (°C) | P (bar) | t (h) | Conv. (%) | Yield (%) | CB (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PhOH | PhCHO | PhCOOH | K1HH | EstαA1HH | D1HH | ||||||
| 1 | 80 | 1 | 24 | 35 | 4.6 | 2.0 | 2.1 | 17 | 9 | 13 | 108 |
| 2a | 80 | 1 | 24 | 9.1 | 1.9 | 2.0 | tr | tr | 0 | 5.2 | 98 |
| 3 | 80 | 5 | 24 | 54 | 16 | 7.1 | 9.8 | 10 | 18 | 21 | 118 |
| 4 | 80 | 5 | 6 | 33 | 3.7 | 2.9 | 3.5 | 8.7 | 9.9 | 10 | 99 |
| 5 | 120 | 5 | 2 | 44 | 7.1 | 6.4 | 7.5 | 2.7 | 11 | 16 | 96 |
| 6 | 120 | 5 | 6 | 90 | 7.6 | 2.6 | 19 | 6.7 | 12 | 12 | 55 |
| Entry | t (h) | Liquid Phase (GC-MS after Silylation) | Residual Lignin (31P NMR) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yield of Monoaromatics (mol%) b | Function Ratio | Monoaromatic Type Proportions b | |||||||||
| Units H | Units G | Units S | Aliph./ArOH/COOH | H | G | S | |||||
| H | HA | V | VA | S | SA | ||||||
| 1 | 2 | 0.4 | 0.1 | 1.2 | 0.3 | 0.2 | 0 | 40/30/30 | 16 | 36 | 48 |
| 2 a | 2 | 0 | 0 | 0.1 | 0 | 0.2 | 0 | 39/21/40 | 12 | 38 | 50 |
| 3 | 6 | 0.6 | 0.3 | 0.7 | 0.4 | 0.3 | 0 | n. d. | n. d. | ||
| WSLp | - | - | - | - | - | - | - | 36/40/24 | 16 | 38 | 46 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hussaini, L.; Launay, F.; Galvez, E. Vanadium-Substituted Phosphomolybdic Acids for the Aerobic Cleavage of Lignin Models—Mechanistic Aspect and Extension to Lignin. Materials 2020, 13, 812. https://doi.org/10.3390/ma13040812
Al-Hussaini L, Launay F, Galvez E. Vanadium-Substituted Phosphomolybdic Acids for the Aerobic Cleavage of Lignin Models—Mechanistic Aspect and Extension to Lignin. Materials. 2020; 13(4):812. https://doi.org/10.3390/ma13040812
Chicago/Turabian StyleAl-Hussaini, Louay, Franck Launay, and Elena Galvez. 2020. "Vanadium-Substituted Phosphomolybdic Acids for the Aerobic Cleavage of Lignin Models—Mechanistic Aspect and Extension to Lignin" Materials 13, no. 4: 812. https://doi.org/10.3390/ma13040812
APA StyleAl-Hussaini, L., Launay, F., & Galvez, E. (2020). Vanadium-Substituted Phosphomolybdic Acids for the Aerobic Cleavage of Lignin Models—Mechanistic Aspect and Extension to Lignin. Materials, 13(4), 812. https://doi.org/10.3390/ma13040812