Advances and Challenges in the Creation of Porous Metal Phosphonates


Abstract
1. Introduction
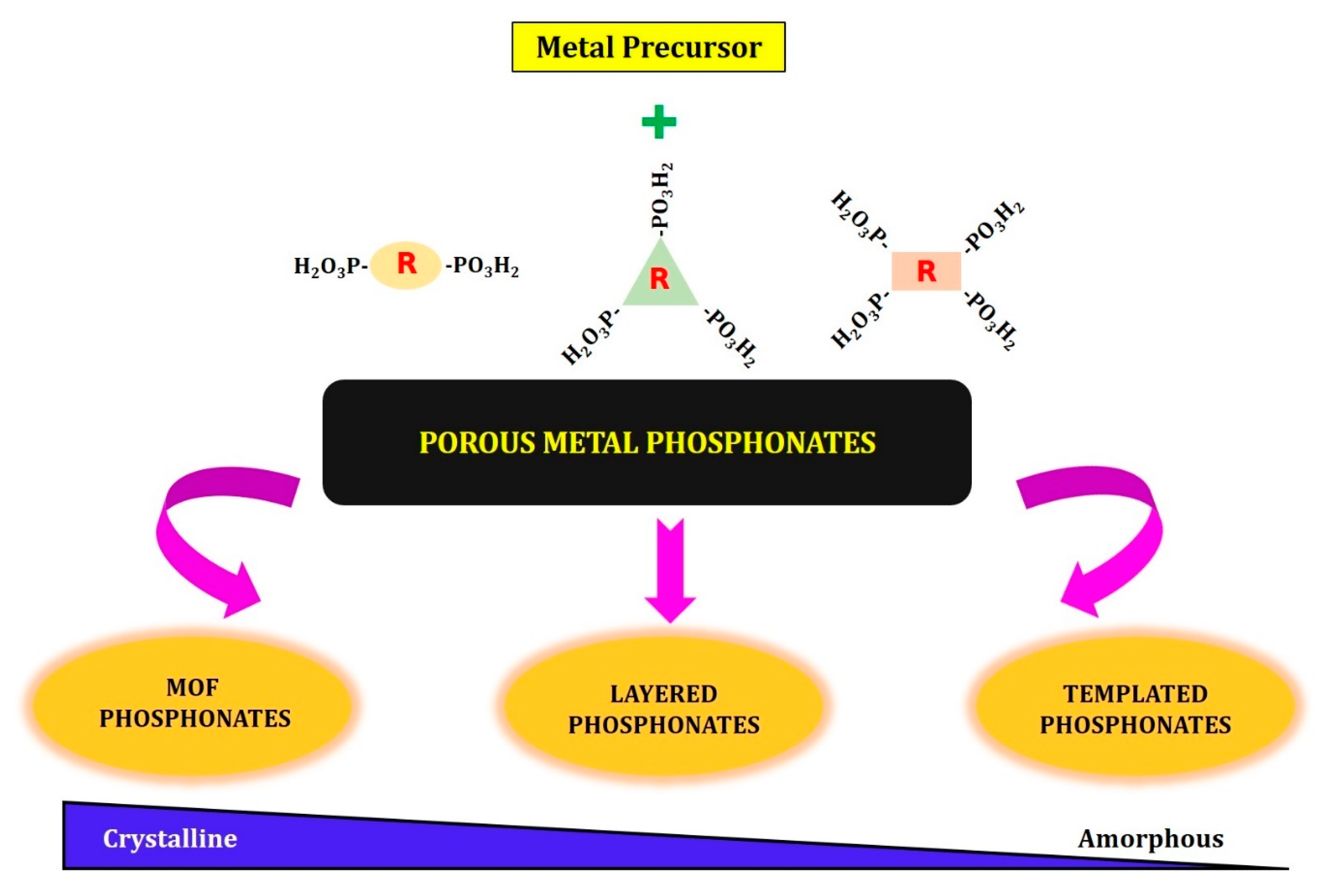
2. Synthesis of Porous Metal Phosphonates
- (1)
- Layered metal phosphonates;
- (2)
- Phosphonate–metal organic frameworks;
- (3)
- Supramolecular templated porous metal phosphonates.
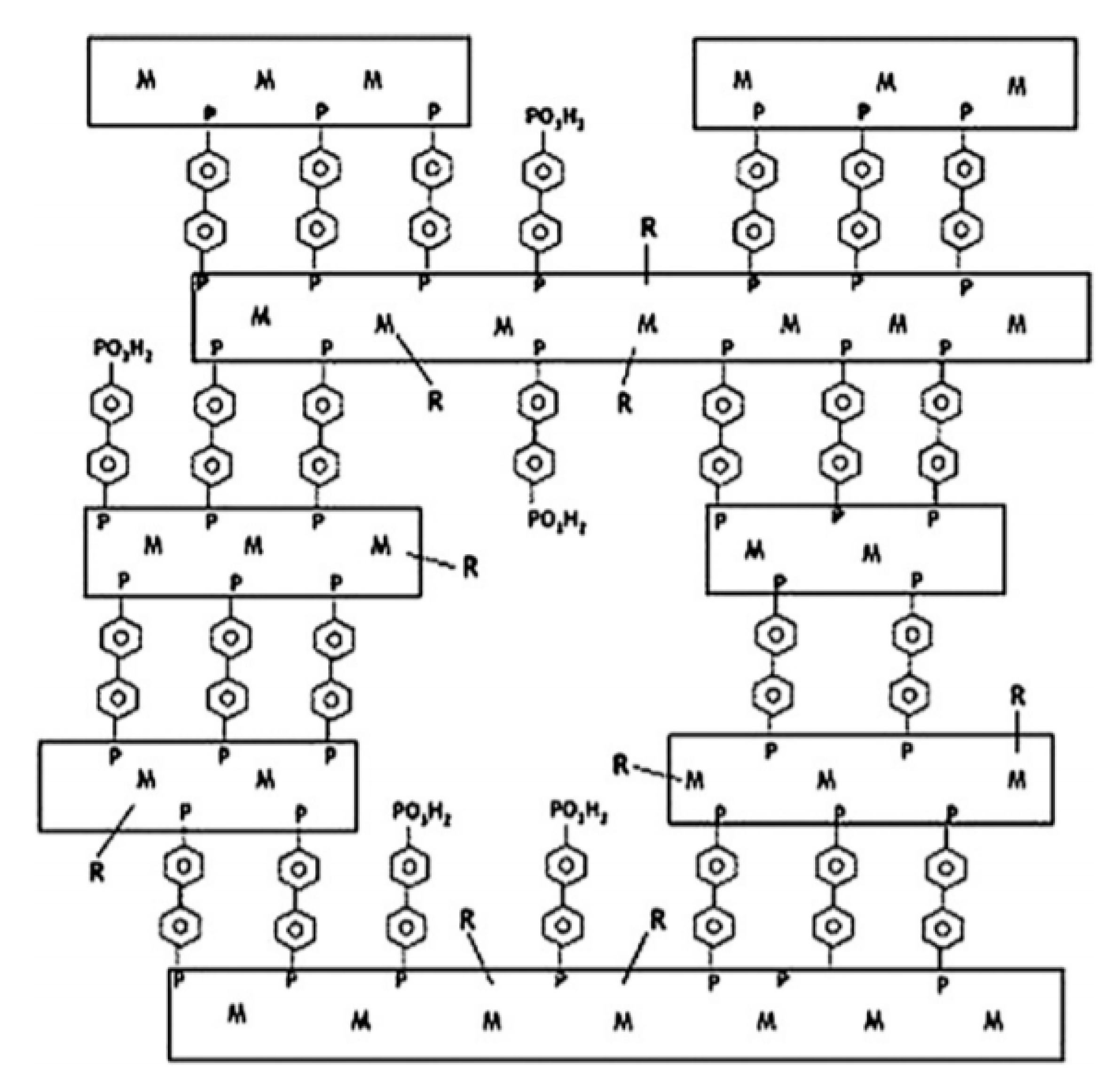
2.1. Layered Metal Phosphonates
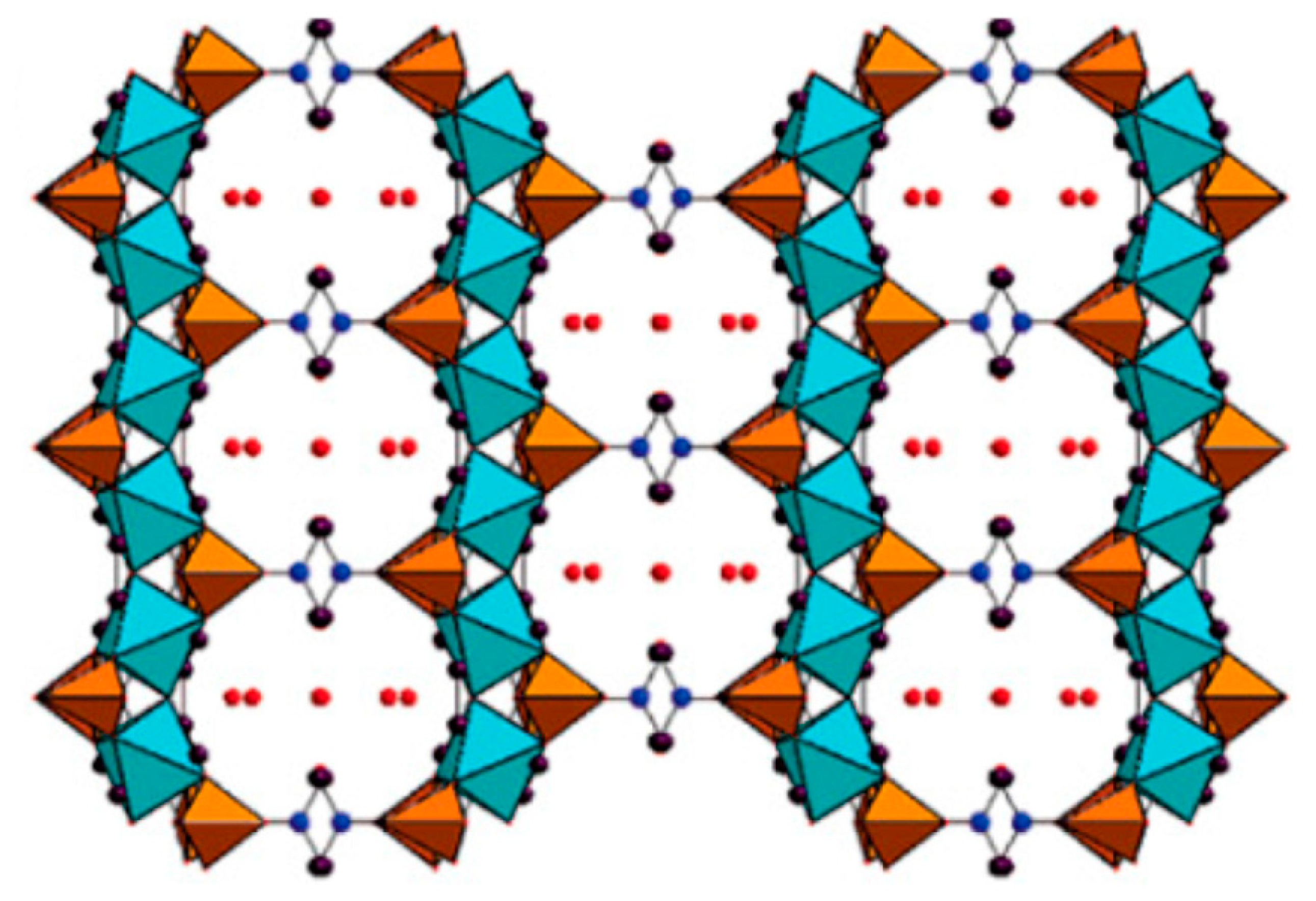
2.2. MOFs–Phosphonates



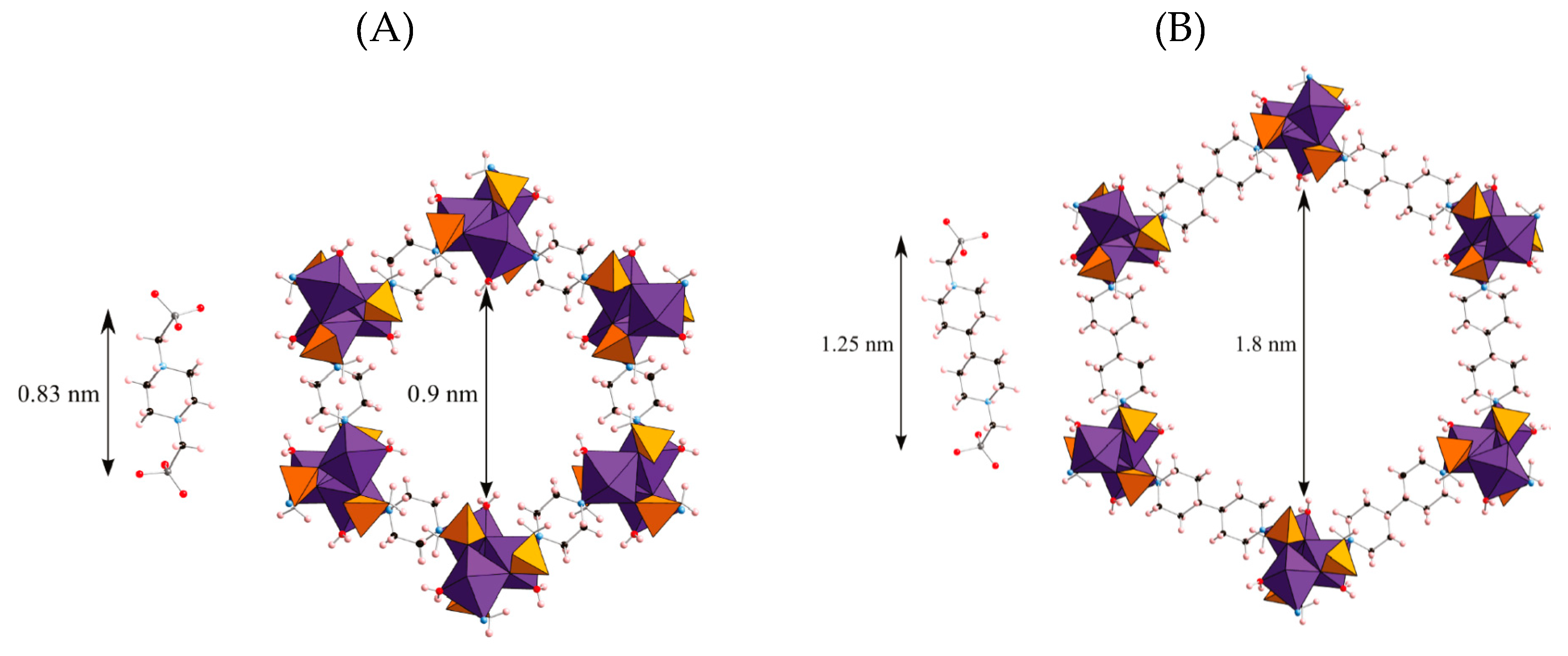
2.3. Templated Porous Metal Phosphonates

3. Applications
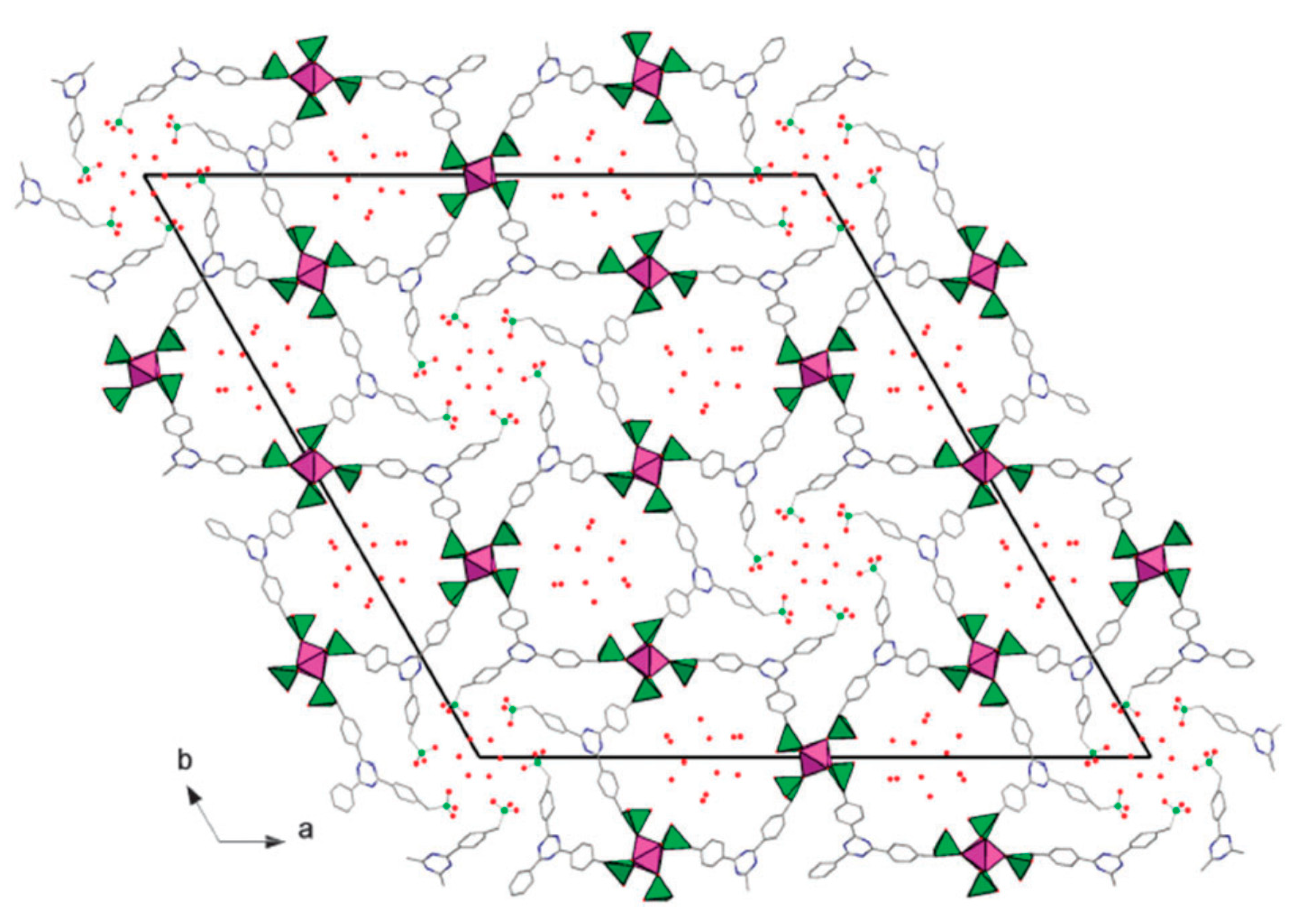
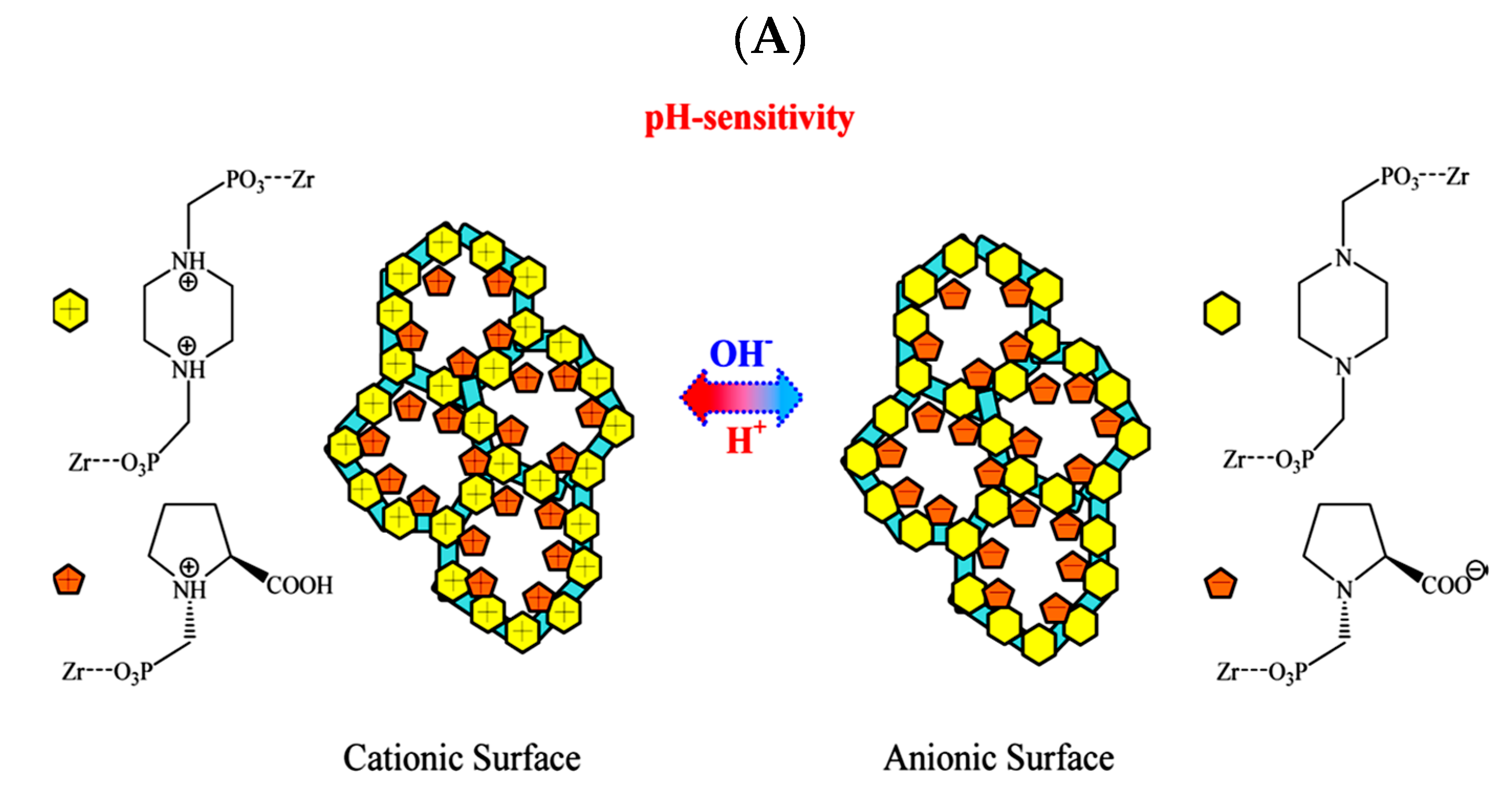
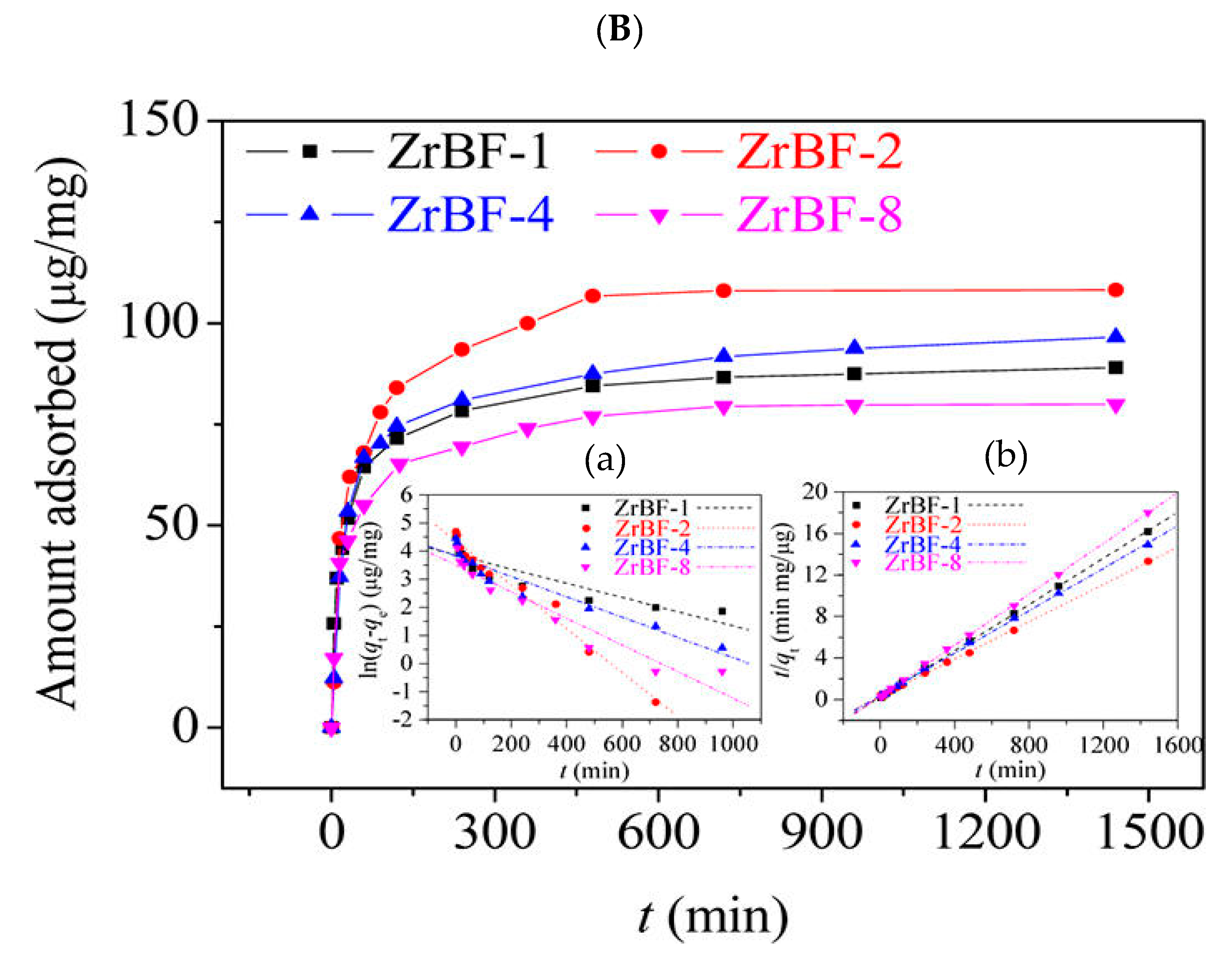
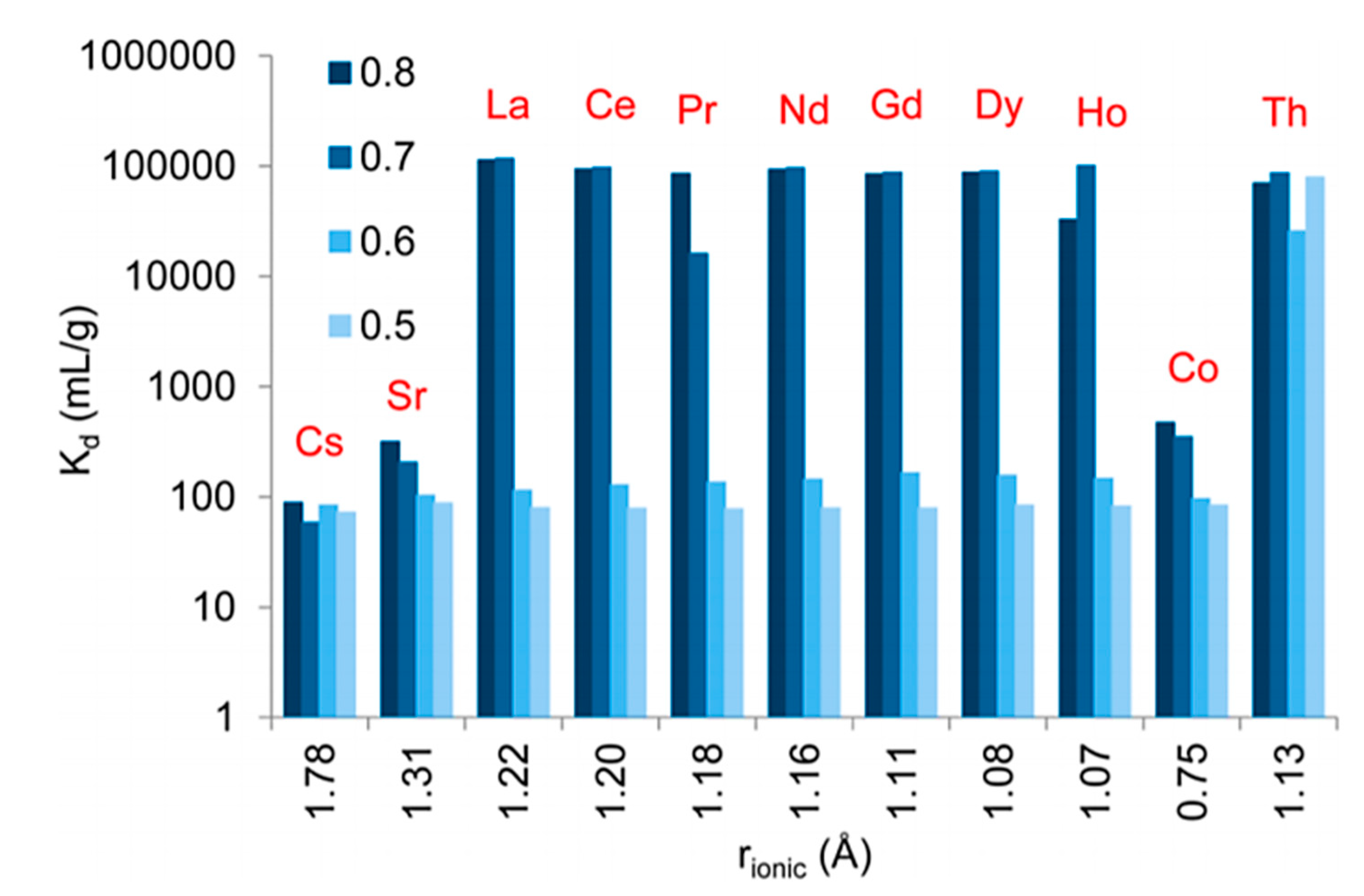
3.1. Separation and Extraction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal Precursor | Phosphonic Linker | Synthesis | Application—Separation/Extraction | Reference |
|---|---|---|---|---|
| Al(OsBu)3 | ATMP BHMT | Hydrothermal | Cu2+ adsorption and lyzozyme adsorption | [94] |
| ZrOCl2 | ATMP | Precipitation | Cation exchange—divalent metal ions | [99] |
| ZrCl4 | H3PMP BPMP | Hydrothermal | Adsorption and delivery of DNA | [101] |
| ZrCl4 | PPA | Hydrothermal | Immobilized metal affinity chromatographic adsorbent for phosphopeptide enrichment | [104] |
| MnCl2 | EDTMP | Hydrothermal | Cu2+ sorption and selective protein adsorption of Cyt–C over BSA | [100] |
| SnCl4 | HEDP ATMP EDTMP | Hydrothermal | Radionuclide separation of Th4+ | [105] |
| ZrOCl2 | BTP BDP BMP | Hydrothermal | Lanthanide and actinide separation | [108] |
| ZrOCl2 | BDPA H3PO4 | Hydrothermal | Ion-exchange metal ions | [106] |
| ZrOCl2 SnCl4 | BDPA H3PO4 | Hydrothermal | Actinides separation | [107] |
| ZrCl4/Zr(OiPr)4 | ATMP | Hydrothermal | Lanthanides extraction | [109] |
3.2. Catalysis

| Metal Precursor | Phosphonic Linker | Synthesis | Application—Catalysis | Reference |
|---|---|---|---|---|
| V(O)(i-PrO)3 | TPPhA | Non-hydrolytic condensation | Oxidation of benzylic alcohols | [81] |
| Ti(SO4)2 | (1R, 2S)-(-)-2-P (1R, 2S)-(+)-2-P | Precipitation (Hydrothermal) | Enantioselective addition of benzaldehyde | [110] |
| Ti(OiPr)4 | Polypeptide capped with phosphonic acid moiety | Non-hydrolytic condensation | Enantioselective hydration of styrene oxide | [111] |
| ZrCl4 | HEDP | Precipitation/Hydrothermal | Solid-acid catalyst for synthesis of methyl-2,3-O-isopropylidene-β-D-ribofuranoside | [112] |
| SnCl4 | PEHMP | Precipitation/Hydrothermal | Oxidation of cyclohexanone to adipic acid in absence of peroxides | [116] |
| FeCl3 | BTP | Precipitation/Hydrothermal | Transesterification for synthesis of biofuels | [115] |
| SnCl4 | BTP | Precipitation/Hydrothermal | Synthesis of 1,4-dihydropyridines | [117] |
| MoCl5 | BTP | Precipitation/Hydrothermal | Synthesis of benzimidazoles | [118] |
| CoCl2 | DPTMP | Hydrothermal | Catalytic oxidation of methylene blue | [119] |
| ZrOCl2 | HEDP ATMP EDTMP | Precipitation/Hydrothermal | Hydrolysis of ethyl acetate; esterification of acetic acid with ethanol and cyclohexanol | [114] |
| ZrOCl2 | HEDP ATMP EDTMP | Precipitation/Hydrothermal | Cycloaddition of aziridnes and CO2 | [121] |
| FeCl3 | HEDP | Precipitation/Hydrothermal | Oxidation of cyclohexanone to adipic acid | [120] |
3.3. Proton Conduction
| Metal Phosphonate | Conductivity (S cm−1) | Activation Energy (eV) | References |
|---|---|---|---|
| ZnCO3·2Zn(OH)2 + BTP | 1.0 × 10−5 (at 98% RH, 25 °C) | 0.17 | [122] |
| Mg(NO3)2 + H8ODTMP | 1.6 × 10−3 (at 100% RH, 19 °C) | 0.31 | [124] |
| β-PCMOF-21/2 | 2.1 × 10−2 (at 90% RH, 85 °C) | 0.21 | [123] |
| CoCa. n H2O (pellet) CoCa. n H2O (SC) | 1.55 × 10−5 (pellet) (at 95% RH, 25°C) 1.00 × 10−3 (SC) (at 95% RH, 25°C) | 0.98 (pellet) 0.90 (SC) | [126] |
| MFM-500-Ni MFM-500-Co | 4.5 × 10−4 (at 98% RH, 25 °C) 4.4 × 10−5 (at 98% RH, 25 °C) | 0.43 | [125] |
| UPG-2 | 5.7 × 10−4 (at 95% RH, 100 °C) | - | [65] |
4. Summary and Future Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, N.W.; Chae, H.K.; Eddaoudi, M.; Kim, J. Reticular synthesis and the design of new materials. Nat. Cell Biol. 2003, 423, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Rowsell, J.L.C.; Millward, A.R.; Park, K.S.; Yaghi, O.M. Hydrogen Sorption in Functionalized Metal−Organic Frameworks. J. Am. Chem. Soc. 2004, 126, 5666–5667. [Google Scholar] [CrossRef] [PubMed]
- Barthelet, K.; Marrot, J.; Riou, D. A breathing hybrid organic-inorganic solid with very large pores and high magnetic characteristics. Angew. Chem. 2002, 41, 281–284. [Google Scholar] [CrossRef]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surblé, S.; Margiolaki, I. A Chromium Terephthalate-Based Solid with Unusually Large Pore Volumes and Surface Area. Science 2005, 309, 2040–2042. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Guan, S.; Fukushima, Y.; Ohsuna, A.T.; Terasaki, O. Novel Mesoporous Materials with a Uniform Distribution of Organic Groups and Inorganic Oxide in Their Frameworks. J. Am. Chem. Soc. 1999, 121, 9611–9614. [Google Scholar] [CrossRef]
- Asefa, T.; MacLachlan, M.J.; Coombs, N.; Ozin, G.A. Periodic mesoporous organosilicas with organic groups inside the channel walls. Nat. Cell Biol. 1999, 402, 867–871. [Google Scholar] [CrossRef]
- Melde, B.J.; Holland, B.T.; Blanford, A.C.F.; Stein, A. Mesoporous Sieves with Unified Hybrid Inorganic/Organic Frameworks. Chem. Mater. 1999, 11, 3302–3308. [Google Scholar] [CrossRef]
- Clearfield, A. Recent advances in metal phosphonate chemistry. Curr. Opin. Solid State Mater. Sci. 1996, 1, 267–278. [Google Scholar] [CrossRef]
- Clearfield, A. Recent advances in metal phosphonate chemistry II. Curr. Opin. Solid State Mater. Sci. 2002, 6, 495–506. [Google Scholar] [CrossRef]
- Shimizu, G.K.H.; Vaidhyanathan, R.; Taylor, J.M. Phosphonate and sulfonate metal organic frameworks. Chem. Soc. Rev. 2009, 38, 1430–1449. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.J.; Perry, H.P.; Clearfield, A. Conventional and Unconventional Metal–Organic Frameworks Based on Phosphonate Ligands: MOFs and UMOFs. Chem. Rev. 2012, 112, 1034–1054. [Google Scholar] [CrossRef]
- Ma, T.-Y.; Yuan, Z.-Y. Metal Phosphonate Hybrid Mesostructures: Environmentally Friendly Multifunctional Materials for Clean Energy and Other Applications. ChemSusChem 2011, 4, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-P.; Ma, T.-Y.; Liu, Y.-L.; Ren, T.-Z.; Yuan, Z.-Y. Metal phosphonate hybrid materials: From densely layered to hierarchically nanoporous structures. Inorg. Chem. Front. 2014, 1, 360–383. [Google Scholar] [CrossRef]
- Bao, S.-S.; Shimizu, G.K.; Zheng, L.-M. Proton conductive metal phosphonate frameworks. Co-ord. Chem. Rev. 2019, 378, 577–594. [Google Scholar] [CrossRef]
- Bhanja, P.; Na, J.; Tang, J.; Lin, J.; Wakihara, T.; Bhaumik, A.; Yamauchi, Y. Nanoarchitectured Metal Phosphates and Phosphonates: A New Material Horizon toward Emerging Applications. Chem. Mater. 2019, 31, 5343–5362. [Google Scholar] [CrossRef]
- Shearan, S.J.; Stock, N.; Emmerling, F.; Demel, J.; Wright, P.A.; Demadis, K.D.; Vassaki, M.; Costantino, F.; Vivani, R.; Sallard, S.; et al. New Directions in Metal Phosphonate and Phosphinate Chemistry. Crystals 2019, 9, 270. [Google Scholar] [CrossRef]
- Alberti, G.; Costantino, U.; Vivani, R.; Zappelli, P. Preparation of Zirconium Diphosphonate-Phosphites With A Narrow Distribution Of Mesopores. MRS Proc. 1991, 233, 101–106. [Google Scholar] [CrossRef]
- Alberti, G.; Casciola, M.; Costantino, U.; Vivani, R. Layered and pillared metal(IV) phosphates and phosphonates. Adv. Mater. 1996, 8, 291–303. [Google Scholar] [CrossRef]
- Clearfield, A. The early history and growth of metal phosphonate chemistry. In Metal Phosphonate Chemistry: From Synthesis to Applications; Clearfield, A., Demadis, K., Eds.; Royal Society of Chemistry Publishing: Cambridge, UK, 2012; pp. 1–44. [Google Scholar]
- Cabeza, A.; Aranda, M.A.G. Metal Carboxyphosphonates. In Metal Phosphonate Chemistry: From Synthesis to Applications; Clearfield, A., Demadis, K., Eds.; Royal Society of Chemistry Publishing: Cambridge, UK, 2012; pp. 107–132. [Google Scholar]
- Dines, M.B.; Digiacomo, P.M. Derivatized lamellar phosphates and phosphonates of M(IV) ions. Inorg. Chem. 1981, 20, 92–97. [Google Scholar] [CrossRef]
- Dines, M.B.; Griffith, P.C. Synthesis and characterization of layered tetravalent metal terphenyl mono- and bis-phosphonates. Polyhedron 1983, 2, 607–611. [Google Scholar] [CrossRef]
- Serre, C.; Mouchaham, G.; Steunou, N.; Devic, T.; Serre, C. Titanium coordination compounds: From discrete metal complexes to metal–organic frameworks. Chem. Soc. Rev. 2017, 46, 3431–3452. [Google Scholar] [CrossRef]
- Poojary, D.M.; Zhang, B.; Bellinghausen, P.; Clearfield, A. Synthesis and X-ray structures of covalently pillared zinc bis(phosphonates). Inorg. Chem. 1996, 35, 5254–5263. [Google Scholar] [CrossRef]
- Poojary, D.M.; Zhang, B.; Bellinghausen, P.; Clearfield, A. Synthesis and X-ray Powder Structures of Two Lamellar Copper Arylenebis(phosphonates). Inorg. Chem. 1996, 35, 4942–4949. [Google Scholar] [CrossRef]
- Poojary, D.M.; Zhang, B.; Clearfield, A. Pillared layered metal phosphonates. Synthesis and X-ray powder structures of copper and zinc alkylenebis(phosphonates). J. Am. Chem. Soc. 1997, 119, 12550–12559. [Google Scholar] [CrossRef]
- Mao, J.G.; Wang, Z.; Clearfield, A. Synthesis, characterization, and crystal structures of to divalent metal diphosphonates with a layered and a 3D network structure. Inorg. Chem. 2002, 41, 2334–2340. [Google Scholar] [CrossRef]
- Bakhmutova, E.V.; Ouyang, X.; Medvedev, D.G.; Clearfield, A. Cobalt Phosphonates: An Unusual Polymeric Cobalt Phosphonate Containing a Clathrated Phosphonate Anion and a Layered Bisphosphonate. Inorg. Chem. 2003, 42, 7046–7051. [Google Scholar] [CrossRef] [PubMed]
- Penicaud, V.; Massiot, D.; Gelbard, G.; Odobel, F.; Bujoli, B. Preparation of structural analogues of divalent metal monophosphonates, using bis(phosphonic) acids: A new strategy to reduce overcrowding of organic groups in the interlayer space. J. Mol. Struct. 1998, 470, 31–38. [Google Scholar] [CrossRef]
- Zhang, B.; Poojary, D.M.; Clearfield, A. Synthesis and Characterization of Layered Zinc Biphenylylenebis(phosphonate) and Three Mixed-Component Arylenebis(phosphonate)/Phosphates. Inorg. Chem. 1998, 37, 1844–1852. [Google Scholar] [CrossRef]
- Cao, G.; Hong, H.G.; Mallouk, T.E. Layered metal phosphates and phosphonates: From crystals to monolayers. Acc. Chem. Res. 1992, 25, 420–427. [Google Scholar] [CrossRef]
- Alberti, G.; Marmottini, F.; Vivani, R.; Zappelli, P. Preparation and Characterization of Pillared Zirconium Phosphite-Diphosphonates with Tuneable Inter-Crystal Mesoporosity. J. Porous Mater. 1998, 5, 221–226. [Google Scholar] [CrossRef]
- Alberti, G.; Marmottini, F.; Murcia-Mascarós, S.; Vivani, R. Preparation and Preliminary Characterization of a Covalently Pillared Zirconium Phosphate- Diphosphonate with Interlayer Microporosity. Angew. Chem. Int. Ed. 1994, 33, 1594–1597. [Google Scholar] [CrossRef]
- Subbiah, A.; Pyle, D.; Rowland, A.; Huang, J.; Narayanan, R.A.; Thiyagarajan, P.; Zoń, J.; Clearfield, A. A Family of Microporous Materials Formed by Sn(IV) Phosphonate Nanoparticles. J. Am. Chem. Soc. 2005, 127, 10826–10827. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, A.; Bhuvanesh, N.; Clearfield, A. A novel inorganic-organic compound: Synthesis and structural characterization of tin(II) phenylbis(phosphonate), Sn2(PO3C6H4PO3). J. Solid State Chem. 2005, 178, 1321–1325. [Google Scholar] [CrossRef]
- Huang, J.; Subbiah, A.; Pyle, D.; Rowland, A.; Smith, B.; Clearfield, A. Globular Porous Nanoparticle Tin(IV) Phenylphosphonates and Mixed Methyl Phenylphosphonates. Chem. Mater. 2006, 18, 5213–5222. [Google Scholar] [CrossRef]
- Gómez-Alcántara, M.D.M.; Cabeza, A.; Olivera-Pastor, P.; Fernández-Moreno, F.; Sobrados, I.; Sanz, J.; Morris, R.E.; Clearfield, A.; Aranda, M.A.G.; Cabeza, A.; et al. Layered microporous tin(iv) bisphosphonates. Dalton Trans. 2007, 23, 2394–2404. [Google Scholar] [CrossRef]
- Kirumakki, S.; Huang, J.; Subbiah, A.; Yao, J.; Rowland, A.; Smith, B.; Mukherjee, A.; Samarajeewa, S.; Clearfield, A. Tin(iv) phosphonates: Porous nanoparticles and pillared materials. J. Mater. Chem. 2009, 19, 2593–2603. [Google Scholar] [CrossRef]
- Perry, H.P.; Law, J.; Zon, J.; Clearfield, A. Porous zirconium and tin phosphonates incorporating 2,2′-bipyridine as supports for palladium nanoparticles. Microporous Mesoporous Mater. 2012, 149, 172–180. [Google Scholar] [CrossRef]
- Cabeza, A.; Gómez-Alcántara, M.D.M.; Olivera-Pastor, P.; Sobrados, I.; Sanz, J.; Xiao, B.; Morris, R.E.; Clearfield, A.; Aranda, M.A.G.; Cabeza, A.; et al. From non-porous crystalline to amorphous microporous metal(IV) bisphosphonates. Microporous Mesoporous Mater. 2008, 114, 322–336. [Google Scholar] [CrossRef]
- Liang, J.; Shimizu, G.K.H. Crystalline Zinc Diphosphonate Metal−Organic Framework with Three-Dimensional Microporosity. Inorg. Chem. 2007, 46, 10449–10451. [Google Scholar] [CrossRef]
- Wu, J.; Hou, H. Highly selective ferric ion sorption and exchange by crystalline metal phosphonates constructured from tetraphosphonic acids. Inorg. Chem. 2007, 46, 7960–7970. [Google Scholar] [CrossRef] [PubMed]
- Vaidhyanathan, R.; Liang, J. A route to functionalized pores in coordination polymers via mixed phosphonate and amino triazole linkers. Supramol. Chem. 2011, 23, 278–282. [Google Scholar] [CrossRef]
- Ma, K.-R.; Zhang, D.-J.; Zhu, Y.-L. Structure and Characterization of a Novel 3D Lead Phosphonate Metal—Organic Framework with Cationic Layer Based on Weak Pb—O(N) Contact. Aust. J. Chem. 2010, 63, 452–457. [Google Scholar] [CrossRef]
- Iremonger, S.S.; Liang, J.; Vaidhyanathan, R.; Shimizu, G.K.H. A permanently porous van der Waals solid by using phosphonate monoester linkers in a metal organic framework. Chem. Commun. 2011, 47, 4430–4432. [Google Scholar] [CrossRef] [PubMed]
- Iremonger, S.S.; Liang, J.; Vaidhyanathan, R.; Martens, I.; Shimizu, G.K.H.; Thomas, D.D.; Aghaji, M.Z.; Yeganegi, S.; Woo, T.K. Phosphonate Monoesters as Carboxylate-like Linkers for Metal Organic Frameworks. J. Am. Chem. Soc. 2011, 133, 20048–20051. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.A.; Miller, S.R.; Warrender, S.J.; Mellot-Draznieks, C.; Lightfoot, P.; Wright, P.A. The first route to large pore metal phosphonates. Chem. Commun. 2006, 3305–3307. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.R.; Pearce, G.M.; Wright, P.A.; Bonino, F.; Chavan, S.M.; Bordiga, S.; Margiolaki, I.; Guillou, N.; Férey, G.; Bourrelly, S.; et al. Structural Transformations and Adsorption of Fuel-Related Gases of a Structurally Responsive Nickel Phosphonate Metal−Organic Framework, Ni-STA-12. J. Am. Chem. Soc. 2008, 130, 15967–15981. [Google Scholar] [CrossRef]
- Pearce, G.M. Synthesis, Adsorption and Catalysis of Large Pore metal Phosphonates. Ph.D. Thesis, University of St Andrews, St Andrews, UK, 2009. [Google Scholar]
- Wharmby, M.T.; Mowat, J.P.S.; Thompson, S.P.; Wright, P.A. Extending the Pore Size of Crystalline Metal Phosphonates toward the Mesoporous Regime by Isoreticular Synthesis. J. Am. Chem. Soc. 2011, 133, 1266–1269. [Google Scholar] [CrossRef]
- Taylor, J.M.; Vaidhyanathan, R. Enhancing the water stability of metal-organic frameworks via phosphonate monoester linkers. J. Am. Chem. Soc. 2012, 134, 14338–14340. [Google Scholar] [CrossRef]
- Mah, R.K.; Lui, M.W. Enhancing order and porosity in a highly robust tin(IV) triphosphonate network. Inorg. Chem. 2013, 52, 7311–7313. [Google Scholar] [CrossRef]
- Mah, R.K.; Gelfand, B.S.; Taylor, J.M.; Shimizu, G.K. Reconciling order, stability, and porosity in phosphonate metal organic frameworks via HF-mediated synthesis. Inorg. Chem. Front. 2015, 2, 273–277. [Google Scholar] [CrossRef]
- Tang, S.F.; Pan, X.B. Fabrication of new metal phosphonates from tritopic triphosphonic acid containing methyl groups and auxillary ligands: Syntheses, structures, and gas adsorption properties. Cryst. Eng. Chem. 2013, 15, 1860–1873. [Google Scholar]
- Serre, C.; Ferey, G. Hybrid open frameworks. 8. Hydrothermal synthesis, crystal structure and thermal behavior of the first three-dimensional titanium(IV) diphosphonate with an open framework structure: Ti3O2(H2O)2(O3P-(CH2)-PO3)2·(H2O)2, or MIL-22. Inorg. Chem. 1999, 38, 5370–5373. [Google Scholar] [CrossRef]
- Riou, D.; Roubeau, O. Composite microporous compounds. Part 1: Synthesis and structure determination of two new vanadium alkyldiphosphonates (MIL-2 and MIL-3) with three-dimensional open frameworks. Microporous Mesoporous Mater. 1998, 23, 23–31. [Google Scholar] [CrossRef]
- Riou, D.; Serre, C. Composite microporous compounds (MIL-n).II: Hydrothermal synthesis and abinitioResolution by X-ray powder diffraction of MIL-5: A vanado diphosphonates with three-dimensional neutral frameworks. J. Solid State Chem. 1998, 141, 89–93. [Google Scholar]
- Riou, D.; Ferey, G. Hybrid open frameworks (MIL-n). Part 3 crystal structures of HT and LT forms of MIL-7: A new vanadium propylenediphosphonate with an open framework. Influence of the synthesis temperature on the oxidation state of vanadium with the same structural type. J. Mater. Chem. 1998, 8, 2733–2735. [Google Scholar] [CrossRef]
- Serre, C.; Groves, J.A. Synthesis, structure and properties of related microporous N,N′-piperazinebismethylenephosphonates of aluminium and titanium. Chem. Mater. 2006, 18, 1451–1457. [Google Scholar] [CrossRef]
- Taddei, M.; Costantino, F.; Vivani, R. Synthesis and Crystal Structure from X-ray Powder Diffraction Data of Two Zirconium Diphosphonates Containing Piperazine Groups. Inorg. Chem. 2010, 49, 9664–9670. [Google Scholar] [CrossRef]
- Taddei, M.; Costantino, F.; Vivani, R.; Sabatini, S.; Lim, S.-H.; Cohen, S.M. The use of a rigid tritopic phosphonic ligand for the synthesis of a robust honeycomb-like layered zirconium phosphonate framework. Chem. Commun. 2014, 50, 5737–5740. [Google Scholar] [CrossRef]
- Taddei, M.; Costantino, F.; Marmottini, F.; Comotti, A.; Sozzani, P.; Vivani, R. The first route to highly stable crystalline microporous zirconium phosphonate metal-organic frameworks. Chem. Commun. 2014, 50, 14831–14834. [Google Scholar] [CrossRef]
- Tang, S.F.; Cai, J.J. A highly porous three-dimensional aluminum phosphonate with hexagonal channels: Synthesis, structure and adsorption properties. Dalton Trans. 2014, 43, 5970–5973. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.; Shearan, S.J.; Donnadio, A.; Casciola, M.; Vivani, R.; Costantino, F. Investigating the effect of positional isomerism on the assembly of zirconium phosphonates based on tritopic linkers. Dalton Trans. 2020, 49, 3662–3666. [Google Scholar] [CrossRef]
- Gu, D.; Schuth, F. Synthesis of non-siliceous mesoporous oxides. Chem. Soc. Rev. 2014, 43, 313–344. [Google Scholar] [CrossRef] [PubMed]
- Soler-Illia, G.J.; Azzaroni, O. Multifunctional hybrids by combining ordered mesoporous materials and macromolecular building blocks. Chem. Soc. Rev. 2011, 40, 1107–1150. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, A.; Inagaki, S. Mesoporous Titanium Phosphate Molecular Sieves with Ion-Exchange Capacity. J. Am. Chem. Soc. 2001, 123, 691–696. [Google Scholar] [CrossRef]
- Guo, X.; Ding, W.; Wang, X.; Yan, Q. Synthesis of a novel mesoporous iron phosphate. Chem. Commun. 2001, 709–710. [Google Scholar] [CrossRef]
- Serre, C.; Auroux, A.; Gervasini, A.; Hervieu, M.; Férey, G. Hexagonal and Cubic Thermally Stable Mesoporous Tin(IV) Phosphates with Acidic and Catalytic Properties. Angew. Chem. Int. Ed. 2002, 41, 1594–1597. [Google Scholar] [CrossRef]
- Ren, T.-Z.; Yuan, Z.-Y.; Azioune, A.; Pireaux, A.J.-J.; Su, B.-L. Tailoring the Porous Hierarchy of Titanium Phosphates. Langmuir 2006, 22, 3886–3894. [Google Scholar] [CrossRef]
- Kimura, T. Synthesis of novel mesoporous aluminum organophosphonate by using a bridged diphosphonic acid. Chem. Mater. 2003, 15, 3742–3744. [Google Scholar] [CrossRef]
- Haskouri, J.E.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Amorós, P. The first pure mesoporous aluminum phosphonates and diphosphonates—New porous hybrid materials. Eur. J. Inorg. Chem. 2004, 1804–1807. [Google Scholar] [CrossRef]
- El Haskouri, J.; Guillem, C.; Latorre, J.; Beltrán, A.; Beltrán, D.; Amoros, P. S+I− Ionic Formation Mechanism to New Mesoporous Aluminum Phosphonates and Diphosphonates. Chem. Mater. 2004, 16, 4359–4372. [Google Scholar] [CrossRef]
- Kimura, T. Oligomeric Surfactant and Triblock Copolymer Syntheses of Aluminum Organophosphonates with Ordered Mesoporous Structures. Chem. Mater. 2005, 17, 5521–5528. [Google Scholar] [CrossRef]
- Kimura, T. Synthesis of Mesostructured and Mesoporous Aluminum Organophosphonates Prepared by Using Diphosphonic Acids with Alkylene Groups. Chem. Mater. 2005, 17, 337–344. [Google Scholar] [CrossRef]
- Mal, N.K.; Fujiwara, M.; Yamada, Y.; Matsukata, M. Synthesis of Surfactant-assisted Microporous Layered Tin Phenylphosphonate. Chem. Lett. 2003, 32, 292–293. [Google Scholar] [CrossRef]
- Sarkar, K.; Yokoi, T.; Tatsumi, T.; Bhaumik, A. Mesoporous hybrid zirconium oxophenylphosphate synthesized in absence of any structure directing agent. Microporous Mesoporous Mater. 2008, 110, 405–412. [Google Scholar] [CrossRef]
- Shi, X.; Yang, J.; Yang, Q. Mesoporous aluminum organophosphonates functionalized with chiral L-proline groups in the pore. Eur. J. Inorg. Chem. 2006, 1936–1939. [Google Scholar] [CrossRef]
- Vasylyev, M.V.; Wachtel, E.J.; Popovitz-Biro, R.; Neumann, R. Titanium Phosphonate Porous Materials Constructed from Dendritic Tetraphosphonates. Chem. A Eur. J. 2006, 12, 3507–3514. [Google Scholar] [CrossRef]
- Vasylyev, M.V.; Neumann, R. Preparation, characterization, and catalytic aerobic oxidation by a vanadium phosphonate mesoporous materials constructed from a dendritic phosphonate. Chem. Mater. 2006, 18, 2781–2783. [Google Scholar] [CrossRef]
- Ma, T.Y.; Zhang, X.J.; Shao, G.S.; Cao, J.L.; Yuan, Z.Y. Ordered microporous titanium phosphonate materials: Synthesis, photocatalytic activity, and heavy metal ion adsorption. J. Phys. Chem. C 2008, 112, 3090–3096. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, X.-J.; Yuan, Z.-Y. High selectivity for metal ion adsorption: From mesoporous phosphonated titanias to meso-/macroporous titanium phosphonates. J. Mater. Sci. 2009, 44, 6775–6785. [Google Scholar] [CrossRef]
- Ma, T.-Y.; Lin, X.-Z.; Zhang, X.-J.; Yuan, Z.-Y. High surface area titanium phosphonate materials with hierarchical porosity for multi-phase adsorption. New J. Chem. 2010, 34, 1209–1216. [Google Scholar] [CrossRef]
- Ma, T.-Y.; Lin, X.-Z.; Yuan, Z.-Y. Periodic mesoporous titanium phosphonate hybrid materials. J. Mater. Chem. 2010, 20, 7406–7415. [Google Scholar] [CrossRef]
- Ma, T.-Y.; Lin, X.-Z.; Yuan, Z.-Y. Cubic mesoporous titanium phosphonates with functionality. Chem. Eur. J. 2010, 16, 8487–8494. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.-Y.; Lin, X.-Z.; Yuan, Z.-Y. Hierarchical meso-/macroporous phosphated and phosphonated titania nanocomposite materials with high photocatalytic activity. Stud. Surf. Sci. Catal. 2010, 175, 571–574. [Google Scholar]
- Zhang, X.J.; Ma, T.Y.; Yuan, Z.-Y. Titania-phosphonate hybrid porous materials: Preparation, photocatalytic activity, and heavy metal ion adsorption. J. Mat. Chem. 2008, 18, 2003–2010. [Google Scholar] [CrossRef]
- Kimura, T. Moleular design of bisphosphonates to adjust their reactivity towards metal sources for the surfactant-assisted synthesis of mesoporous films. Angew. Chem. Int. Ed. 2017, 56, 13459–13463. [Google Scholar] [CrossRef]
- Wakabayashi, R.; Kimura, T. Further Understanding of the Reactivity Control of Bisphosphonates to a Metal Source for Fabricating Highly Ordered Mesoporous Films. Chem. Eur. J. 2019, 25, 5971–5977. [Google Scholar] [CrossRef]
- Wang, Y.; Alauzun, J.G.; Mutin, P.H. Water-Stable, Nonsiliceous Hybrid Materials with Tunable Porosity and Functionality: Bridged Titania-Bisphosphonates. Chem. Mater. 2020, 32, 2910–2918. [Google Scholar] [CrossRef]
- Hossain, K.; Mercier, L. Intraframework Metal Ion Adsorption in Ligand-Functionalized Mesoporous Silica. Adv. Mater. 2002, 14, 1053–1056. [Google Scholar] [CrossRef]
- Zhu, H.; Jones, D.J.; Zajac, J.; Dutartre, R.; Rhomari, A.M.; Rozière, J. Synthesis of Periodic Large Mesoporous Organosilicas and Functionalization by Incorporation of Ligands into the Framework Wall. Chem. Mater. 2002, 14, 4886–4894. [Google Scholar] [CrossRef]
- Ma, T.; Zhang, X.-J.; Yuan, Z.-Y. Hierarchical Meso-/Macroporous Aluminum Phosphonate Hybrid Materials as Multifunctional Adsorbents. J. Phys. Chem. C 2009, 113, 12854–12862. [Google Scholar] [CrossRef]
- Salis, A.; Bhattacharyya, M.S.; Monduzzi, M. Specific Ion Effects on Adsorption of Lysozyme on Functionalized SBA-15 Mesoporous Silica. J. Phys. Chem. B 2010, 114, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Steri, D.; Monduzzi, M.; Salis, A. Ionic strength affects lysozyme adsorption and release from SBA-15 mesoporous silica. Microporous Mesoporous Mater. 2013, 170, 164–172. [Google Scholar] [CrossRef]
- Ding, Y.; Yin, G.; Liao, X.; Huang, Z.; Chen, X.; Yao, Y.; Li, J. A convenient route to synthesize SBA-15 rods with tunable pore length for lysozyme adsorption. Microporous Mesoporous Mater. 2013, 170, 45–51. [Google Scholar] [CrossRef]
- Bhattacharyya, M.S.; Hiwale, P.; Piras, M.; Medda, L.; Steri, D.; Piludu, M.; Salis, A.; Monduzzi, M. Lysozyme Adsorption and Release from Ordered Mesoporous Materials. J. Phys. Chem. C 2010, 114, 19928–19934. [Google Scholar] [CrossRef]
- Shah, B.; Chudasam, U. Application of zirconium phosphonate-a novel hybrid material as an ion exchanger, Desalin. Water Treat. 2012, 38, 227–235. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Liu, Y.-L.; Ren, T.-Z.; Yuan, Z.-Y. Hollow manganese phosphonate microspheres with hierarchical porosity for efficient adsorption and separation. Nanoscale 2014, 6, 6627–6636. [Google Scholar] [CrossRef]
- Tang, Y.; Ren, Y.; Shi, X. Bifunctional mesoporous zirconium phosphonates for delivery of nuclei acids. Inorg. Chem. 2013, 52, 1388–1397. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Rosenholm, J.M.; Gu, H. Synthesis and characterization of pore size-tunable magnetic mesoporous silica nanoparticles. J. Colloid Interface Sci. 2011, 361, 16–24. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, W.; Bergman, L.; Rosenholm, J.M.; Lindén, M.; Wu, G.; Xu, H.; Gu, H.-C. Magnetic mesoporous silica nanospheres as DNA/drug carrier. Mater. Lett. 2012, 67, 379–382. [Google Scholar] [CrossRef]
- Wei, X.; Shi, X. Mesoporous Zirconium Phenylphosphonates for Selective Enrichment of Phosphopeptides. J. Phys. Chem. C 2014, 118, 4213–4221. [Google Scholar] [CrossRef]
- Lv, K.; Han, J.; Yang, C.-T.; Cheng, C.-M.; Luo, Y.-M.; Wang, X.-L. A category of hierarchically porous tin (IV) phosphonate backbone with the implication for radioanalytical separation. Chem. Eng. J. 2016, 302, 368–376. [Google Scholar] [CrossRef]
- Sibernagel, R.; Martin, C.H.; Clearfield, A. Zirconium(IV) phosphonate-phosphate as efficient ion-exchange materials. Inorg. Chem. 2016, 55, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Silbernagel, R.; Shehee, T.C.; Martin, C.H.; Hobbs, D.T.; Clearfield, A. Zr/Sn(IV) Phosphonates as Radiolytically Stable Ion-Exchange Materials. Chem. Mater. 2016, 28, 2254–2259. [Google Scholar] [CrossRef]
- Luca, V.; Tejada, J.J.; Vega, D.; Arrachart, G.; Rey, C. Zirconium(IV)–Benzene Phosphonate Coordination Polymers: Lanthanide and Actinide Extraction and Thermal Properties. Inorg. Chem. 2016, 55, 7928–7943. [Google Scholar] [CrossRef]
- Veliscek-Carolan, J.; Rawal, A.; Luca, V.; Hanley, T. Zirconium phosphonate sorbents with tunable structure and function. Microporous Mesoporous Mater. 2017, 252, 90–104. [Google Scholar] [CrossRef]
- Milo, A.; Neumann, R. A Tripodal Peptidic Titanium Phosphonate as a Homochiral Porous Solid Medium for the Heterogeneous Enantioselective Hydration of Epoxides. Adv. Synth. Catal. 2010, 352, 2159–2165. [Google Scholar] [CrossRef]
- Ji, Y.; Ma, X.; Wu, X.; Wang, N.; Wang, Q.; Zhou, X. A novel type of chiral titanium phosphonates and hybrid titanium phosphonates for heterogeneous asymmetric catalysis. Catal. Lett. 2007, 118, 187–194. [Google Scholar] [CrossRef]
- Lin, X.-Z.; Yuan, Z.-Y. Synthesis of Mesoporous Zirconium Organophosphonate Solid-Acid Catalysts. Eur. J. Inorg. Chem. 2012, 2012, 2661–2664. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. “Turning over” Definitions in Catalytic cycles. ACS Catal. 2012, 2, 2787–2794. [Google Scholar] [CrossRef]
- Lin, X.-Z.; Ren, T.-Z.; Yuan, Z. Mesoporous zirconium phosphonate materials as efficient water-tolerable solid acid catalysts. Catal. Sci. Technol. 2015, 5, 1485–1494. [Google Scholar] [CrossRef]
- Pramanik, M.; Bhaumik, A. Organic-Inorganic Hybrid Supermicroporous Iron(III) Phosphonate Nanoparticles as an Efficient Catalyst for the Synthesis of Biofuels. Chem. Eur. J. 2013, 19, 8507–8514. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Pramanik, M.; Patra, A.K.; Nandi, M.; Uyama, H.; Bhaumik, A. Hybrid porous tin(iv) phosphonate: An efficient catalyst for adipic acid synthesis and a very good adsorbent for CO2 uptake. Chem. Commun. 2012, 48, 6738. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, M.; Bhaumik, A. Self-assembled hybrid tin phosphonate nanoparticles with bimodal porosity: An insight towards the efficient and selective catalytic process for the synthesis of bioactive 1,4-dihydropyridines under solvent-free conditions. J. Mater. Chem. A 2013, 1, 11210–11220. [Google Scholar] [CrossRef]
- Pramanik, M.; Bhaumik, A. Self-Assembled Hybrid Molybdenum Phosphonate Porous Nanomaterials and Their Catalytic Activity for the Synthesis of Benzimidazoles. ChemCatChem 2014, 6, 2577–2586. [Google Scholar] [CrossRef]
- Zhu, Y.-P.; Ren, T.-Z.; Yuan, Z.-Y. Hollow cobalt phosphonate spherical hybrid as high-efficiency Fenton catalyst. Nanoscale 2014, 6, 11395–11402. [Google Scholar] [CrossRef]
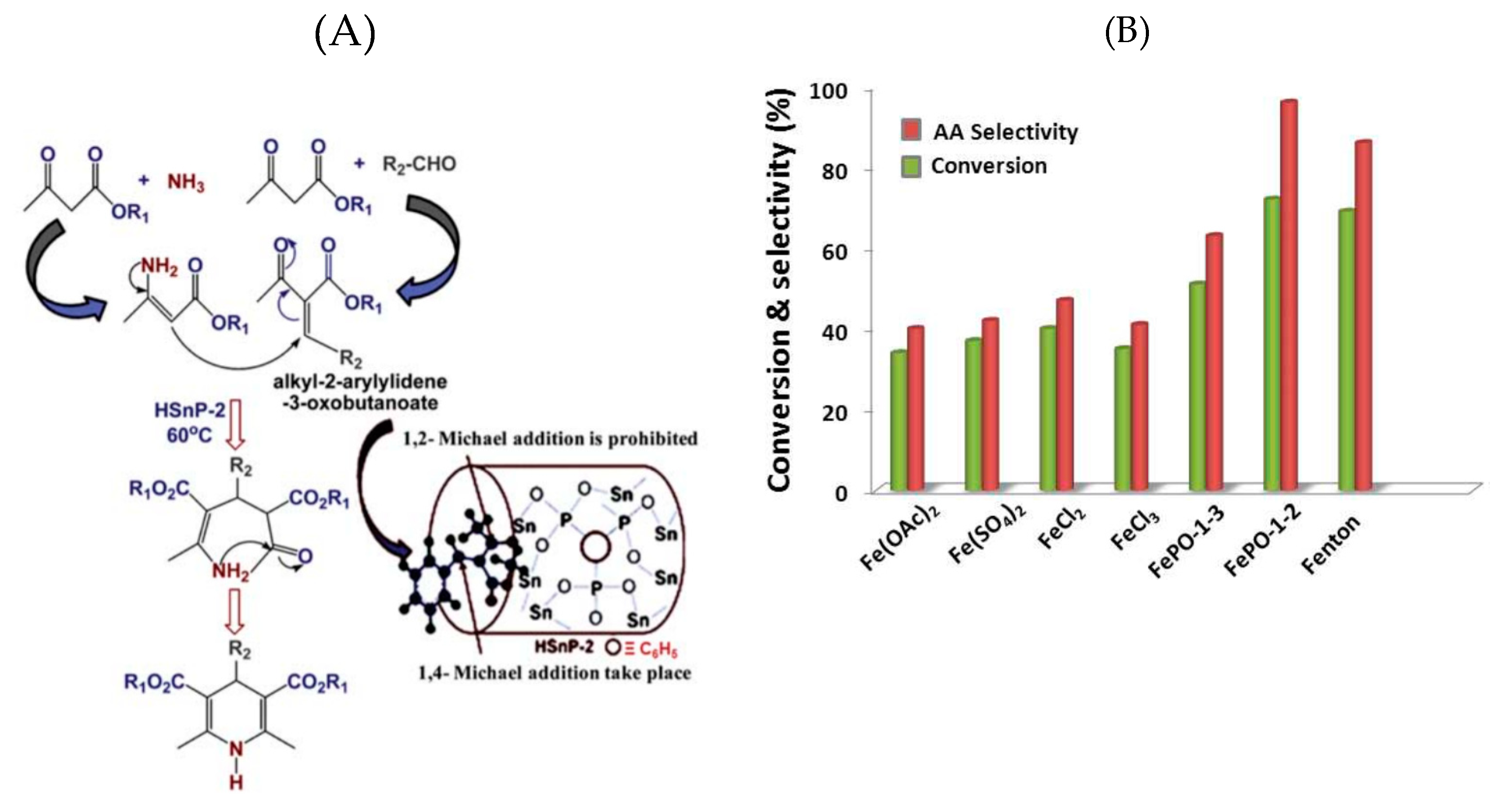
- Bhanja, P.; Ghosh, K.; Islam, S.S.; Patra, A.K.; Islam, S.M.; Bhaumik, A. A new hybrid iron phosphonate materials as an efficient catalyst for the synthesis of adipic acid in air and water. ACS Sustain. Chem. Eng. 2016, 4, 7147–7157. [Google Scholar] [CrossRef]
- Lin, X.-Z.; Yang, Z.-Z.; He, L.-N.; Yuan, Z. Mesoporous zirconium phosphonates as efficient catalysts for chemical CO2 fixation. Green Chem. 2015, 17, 795–798. [Google Scholar] [CrossRef]
- Taylor, J.M.; Mah, R.K.; Moudrakovski, I.L.; Ratcliffe, C.I.; Vaidhyanathan, R.; Shimizu, G.K.H. Facile Proton Conduction via Ordered Water Molecules in a Phosphonate Metal−Organic Framework. J. Am. Chem. Soc. 2010, 132, 14055–14057. [Google Scholar] [CrossRef]
- Kim, S.; Dawson, K.W.; Gelfand, B.S.; Taylor, J.M.; Shimizu, G.K.H. Enhancing Proton Conduction in a Metal-Organic Framework by Isomorphous Ligand Replacement. J. Am. Chem. Soc. 2013, 135, 963–966. [Google Scholar] [CrossRef]
- Colodrero, R.M.P.; Olivera-Pastor, P.; Losilla, E.R.; Hernández-Alonso, D.; Aranda, M.A.G.; Leon-Reina, L.; Rius, J.; Demadis, K.D.; Moreau, B.; Villemin, D.; et al. High Proton Conductivity in a Flexible, Cross-Linked, Ultramicroporous Magnesium Tetraphosphonate Hybrid Framework. Inorg. Chem. 2012, 51, 7689–7698. [Google Scholar] [CrossRef] [PubMed]
- Pili, S.; Argent, S.P.; Morris, C.G.; Rought, P.; Garcia-Sakai, V.; Silverwood, I.P.; Easun, T.L.; Li, M.; Warren, M.R.; Murray, C.A.; et al. Proton conduction in a phosphonate-based metal-organic framework mediated by intrinsic “Free diffusion inside a sphere”. J. Am. Chem. Soc. 2016, 138, 6352–6355. [Google Scholar] [CrossRef] [PubMed]
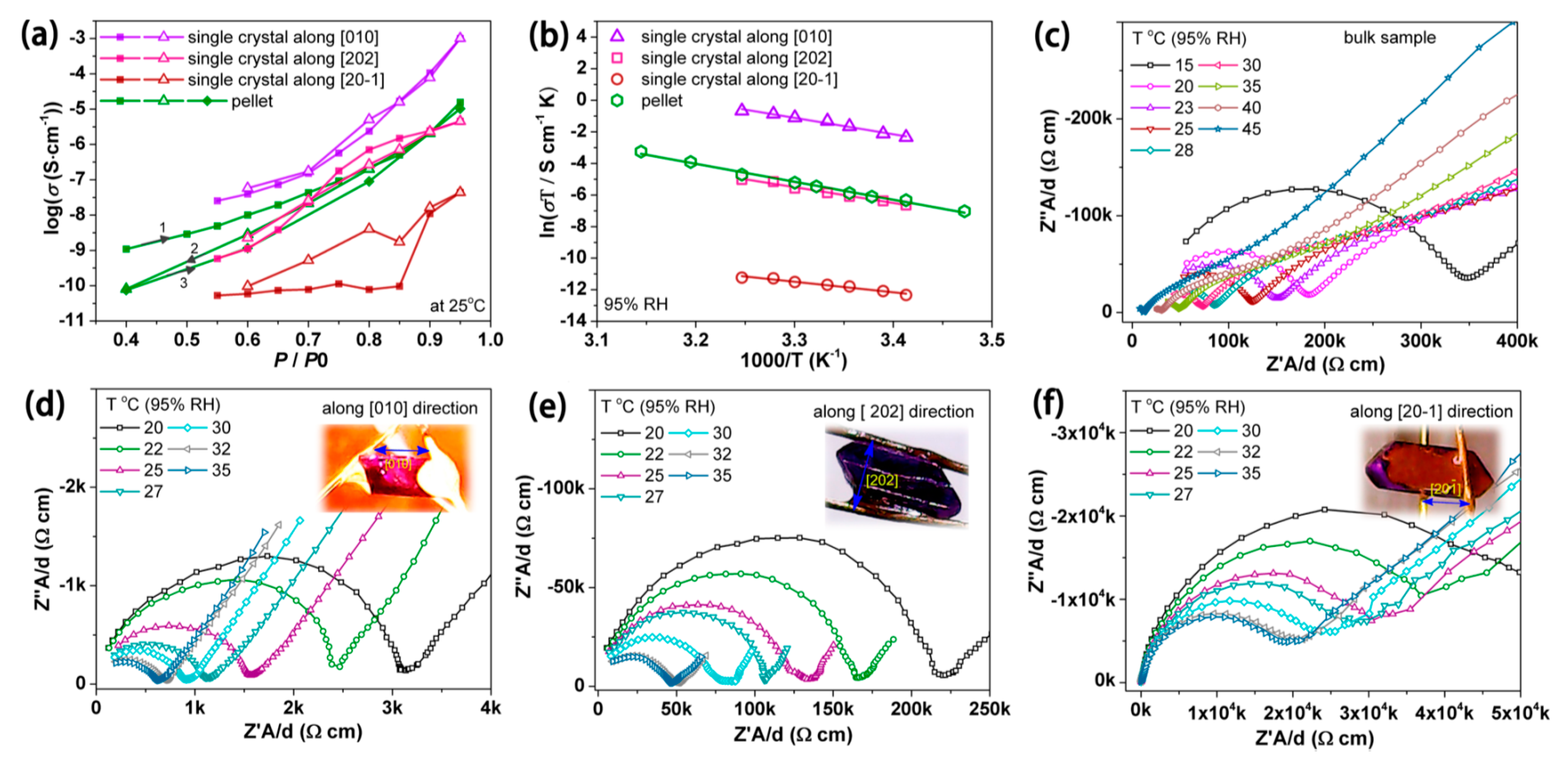
- Bao, S.-S.; Li, N.-Z.; Taylor, J.M.; Shen, Y.; Kitagawa, H.; Zheng, L.-M. Co-Ca Phosphonate Showing Humidity-Sensitive Single Crystal to Single Crystal Structural Transformation and Tunable Proton Conduction Properties. Chem. Mater. 2015, 27, 8116–8125. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mysore Ramesha, B.; Meynen, V. Advances and Challenges in the Creation of Porous Metal Phosphonates. Materials 2020, 13, 5366. https://doi.org/10.3390/ma13235366
Mysore Ramesha B, Meynen V. Advances and Challenges in the Creation of Porous Metal Phosphonates. Materials. 2020; 13(23):5366. https://doi.org/10.3390/ma13235366
Chicago/Turabian StyleMysore Ramesha, Bharadwaj, and Vera Meynen. 2020. "Advances and Challenges in the Creation of Porous Metal Phosphonates" Materials 13, no. 23: 5366. https://doi.org/10.3390/ma13235366
APA StyleMysore Ramesha, B., & Meynen, V. (2020). Advances and Challenges in the Creation of Porous Metal Phosphonates. Materials, 13(23), 5366. https://doi.org/10.3390/ma13235366