Transition Metal Coordination Polymers with Trans-1,4-Cyclohexanedicarboxylate: Acidity-Controlled Synthesis, Structures and Properties


 ,
,  ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization Techniques
2.3. Synthesis
2.3.1. Synthesis of [Co(H2O)4(chdc)]n (1)
2.3.2. Synthesis of [Fe(H2O)4(chdc)]n (2)
2.3.3. Synthesis of [Cd(H2O)(chdc)]n∙0.5nCH3CN (3)
2.3.4. Synthesis of [Mn4(H2O)3(chdc)4]n (4)
2.3.5. Synthesis of [Mn2(Hchdc)2(chdc)]n (5)
2.3.6. Synthesis of Oxides
2.4. X-ray Crystallography
3. Results and Discussion
3.1. Synthesis
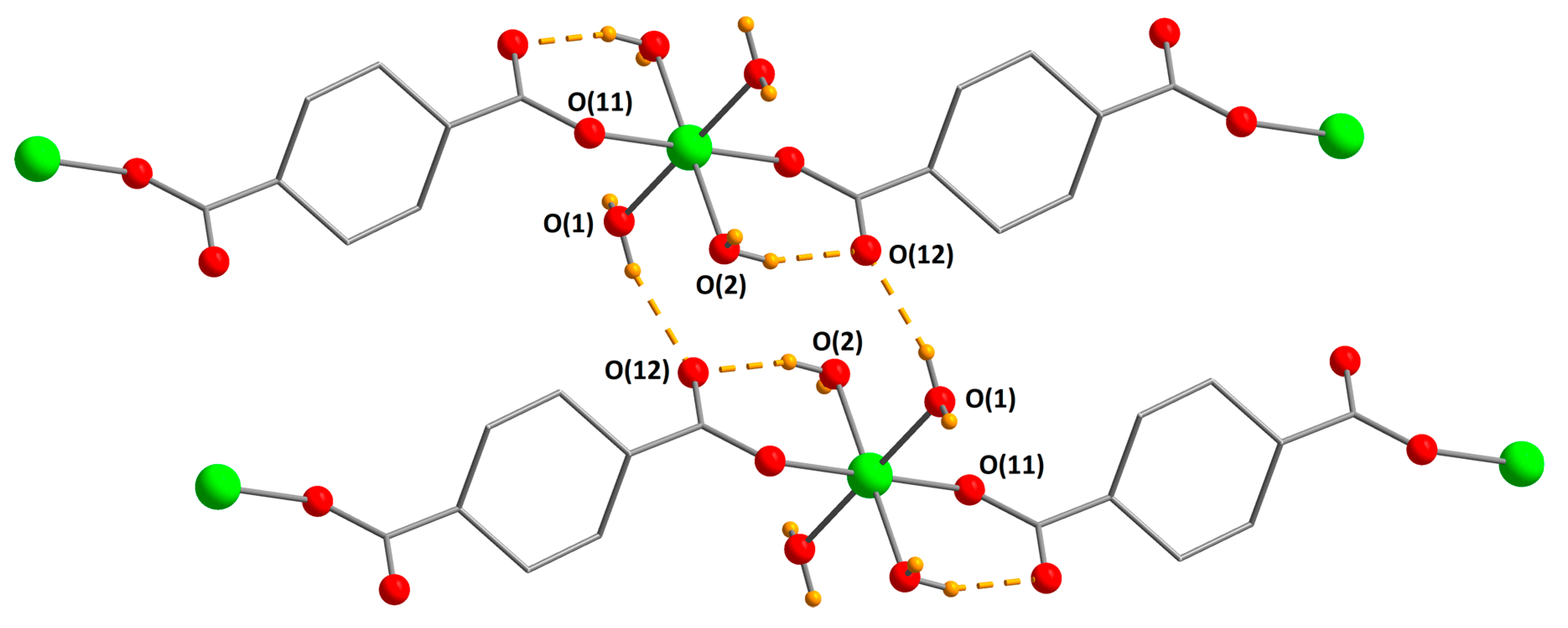
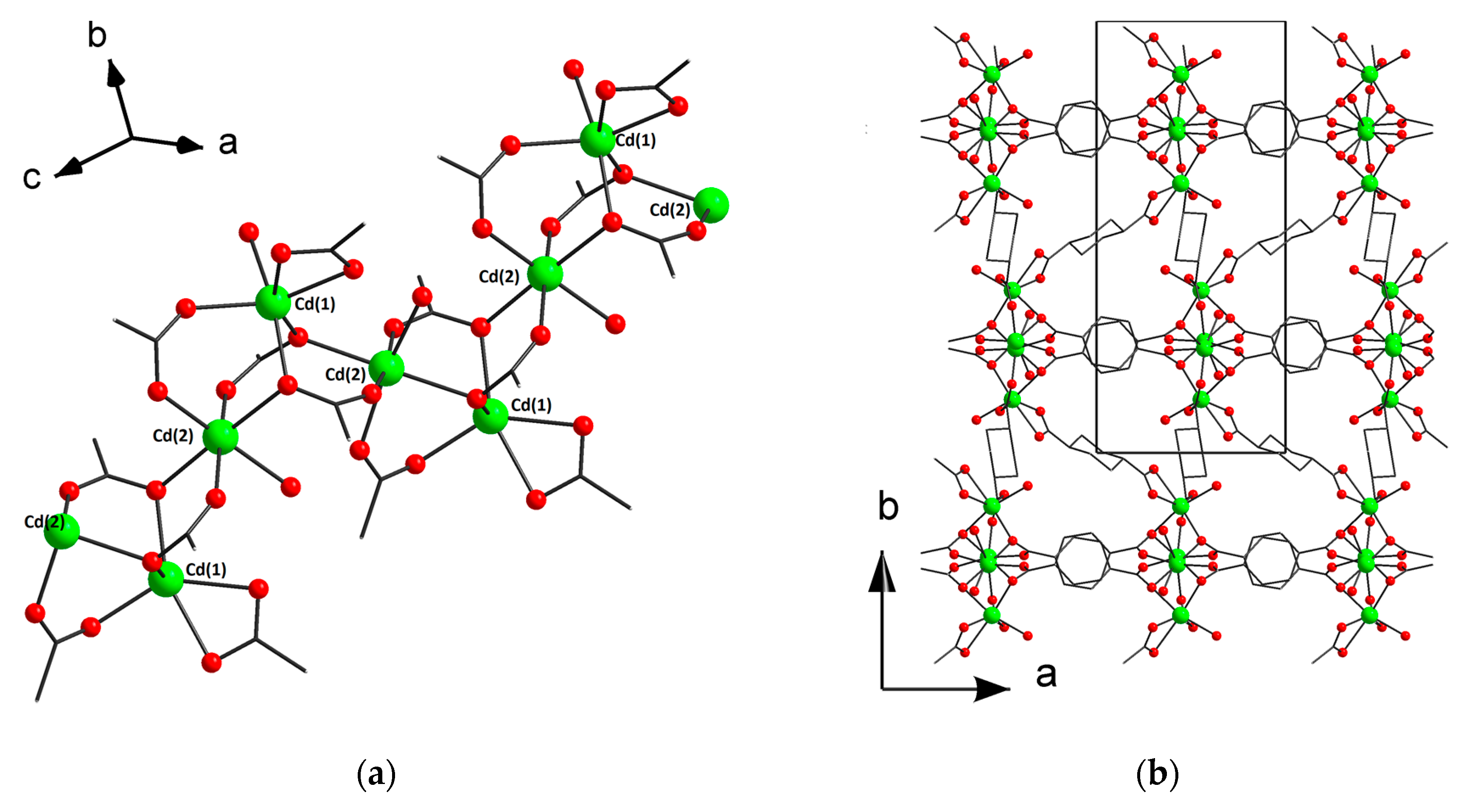
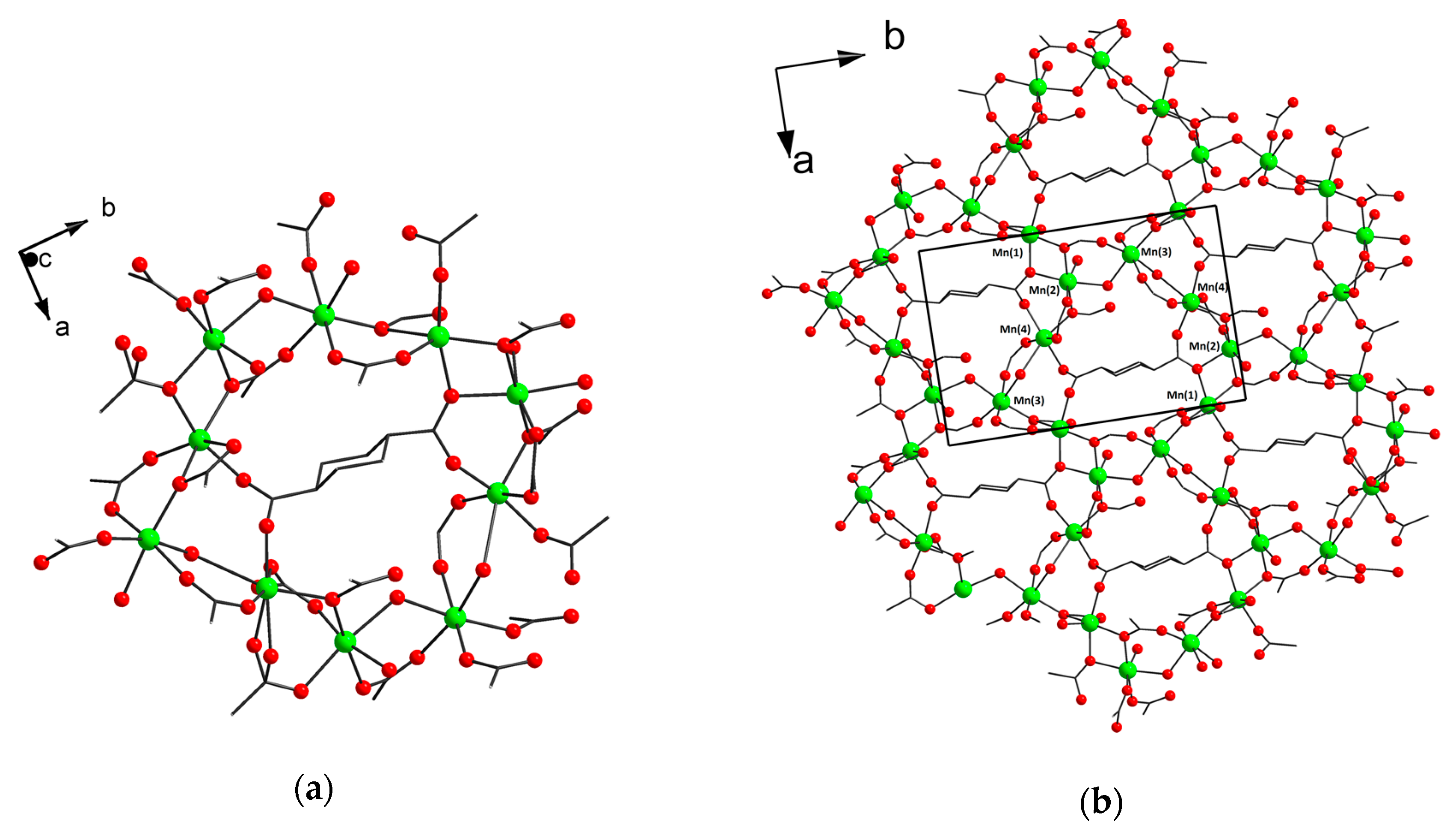
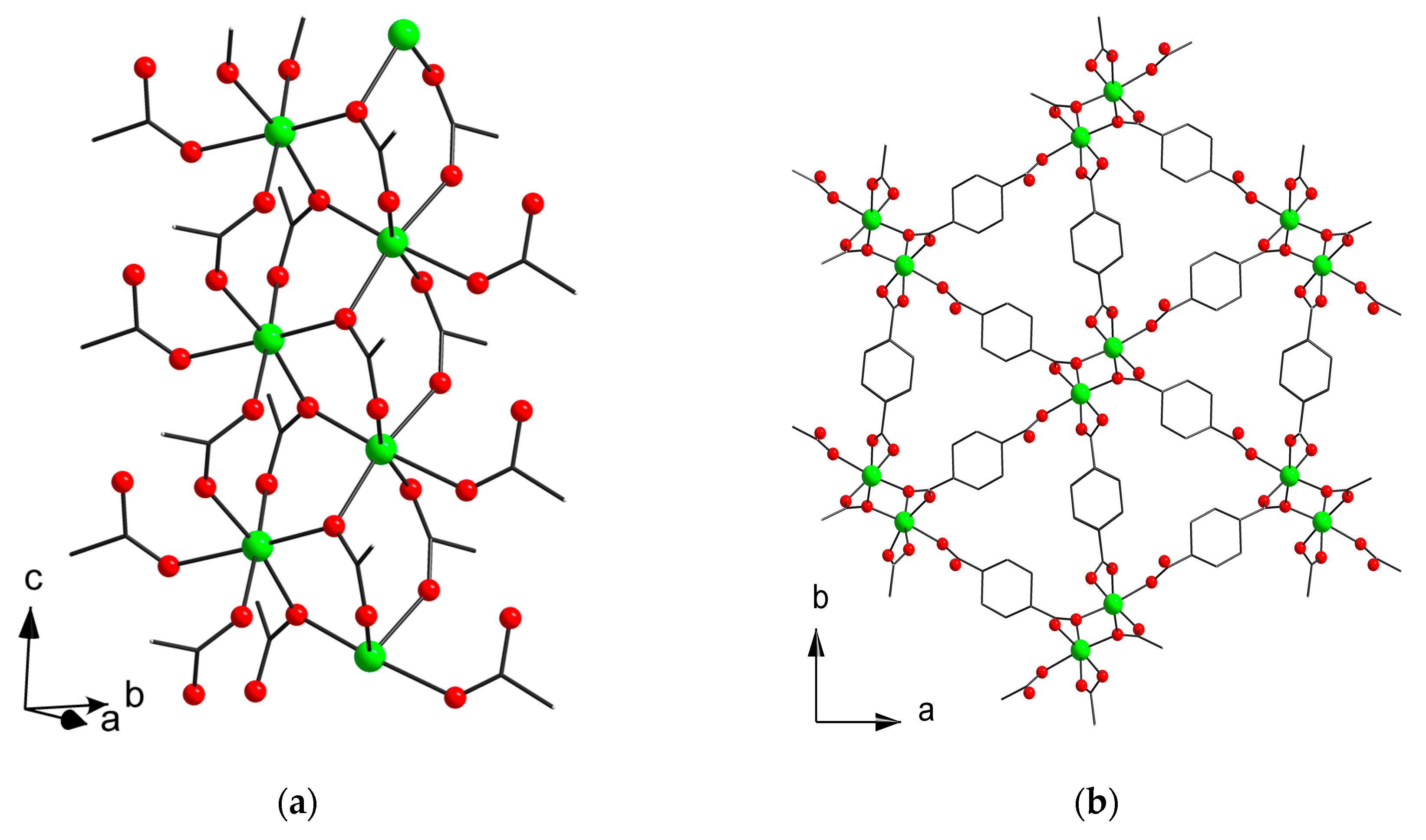
3.2. Structure Descriptions and Infrared Spectroscopy
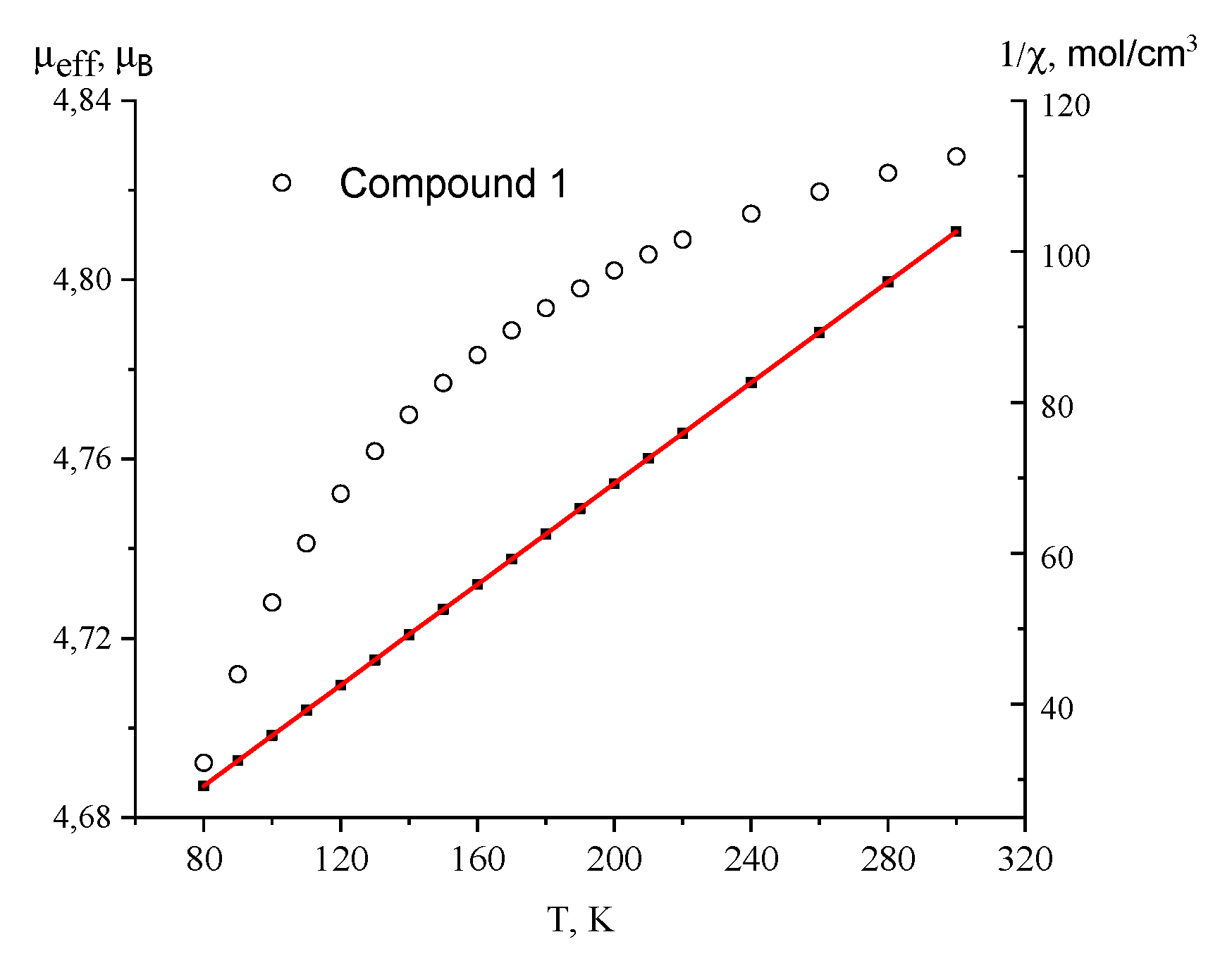
3.3. Magnetochemical Analysis
3.4. Thermal Stability and Thermolysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A. New Polymorph of (e,e)-H2chdc


Appendix B. Crystallographic Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | 1 | 2 | 3 | 4 | 5o | 5m | 6 |
|---|---|---|---|---|---|---|---|
| Chemical formula | C8H18CoO8 | C8H18FeO8 | C9H13.5CdN0.5O5 | C32H46Mn4O19 | C24H32Mn2O12 | C24H32Mn2O12 | C8H12O4 |
| Mr, g∙mol−1 | 301.15 | 298.07 | 321.10 | 954.45 | 622.37 | 622.37 | 172.18 |
| Crystal system, space group | Triclinic, P¯1 | Triclinic, P¯1 | Monoclinic, P21/c | Monoclinic, 21/c | Orthorhombic, Fdd2 | Monoclinic, P21 | Triclinic, P¯1 |
| Temperature (K) | 130 | 130 | 130 | 130 | 130 | 130 | 130 |
| a, b, c (Å) | 4.9320(4), 6.3130(5), 9.5216(7) | 4.9361(4), 6.3194(6), 9.5296(9) | 10.6697(5), 22.4618(11), 9.6558(5) | 10.9672(2), 16.7768(3), 19.6763(4) | 42.792(3), 23.7832(12), 4.8132(2) | 23.7915(19), 4.81470(19), 24.4802(18) | 5.2936(7), 5.6436(8), 7.2022(12) |
| α, β, γ (°) | 80.347(6), 79.008(6), 77.250(7) | 80.655(8), 79.183(8), 77.743(8) | 90.0000, 113.017(6), 90.0000 | 90.0000, 90.2332(18), 90.0000 | 90.0000, 90.0000, 90.0000 | 90.0000, 119.049(10), 90.0000 | 71.861(14), 78.609(13), 79.854(12) |
| V (Å3) | 281.39(4) | 282.99(5) | 2129.9(2) | 3620.29(13) | 4898.5(5) | 2451.4(4) | 198.92(5) |
| Z | 1 | 1 | 8 | 4 | 8 | 4 | 1 |
| μ (mm−1) | 1.55 | 1.36 | 2.05 | 1.45 | 1.10 | 1.10 | 0.12 |
| Crystal size (mm) | 0.50 × 0.31 × 0.15 | 0.23 × 0.07 × 0.07 | 0.58 × 0.47 × 0.16 | 0.55 × 0.30 × 0.22 | 0.29 × 0.05 × 0.05 | 0.29 × 0.05 × 0.05 | 0.13 × 0.08 × 0.07 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 4449, 1382, 1351 | 2138, 1302, 1220 | 10553, 4912, 4554 | 30543, 8653, 7694 | 5975, 2077, 1837 | 12172, 7973, 5190 | 1493, 918, 722 |
| Rint | 0.026 | 0.020 | 0.017 | 0.024 | 0.044 | 0.045 | 0.013 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.021, 0.057, 1.13 | 0.026, 0.061, 1.05 | 0.022, 0.048, 1.11 | 0.030, 0.069, 1.02 | 0.037, 0.074, 1.04 | 0.059, 0.105, 0.98 | 0.040, 0.089, 1.03 |
| No. of parameters | 91 | 91 | 293 | 533 | 220 | 698 | 57 |
| No. of restraints | 4 | 4 | 5 | 69 | 103 | 453 | 0 |
| Δρmax, Δρmin (e∙Å-3) | 0.36, −0.47 | 0.39, −0.30 | 0.63, −0.63 | 1.23, −1.26 | 0.44, −0.36 | 0.49, −0.57 | 0.28, −0.20 |
References
- Zhou, H.C.; Long, J.R.; Yaghi, O.M. Introduction to Metal−Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Howarth, A.J.; Peters, A.W.; Vermeulen, N.A.; Wang, T.C.; Hupp, J.T.; Farha, O.K. Best practices for the synthesis, activation, and characterization of metal−organic frameworks. Chem. Mater. 2017, 29, 26–39. [Google Scholar] [CrossRef]
- Pascanu, V.; González Miera, G.; Ken Inge, A.; Martín-Matute, B. Metal–Organic Frameworks as Catalysts for Organic Synthesis: A Critical Perspective. J. Am. Chem. Soc. 2019, 141, 7223–7234. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Shi, W.; Cheng, P. Synthetic strategies for chiral metal-organic frameworks. Chin. Chem. Lett. 2017, 29, 819–822. [Google Scholar] [CrossRef]
- Sun, D.; Han, L.-L.; Yuan, S.; Deng, Y.-K.; Xu, M.-Z.; Sun, D.-F. Four New Cd(II) Coordination Polymers with Mixed Multidentate N-Donors and Biphenyl-Based Polycarboxylate Ligands: Syntheses, Structures, and Photoluminescent Properties. Cryst. Growth Des. 2013, 13, 377–385. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jung, D.Y. Conformation change of the cyclohexanedicarboxylate ligand toward 2D and 3D La(iii)-organic coordination networks. Chem. Commun. 2002, 8, 908–909. [Google Scholar]
- Chen, J.; Ohba, M.; Zhao, D.; Kaneko, W.; Kitagawa, S. Polynuclear core-based nickel 1,4-cyclohexanedicarboxylate coordination polymers as temperature-dependent hydrothermal reaction products. Cryst. Growth Des. 2006, 6, 664–668. [Google Scholar] [CrossRef]
- Chi-Duran, I.; Enríquez, J.; Manquian, C.; Wrighton-Araneda, K.; Cañon-Mancisidor, W.; Venegas-Yazigi, D.; Herrera, F.; Pratap Singh, D. pH-Controlled Assembly of 3D and 2D Zinc-Based Metal-Organic Frameworks with Tetrazole Ligands. ACS Omega 2018, 3, 801–807. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jung, D.Y. Hydrothermal Synthesis and Magnetic Behavior of a Novel Layered Coordination Polymer Generated from Manganese (II) Adipate. Inorg. Chem. 2000, 39, 1470–1475. [Google Scholar] [CrossRef]
- Zhou, G.J.; Richter, J.; Schnack, J.; Zheng, Y.Z. Hydrophobicity-Driven Self-Assembly of an Eighteen-Membered Honeycomb Lattice with Almost Classical Spins. Chem. Eur. J. 2016, 22, 14846–14850. [Google Scholar] [CrossRef]
- Martínez Casado, F.J.; Fabelo, O.; Rodríguez-Velamazan, J.A.; Ramos Riesco, M.; Rodríguez Cheda, J.A.; Labrador, A.; Rodríguez-Blanco, C.; Campo, J.; Sanchez-Alarcos, V.; Muller, H. Manganese(II) Butyrate-Based MOFs: Structures, Thermal and Magnetic Properties. Cryst. Growth Des. 2011, 11, 4080–4089. [Google Scholar] [CrossRef]
- Reinsch, H.; De Vos, D. Structures and properties of gallium-MOFs with MIL-53-topology based on aliphatic linker molecules. Microporous Mesoporous Mater. 2014, 200, 311–316. [Google Scholar] [CrossRef]
- Reinsch, H.; Pillai, R.S.; Siegel, R.; Senker, J.; Lieb, A.; Maurin, G.; Stock, N. Structure and properties of Al-MIL-53-ADP, a breathing MOF based on the aliphatic linker molecule adipic acid. Dalton Trans. 2016, 45, 4179–4186. [Google Scholar] [CrossRef]
- Bueken, B.; Vermoortele, F.; Vanpoucke, D.E.P.; Reinsch, H.; Tsou, C.-C.; Valvekens, P.; De Baerdemaeker, T.; Ameloot, R.; Kirschhock, C.E.A.; Van Speybroeck, V.; et al. A Flexible Photoactive Titanium Metal–Organic Framework Based on a [TiIV3(μ3-O)(O)2(COO)6] Cluster. Angew. Chem. Int. Ed. 2015, 54, 13912–13917. [Google Scholar] [CrossRef] [PubMed]
- Niekiel, F.; Lannoeye, J.; Reinsch, H.; Munn, A.S.; Heerwig, A.; Zizak, I.; Kaskel, S.; Walton, R.I.; De Vos, D.; Llewellyn, P.; et al. Conformation-controlled sorption properties and breathing of the aliphatic Al-MOF [Al(OH)(CDC)]. Inorg. Chem. 2014, 53, 4610–4620. [Google Scholar] [CrossRef] [PubMed]
- Bueken, B.; Vermoortele, F.; Cliffe, M.J.; Wharmby, M.T.; Foucher, D.; Wieme, J.; Vanduyfhuys, L.; Martineau, C.; Stock, N.; Taulelle, F.; et al. A Breathing Zirconium Metal-Organic Framework with Reversible Loss of Crystallinity by Correlated Nanodomain Formation. Chem. Eur. J. 2016, 22, 3264–3267. [Google Scholar] [CrossRef]
- Bueken, B.; Van Velthoven, N.; Krajnc, A.; Smolders, S.; Taulelle, F.; Mellot-Draznieks, C.; Mali, G.; Bennett, T.D.; De Vos, D. Tackling the Defect Conundrum in UiO-66: A Mixed-Linker Approach to Engineering Missing Linker Defects. Chem. Mater. 2017, 29, 10478–10486. [Google Scholar] [CrossRef]
- Kim, T.K.; Lee, K.J.; Cheon, J.Y.; Lee, J.H.; Joo, S.H.; Moon, H.R. Nanoporous Metal Oxides with Tunable and Nanocrystalline Frameworks via Conversion of Metal–Organic Frameworks. J. Am. Chem. Soc. 2013, 135, 8940–8946. [Google Scholar] [CrossRef]
- Lim, S.; Suh, K.; Kim, Y.; Yoon, M.; Park, H.; Dybtsev, D.N.; Kim, K. Porous carbon materials with a controllable surface area synthesized from metal-organic frameworks. Chem. Commun. 2012, 48, 7447–7449. [Google Scholar] [CrossRef]
- Xi, K.; Cao, S.; Peng, X.; Ducati, C.; Kumar, V.; Cheetham, A.K. Carbon with hierarchical pores from carbonized metal–organic frameworks for lithium sulphur batteries. Chem. Commun. 2013, 49, 2192–2194. [Google Scholar] [CrossRef]
- Kurmoo, M.; Kumagai, H.; Hughes, S.M.; Kepert, C.J. Reversible guest exchange and ferrimagnetism (T(C) = 60.5 K) in a porous cobalt(II)-hydroxide layer structure pillared with trans-1,4-cyclohexanedicarboxylate. Inorg. Chem. 2003, 42, 6709–6722. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Sun, H.J.; Lee, D.H.; Park, G. Synthesis and Structure of a 3-D Metal-Organic Framework, [Cd2(1,4-cyclohexanedicarboxylate)2·DMF], Comprising Unusual Two Different Ligand Conformations. Bull. Korean Chem. Soc. 2012, 33, 3111–3114. [Google Scholar] [CrossRef]
- Thirumurugan, A.; Avinash, M.B.; Rao, C.N.R. 1, 2-, 1, 3- and 1, 4-Cyclohexanedicarboxylates of Cd and Mn with chain and layered structures. Dalton Trans. 2006, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Z.; Xue, W.; Zhang, W.X.; Tong, M.L.; Chen, X.M.; Grandjean, F.; Long, G.J.; Ng, S.W.; Panissod, P.; Drillon, M. Spin-Frustrated Complex, [FeIIFeIII(trans-1,4-cyclohexanedicarboxylate)1.5]∞: Interplay between Single-Chain Magnetic Behavior and Magnetic Ordering. Inorg. Chem. 2009, 48, 2028–2042. [Google Scholar] [CrossRef]
- Demakov, P.A.; Sapchenko, S.A.; Samsonenko, D.G.; Dybtsev, D.N.; Fedin, V.P. Coordination polymers based on zinc(II) and manganese(II) with 1,4-cyclohexanedicarboxylic acid. Russ. Chem. Bull. 2018, 67, 490–496. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro 1.171.38.46; Rigaku Oxford Diffraction: The Woodlands, TX, USA, 2015. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3. [Google Scholar]
- Kurmoo, M.; Kumagai, H.; Akita-Tanaka, M.; Inoue, K.; Takagi, S. Metal-organic frameworks from homometallic chains of nickel(II) and 1,4-cyclohexanedicarboxylate connectors: Ferrimagnet-ferromagnet transformation. Inorg. Chem. 2006, 45, 1627–1637. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Koyioni, M.; Michaelidou, S.S. Synthesis of [(4-Chloro-5H-1,2,3-dithiazol-5-ylidene)amino]azines. Molecules 2011, 16, 8992–9002. [Google Scholar] [CrossRef]
- Li, Z.Y.; Cao, Y.Q.; Zhang, X.M.; Xu, Y.L.; Cao, G.X.; Zhang, F.L.; Li, S.Z.; Zhanga, F.Q.; Zhai, B. Linking cobalt-water chains with cis-1,4-cyclohexanedicarboxylate bridges to form a 2D network exhibiting spin-canted antiferromagnetism. New J. Chem. 2017, 41, 457–461. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Liu, S.; Jeppson, P.; Sandstrom, J.; Caruso, A.N.; Schulz, D.L. Synthesis, structure and magnetic properties of {Mn5(OC(O)CH3)6(OC(O)C6H5)4}∞. Polyhedron 2007, 26, 2235–2242. [Google Scholar] [CrossRef]
- Liu, S.; Bremer, M.T.; Lovaasen, J.; Caruso, A.N.; O’Neill, K.; Simpson, L.; Parilla, P.A.; Heben, M.J.; Schulz, D.L. Structural and magnetic studies of two-dimensional solvent-free manganeseII complexes prepared via ligand exchange reaction under solvothermal conditions. Inorg. Chem. 2008, 47, 1568–1575. [Google Scholar] [CrossRef]
- Bushuev, M.B.; Virovets, A.V.; Garcia, Y.; Gieck, C.; Sheludyakova, L.A.; Ikorskii, V.N.; Tremel, W.; Gütlich, P.; Lavrenova, L.G. Mononuclear coordination compounds based on a novel chelating triazole ligand: 1-vinyl-3-acetylamino-1,2,4-triazole. Polyhedron 2002, 21, 797–804. [Google Scholar] [CrossRef]
- Lavrenova, L.G.; Bushuev, M.B.; Virovets, A.V.; Naumov, D.Y.; Sheludyakova, L.A.; Shvedenkov, Y.G. Coordination Compounds—Synthesis and Study of Cobalt(II), Nickel(II), and Copper(II) Nitrate Complexes with 3-Acetylamino-1,2,4-triazole. Russ. J. Inorg. Chem. 2000, 45, 1658–1663. [Google Scholar]
- Peng, L.; Zhang, J.; Xue, Z.; Han, B.; Li, J.; Yang, G. Large-pore mesoporous Mn3O4 crystals derived from metal–organic frameworks. Chem. Commun. 2013, 49, 11695–11697. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Low, Z.-X.; Feng, Y.; Leong, S.; Zhong, Z.; Yao, J.; Hapgood, K.; Wang, H. Direct conversion of two-dimensional ZIF-L film to porous ZnO nano-sheet film and its performance as photoanode in dye-sensitized solar cell. Microporous Mesoporous Mater. 2014, 194, 1–7. [Google Scholar] [CrossRef]
- Wu, R.; Qian, X.; Rui, X.; Liu, H.; Yadian, B.; Zhou, K.; Wei, J.; Yan, Q.; Feng, X.-Q.; Long, Y.; et al. Zeolitic Imidazolate Framework 67-Derived High Symmetric Porous Co3O4 Hollow Dodecahedra with Highly Enhanced Lithium Storage Capability. Small 2014, 10, 1932–1938. [Google Scholar] [CrossRef]
- Gan, X.; Zheng, R.; Liu, T.; Meng, J.; Chen, R.; Sun, X.; Sun, X. N-Doped Mesoporous In2O3 for Photocatalytic Oxygen Evolution from the In-based Metal–Organic Frameworks. Chem. Eur. J. 2017, 23, 7264–7271. [Google Scholar] [CrossRef]
- Xiao, J.; Diao, K.; Zheng, Z.; Cui, X. MOF-derived porous ZnO/Co3O4 nanocomposites for high performance acetone gas sensing. J. Mater. Sci. Mater. Electron. 2018, 29, 8535–8546. [Google Scholar] [CrossRef]
- Dong, X.; Su, Y.; Lu, T.; Zhang, L.; Wu, L.; Lv, Y. MOFs-derived dodecahedra porous Co3O4: An efficient cataluminescence sensing material for H2S. Sens. Actuators B 2018, 258, 349–357. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Strickler, P. Die Kristallstruktur von 1,4-trans-Cyclohexan-dicarbonsäure. Helv. Chim. Acta 1966, 49, 2505–2515. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Weng, J.B. Cis-Cyclohexane-1,4-dicarboxylic acid. Acta Crystallogr. 2009, E65, o1293. [Google Scholar] [CrossRef] [PubMed]







| Compound Number | Compound Formula | M2+: H2chdc: Base (Molar Ratio) | T, °C | pHstart | pHfinal | Product at Higher pH | Product at Lower pH |
|---|---|---|---|---|---|---|---|
| 1 | [Co(H2O)4(chdc)]n | 1: 1: 0.5 | 80 | 4.5 | 5.0 | unknown | H2chdc |
| 2 | [Fe(H2O)4(chdc)]n | 1: 1: 0.75 | 80 | - | - | unknown | H2chdc |
| 3 | [Cd(H2O)(chdc)]n0.5nCH3CN | 1: 1.3: 1.2 | 100 | 4.8 | 4.5 | [Cd(H2O)2(chdc)]n | H2chdc |
| 4 | [Mn4(H2O)3(chdc)4]n | 1: 0.8: 1 | 80 | 4.9 | 5.2 | - | H2chdc |
| 5 | [Mn2(Hchdc)2(chdc)]n | 1: 2: 1 | 80 | 4.6 | 4.9 | - | H2chdc |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demakov, P.A.; Bogomyakov, A.S.; Urlukov, A.S.; Andreeva, A.Y.; Samsonenko, D.G.; Dybtsev, D.N.; Fedin, V.P. Transition Metal Coordination Polymers with Trans-1,4-Cyclohexanedicarboxylate: Acidity-Controlled Synthesis, Structures and Properties. Materials 2020, 13, 486. https://doi.org/10.3390/ma13020486
Demakov PA, Bogomyakov AS, Urlukov AS, Andreeva AY, Samsonenko DG, Dybtsev DN, Fedin VP. Transition Metal Coordination Polymers with Trans-1,4-Cyclohexanedicarboxylate: Acidity-Controlled Synthesis, Structures and Properties. Materials. 2020; 13(2):486. https://doi.org/10.3390/ma13020486
Chicago/Turabian StyleDemakov, Pavel A., Artem S. Bogomyakov, Artem S. Urlukov, Aleksandra Yu. Andreeva, Denis G. Samsonenko, Danil N. Dybtsev, and Vladimir P. Fedin. 2020. "Transition Metal Coordination Polymers with Trans-1,4-Cyclohexanedicarboxylate: Acidity-Controlled Synthesis, Structures and Properties" Materials 13, no. 2: 486. https://doi.org/10.3390/ma13020486
APA StyleDemakov, P. A., Bogomyakov, A. S., Urlukov, A. S., Andreeva, A. Y., Samsonenko, D. G., Dybtsev, D. N., & Fedin, V. P. (2020). Transition Metal Coordination Polymers with Trans-1,4-Cyclohexanedicarboxylate: Acidity-Controlled Synthesis, Structures and Properties. Materials, 13(2), 486. https://doi.org/10.3390/ma13020486