Preparation and Characterization of Electrospun Collagen Based Composites for Biomedical Applications

 , ,
, , 

Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Fabrication of Electrospun Fibers
2.3. Methods and Methodologies
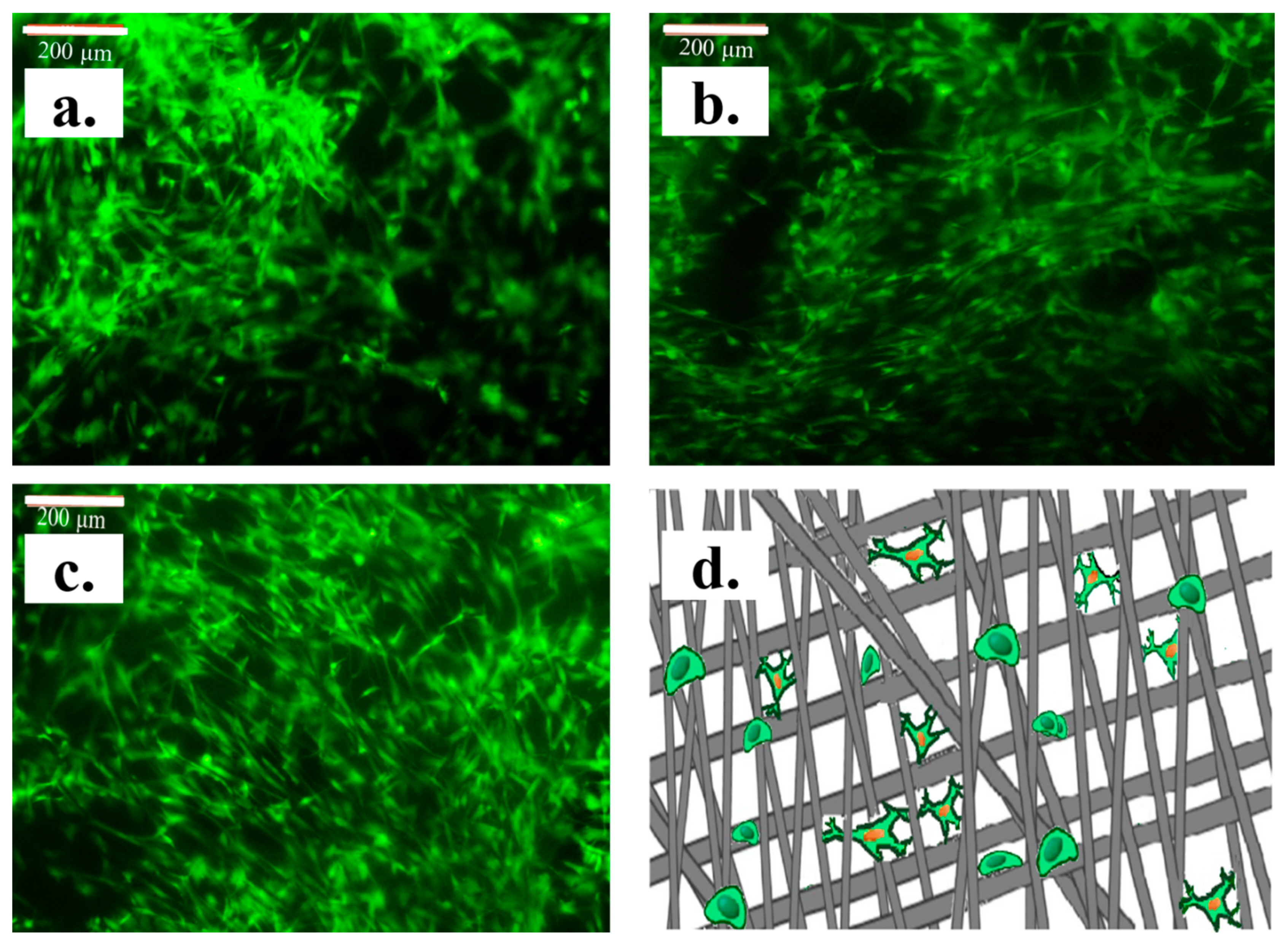
3. Results and Discussion
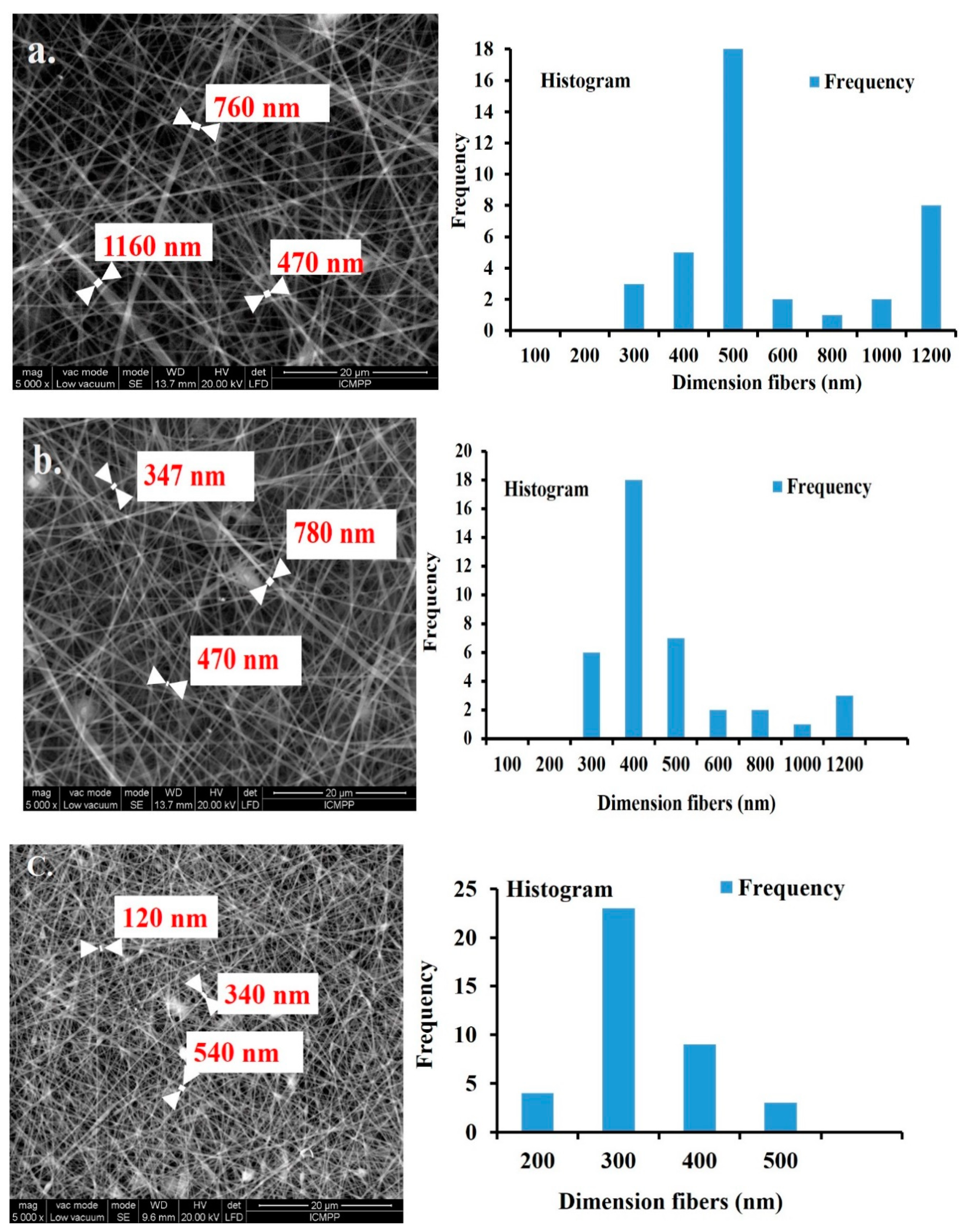
Structural Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Song, J.H.; Kim, Y.-T.; Cho, S.; Song, W.-J.; Moon, S.; Park, C.-G.; Park, S.; Myoung, J.-M.; Jeong, U. Surface-Embedded Stretchable Electrodes by Direct Printing and their Uses to Fabricate Ultrathin Vibration Sensors and Circuits for 3D Structures. Adv. Mater. 2017, 29, 1702625. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Niu, J.; Liu, J.; Yin, L.; Xu, J. In situ encapsulation of laccase in microfibers by emulsion electrospinning: Preparation, characterization, and application. Bioresour. Technol. 2010, 101, 8942–8947. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.N.; Balkus, K.J. Enzyme Immobilization via Electrospinning. Top. Catal. 2012, 55, 1057–1069. [Google Scholar] [CrossRef]
- Zdarta, J.; Jankowska, K.; Bachosz, K.; Kijeńska-Gawrońska, E.; Zgoła-Grześkowiak, A.; Kaczorek, E.; Jesionowski, T. A promising laccase immobilization using electrospun materials for biocatalytic degradation of tetracycline: Effect of process conditions and catalytic pathways. Catal. Today 2020, 348, 127–136. [Google Scholar] [CrossRef]
- Esmaeili, E.; Deymeh, F.; Rounaghi, S.A. Synthesis and characterization of the electrospun fibers prepared from waste polymeric materials. Int. J. Nano Dimens. 2017, 8, 171–181. [Google Scholar]
- Bhardwaj, N.; Kundu, S.C. Electrospinning: A fascinating fiber fabrication technique. Biotechnol. Adv. 2010, 28, 325–347. [Google Scholar] [CrossRef]
- Frenot, A.; Chronakis, I.S. Polymer nanofibers assembled by electrospinning. Curr. Opin. Colloid Interface Sci. 2003, 8, 64–75. [Google Scholar] [CrossRef]
- Kijeńska, E.; Bolek, T.; Bil, M.; Święszkowski, W. Alignment and bioactive molecule enrichment of bio-composite scaffolds towards peripheral nerve tissue engineering. J. Mater. Chem. B 2019, 7, 4509–4519. [Google Scholar] [CrossRef]
- Kijeńska, E.; Swieszkowski, W. Chapter 2–General Requirements of Electrospun Materials for Tissue Engineering: Setups and Strategy for Successful Electrospinning in Laboratory and Industry. In Electrospun Materials for Tissue Engineering and Biomedical Applications; Uyar, T., Kny, E., Eds.; Woodhead Publishing: Sawston, UK, 2017; pp. 43–56. [Google Scholar]
- Matthews, J.A.; Wnek, G.E.; Simpson, D.G.; Bowlin, G.L. Electrospinning of Collagen Nanofibers. Biomacromolecules 2002, 3, 232–238. [Google Scholar] [CrossRef]
- Okuyama, K.; Miyama, K.; Mizuno, K.; Bächinger, H.P. Crystal structure of (Gly-Pro-Hyp)9: Implications for the collagen molecular model. Biopolymers 2012, 97, 607–616. [Google Scholar] [CrossRef]
- Huang, L.; Nagapudi, K.; Apkarian, P.R.; Chaikof, E.L. Engineered collagen–PEO nanofibers and fabrics. J. Biomater. Sci. Polym. Ed. 2001, 12, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-G.; Wan, L.-S.; Xu, Z.-K. Immobilization of catalase on electrospun nanofibrous membranes modified with bovine serum albumin or collagen: Coupling site-dependent activity and protein-dependent stability. Soft Matter 2009, 5, 4161–4168. [Google Scholar] [CrossRef]
- Khadka, D.B.; Haynie, D.T. Protein-and peptide-based electrospun nanofibers in medical biomaterials. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 1242–1262. [Google Scholar] [CrossRef]
- Fiorani, A.; Gualandi, C.; Panseri, S.; Montesi, M.; Marcacci, M.; Focarete, M.L.; Bigi, A. Comparative performance of collagen nanofibers electrospun from different solvents and stabilized by different crosslinkers. J. Mater. Sci. Mater. Electron. 2014, 25, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Yong, T.; Teo, W.E.; Ma, Z.; Ramakrishna, S. Fabrication and Endothelialization of Collagen-Blended Biodegradable Polymer Nanofibers: Potential Vascular Graft for Blood Vessel Tissue Engineering. Tissue Eng. 2005, 11, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, L.; Imran, M.; Cho, B.G.; Al Masry, W.A.; Kim, D.H. Material Recycling—Trends and Perspectives; Achilias, D.S., Ed.; InTech Open: Rijeka, Croatia, 2012. [Google Scholar]
- Khoonkari, M.; Haghighi, A.H.; Sefidbakht, Y.; Shekoohi, K.; Ghaderian, A. Chemical Recycling of PET Wastes with Different Catalysts. Int. J. Polym. Sci. 2015, 2015, 124524. [Google Scholar] [CrossRef]
- May-Pat, A. Mechanical properties of PET composites using multi-walled carbon nanotubes functionalized by inorganic and itaconic acids. Express Polym. Lett. 2012, 6, 96–106. [Google Scholar] [CrossRef]
- Ma, Z.; Kotaki, M.; Yong, T.; He, W.; Ramakrishna, S. Surface engineering of electrospun polyethylene terephthalate (PET) nanofibers towards development of a new material for blood vessel engineering. Biomaterials 2005, 26, 2527–2536. [Google Scholar] [CrossRef]
- Zander, N.E.; Gillan, M.; Sweetser, D. Recycled PET Nanofibers for Water Filtration Applications. Materials 2016, 9, 247. [Google Scholar] [CrossRef]
- Wu, C.-M.; Chiou, H.-G.; Lin, S.-L.; Lin, J.-M. Effects of electrostatic polarity and the types of electrical charging on electrospinning behavior. J. Appl. Polym. Sci. 2012, 126, E89–E97. [Google Scholar] [CrossRef]
- Jung, K.-H.; Huh, M.-W.; Meng, W.; Yuan, J.; Hyun, S.H.; Bae, J.-S.; Hudson, S.M.; Kang, I.-K. Preparation and antibacterial activity of PET/chitosan nanofibrous mats using an electrospinning technique. J. Appl. Polym. Sci. 2007, 105, 2816–2823. [Google Scholar] [CrossRef]
- Nayak, R.; Padhye, R.; Kyratzis, I.L.; Truong, Y.B.; Arnold, L. Recent advances in nanofibre fabrication techniques. Text. Res. J. 2011, 82, 129–147. [Google Scholar] [CrossRef]
- Drobota, M.; Vlad, S.; Gradinaru, L.; Butnaru, M. Investigation of properties of nanofibers from collagen and polyethylene terephthalate using a natural cross-linker. Cellul. Chem. Technol. 2019, 53, 211–218. [Google Scholar] [CrossRef]
- Abraham, L.C.; Vorrasi, J.; Kaplan, D.L. Impact of collagen structure on matrix trafficking by human fibroblasts. J. Biomed. Mater. Res. 2004, 70, 39–48. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Ajji, A.; Cole, K.; Dumoulin, M.; Brisson, J. Amorphous orientation of poly(ethylene terephthalate) by X-ray diffraction in combination with Fourier transform infra-red spectroscopy. Polymer 1995, 36, 4023–4030. [Google Scholar] [CrossRef]
- Kirov, K.R.; Assender, H.E. Quantitative ATR-IR Analysis of Anisotropic Polymer Films: Surface Structure of Commercial PET. Macromolecules 2005, 38, 9258–9265. [Google Scholar] [CrossRef]
- Rusu, E.; Drobota, M.; Barboiu, V. Structural investigations of amines treated polyester thin films by FTIR-ATR spectroscopy. J. Optoelectron. Adv. Mater. 2008, 10, 377–381. [Google Scholar]
- Park, S.C.; Liang, Y.; Lee, H.S. Quantitative Analysis Method for Three-Dimensional Orientation of PTT by Polarized FTIR-ATR Spectroscopy. Macromolecules 2004, 37, 5607–5614. [Google Scholar] [CrossRef]
- Duan, L.; Yuan, J.; Yang, X.; Cheng, X.; Li, J. Interaction study of collagen and sericin in blending solution. Int. J. Biol. Macromol. 2016, 93, 468–475. [Google Scholar] [CrossRef]
- Drobota, M.; Aflori, M.; Barboiu, V. Protein immobilization on Poly(ethylene terephthalate) films modified by plasma and chemical treatments. Dig. J. Nanomater. Biostruct. 2010, 5, 35–42. [Google Scholar]
- Susi, H.; Byler, D.M. Resolution-enhanced fourier transform infrared spectroscopy of enzymes. Methods Enzymol. 1986, 130, 290–311. [Google Scholar] [CrossRef] [PubMed]
- Arrondo, J.L.R.; Muga, A.; Castresana, J.; Goñi, F.M. Quantitative studies of the structure of proteins in solution by fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 1993, 59, 23–56. [Google Scholar] [CrossRef]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef] [PubMed]
- Oberg, A.K.; Ruysschaert, J.-M.; Goormaghtigh, E. The optimization of protein secondary structure determination with infrared and circular dichroism spectra. JBIC J. Biol. Inorg. Chem. 2004, 271, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. Evaluation of the Information Content in Infrared Spectra for Protein Secondary Structure Determination. Biophys. J. 2006, 90, 2946–2957. [Google Scholar] [CrossRef] [PubMed]
- Shivu, B.; Seshadri, S.; Li, J.; Oberg, K.A.; Uversky, V.N.; Fink, A.L. Distinct β-Sheet Structure in Protein Aggregates Determined by ATR–FTIR Spectroscopy. Biochemistry 2013, 52, 5176–5183. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, M.; Meiyappan, M.; Todd, P.; Hudson, B.G. Crystal Structure of NC1 Domains. J. Biol. Chem. 2002, 277, 31142–31153. [Google Scholar] [CrossRef]
- Payne, K.J.; Veis, A. Fourier transform ir spectroscopy of collagen and gelatin solutions: Deconvolution of the amide I band for conformational studies. Biopolymers 1988, 27, 1749–1760. [Google Scholar] [CrossRef]
- Tian, Z.; Wu, K.; Liu, W.; Shen, L.; Li, G. Two-dimensional infrared spectroscopic study on the thermally induced structural changes of glutaraldehyde-crosslinked collagen. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 140, 356–363. [Google Scholar] [CrossRef]
- Al Hodali, R. Numerical Simulation of an Agricultural Foodstuffs Drying Unit Using Solar Energy and Adsorption Process. Ph.D. Thesis, Universite’ Libre de Bruxelles, Brussels, Belgium, 1997. [Google Scholar]
- Medeiros, E.S.; Mattoso, L.H.C.; Offeman, R.D.; Wood, D.F.; Orts, W.J. Effect of relative humidity on the morphology of electrospun polymer fibers. Can. J. Chem. 2008, 86, 590–599. [Google Scholar] [CrossRef]
- Huang, L.; Bui, N.; Manickam, S.S.; McCutcheon, J.R. Controlling electrospun nanofiber morphology and mechanical properties using humidity. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1734–1744. [Google Scholar] [CrossRef]
- Hardick, O.; Stevens, B.; Bracewell, D.G. Nanofibre fabrication in a temperature and humidity controlled environment for improved fibre consistency. J. Mater. Sci. 2011, 46, 3890–3898. [Google Scholar] [CrossRef]
- Purushothamana, A.E.; Thakurb, K.; Kandasubramanian, B. Development of highly porous, Electrostatic force assisted nanofiber fabrication for biological applications. Int. J. Polym. Mater. 2020, 69, 474–504. [Google Scholar]
- Badii, F.; Macnaughtan, W.; Mitchell, J.R.; Farhat, I.A. The Effect of Drying Temperature on Physical Properties of Thin Gelatin Films. Dry Technol. 2013, 32, 30–38. [Google Scholar] [CrossRef]
- Chang, L.; Pikal, M.J. Mechanisms of protein stabilization in the solid state. J. Pharm. Sci. 2009, 98, 2886–2908. [Google Scholar] [CrossRef]
- Luthra, S.; Kalonia, D.S.; Pikal, M.J.; Lechuga-Ballesteros, D. Simultaneous Measurement of Water Desorption Isotherm and Heats of Water Desorption of Proteins Using Perfusion Isothermal Microcalorimetry. J. Pharm. Sci. 2007, 96, 1974–1982. [Google Scholar] [CrossRef]
- Luo, X.; Guo, Z.; He, P.; Chen, T.; Li, L.; Ding, S.; Li, H. Study on structure, mechanical property and cell cytocompatibility of electrospun collagen nanofibers crosslinked by common agents. Int. J. Biol. Macromol. 2018, 113, 476–486. [Google Scholar] [CrossRef]
- Yasuda, H.; Stannett, V. Permeation, solution, and diffusion of water in some high polymers. J. Polym. Sci. 1962, 57, 907–923. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, H.; Guo, M. Relaxation of a hydrophilic polymer induced by moisture desorption through the glass transition. Phys. Chem. Chem. Phys. 2015, 17, 3186–3195. [Google Scholar] [CrossRef]
- Burgess, S.K.; Mikkilineni, D.; Yu, D.; Kim, D.; Mubarak, C.; Kriegel, R.; Koros, W. Water sorption in poly(ethylene furanoate) compared to poly(ethylene terephthalate). Polymer 2014, 55, 6870–6882. [Google Scholar] [CrossRef]
- Langevin, D.; Grenet, J.; Saiter, J. Moisture sorption in pet influence on the thermokinetic parameters. Eur. Polym. J. 1994, 30, 339–345. [Google Scholar] [CrossRef]
- Alessi, M.; Conzatti, L.; Hodge, P.; Scafati, S.T.; Stagnaro, P. A Possible Means to Assist the Processing of PET, PTT and PBT. Macromol. Mater. Eng. 2010, 295, 374–380. [Google Scholar] [CrossRef]
- Jabarin, S.A.; Lofgren, E.A. Effects of water absorption on physical properties and degree of molecular orientation of poly (ethylene terephthalate). Polym. Eng. Sci. 1986, 26, 620–625. [Google Scholar] [CrossRef]
- Serbezeanu, D.; Vlad-Bubulac, T.; Rusu, D.; Pîrcălăbioru, G.G.; Samoilă, I.; Dinescu, S.; Aflori, M. Functional Polyimide-Based Electrospun Fibers for Biomedical Application. Materials 2019, 12, 3201. [Google Scholar] [CrossRef] [PubMed]
- Nistor, A.; Stiubianu, G.; Racles, C.; Cazacu, M. Evaluation of the water sorption capacity of some polymeric materials by Dynamic Vapour Sorption. Mater. Plast. 2011, 48, 33–37. [Google Scholar]
- Xu, C.; Willför, S.M.; Holmlund, P.; Holmbom, B. Rheological properties of water-soluble spruce O-acetyl galactoglucomannans. Carbohydr. Polym. 2009, 75, 498–504. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Nistor, M.T.; Vasile, C.; Chiriac, A.P.; Tartau, L.M. Biocompatibility, biodegradability, and drug carrier ability of hybrid collagen-based hydrogel nanocomposites. J. Bioact. Compat. Polym. 2013, 28, 540–556. [Google Scholar] [CrossRef]
- Stonadge, P.R.; Benham, M.J.; Ross, D.K.; Manwaring, C.; Harris, I.R. The Measurement of Concentration Dependent Diffusion Coefficients in the Solid-solution Alloy Pd-Y*. Z. Phys. Chem. 1993, 181, 125–131. [Google Scholar] [CrossRef]
- Khorshidi, S.; Solouk, A.; Mirzadeh, H.; Mazinani, S.; Lagarón, J.M.; Sharifi, S.; Ramakrishna, S. A review of key challenges of electrospun scaffolds for tissue-engineering applications. J. Tissue Eng. Regen. Med. 2015, 10, 715–738. [Google Scholar] [CrossRef]
- Teixeira, A.I.; Abrams, G.A.; Bertics, P.J.; Murphy, C.J.; Nealey, P.F. Epithelial contact guidance on well-defined micro- and nanostructured substrates. J. Cell Sci. 2003, 116, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.B.; Guido, S.; Tranquillo, R.T. Biased cell migration of fibroblasts exhibiting contact guidance in oriented collagen gels. Ann. Biomed. Eng. 1994, 22, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Gee, A.O.; Baker, B.M.; Silverstein, A.M.; Montero, G.; Esterhai, J.L.; Mauck, R.L. Fabrication and evaluation of biomimetic-synthetic nanofibrous composites for soft tissue regeneration. Cell Tissue Res. 2012, 347, 803–813. [Google Scholar] [CrossRef]
- Londono, C.; Loureiro, M.J.; Slater, B.; Lücker, P.B.; Soleas, J.; Sathananthan, S.; Aitchison, J.S.; Kabla, A.J.; McGuigan, A.P. Nonautonomous contact guidance signaling during collective cell migration. Proc. Natl. Acad. Sci. USA 2014, 111, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Wendorff, J.H.; Greiner, A. Use of electrospinning technique for biomedical applications. Polymer 2008, 49, 5603–5621. [Google Scholar] [CrossRef]
- Mehnath, S.; Chitra, K.; Karthikeyan, K.; Jeyaraj, M. Localized delivery of active targeting micelles from nanofibers patch for effective breast cancer therapy. Int. J. Pharm. 2020, 584, 119412. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Weight (%) | BET | |
|---|---|---|---|
| Area (m2/g) | Monolayer (g/g) | ||
| PET | 1.4341 | 22.91 | 0.01183 |
| C | 20.6061 | 221.08 | 0.06295 |
| C-PET | 17.0599 | 143.852 | 0.04097 |
| Sample | Mt/M∞ < 0.5 | Mt/M∞ > 0.5 |
|---|---|---|
| D1 (cm2/s) × 10−6 | D2 (cm2/s) × 10−6 | |
| PET fibers | 1.15 | 6.41 |
| C fibers | 0.409 | 4.87 |
| C-PET fibers | 0.292 | 4.86 |
| PET Film | 0.0372 | 0.22 |
| C Film | 0.0298 | 0.38 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drobota, M.; Gradinaru, L.M.; Vlad, S.; Bargan, A.; Butnaru, M.; Angheloiu, M.; Aflori, M. Preparation and Characterization of Electrospun Collagen Based Composites for Biomedical Applications. Materials 2020, 13, 3961. https://doi.org/10.3390/ma13183961
Drobota M, Gradinaru LM, Vlad S, Bargan A, Butnaru M, Angheloiu M, Aflori M. Preparation and Characterization of Electrospun Collagen Based Composites for Biomedical Applications. Materials. 2020; 13(18):3961. https://doi.org/10.3390/ma13183961
Chicago/Turabian StyleDrobota, Mioara, Luiza Madalina Gradinaru, Stelian Vlad, Alexandra Bargan, Maria Butnaru, Marian Angheloiu, and Magdalena Aflori. 2020. "Preparation and Characterization of Electrospun Collagen Based Composites for Biomedical Applications" Materials 13, no. 18: 3961. https://doi.org/10.3390/ma13183961
APA StyleDrobota, M., Gradinaru, L. M., Vlad, S., Bargan, A., Butnaru, M., Angheloiu, M., & Aflori, M. (2020). Preparation and Characterization of Electrospun Collagen Based Composites for Biomedical Applications. Materials, 13(18), 3961. https://doi.org/10.3390/ma13183961