Novel III-V Nitride Polymorphs in the P42/mnm and Pbca Phases

Abstract
1. Introduction
2. Theoretical Methods
3. Results and Discussion
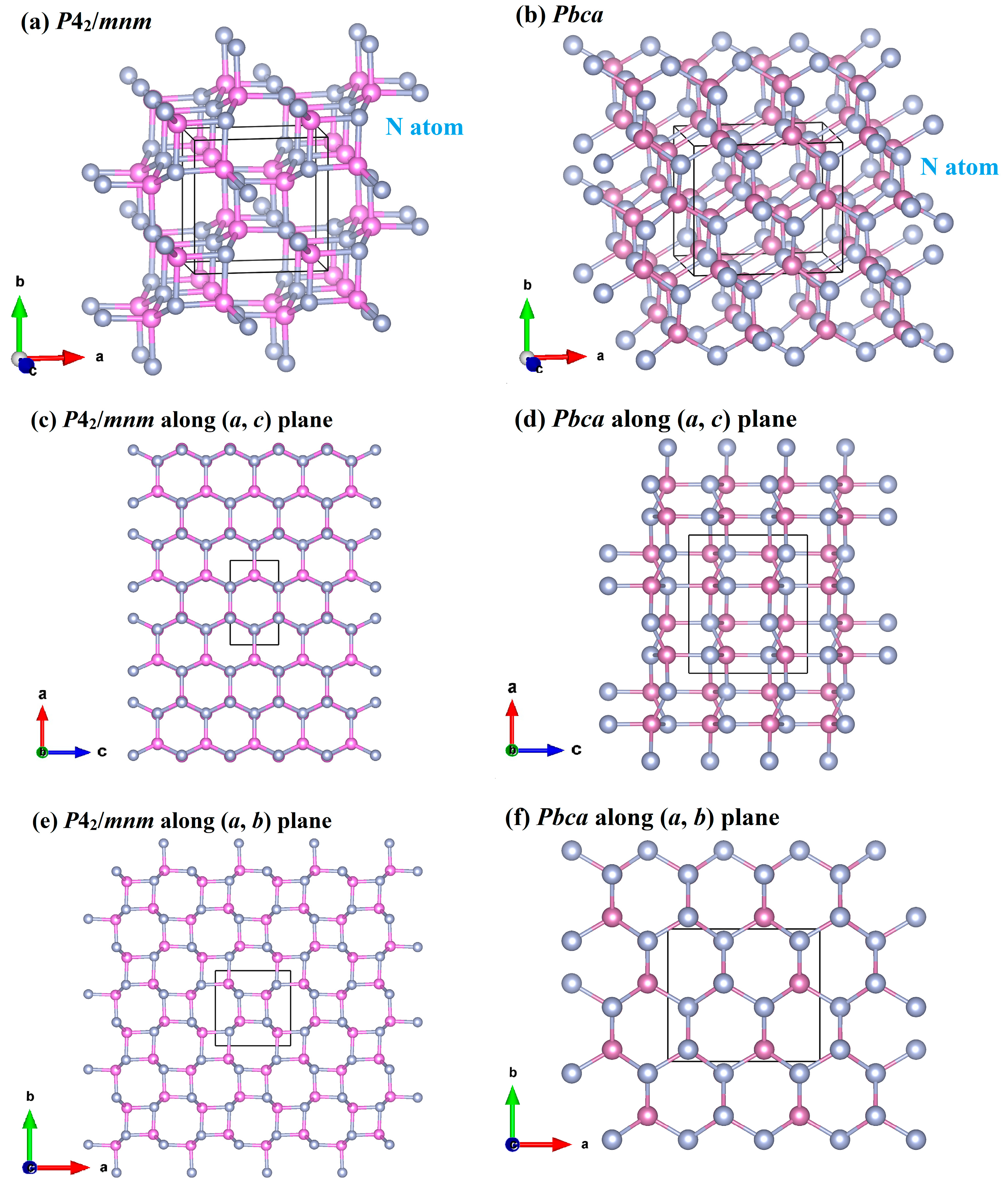
3.1. Structural Properties
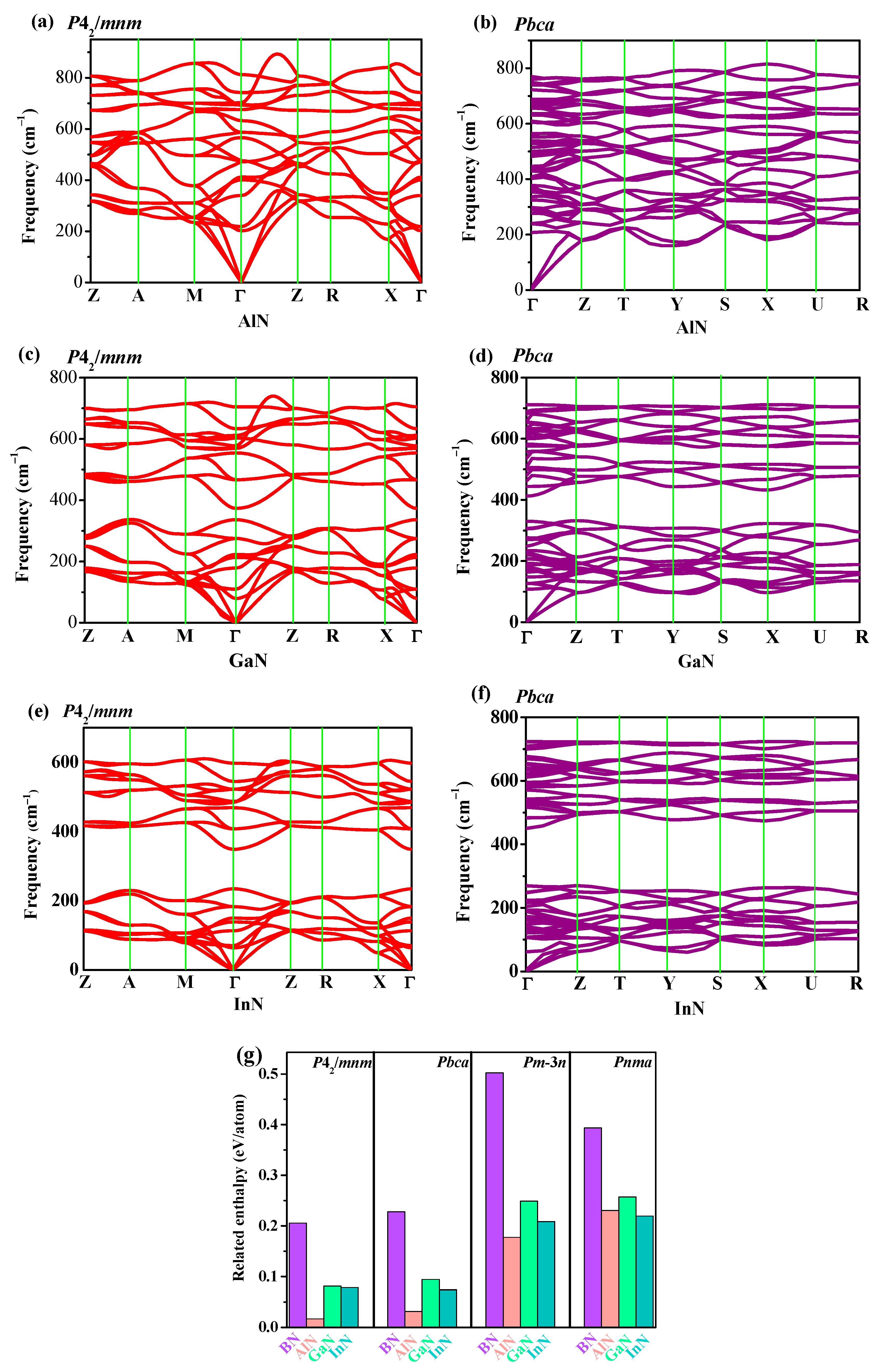
3.2. Stability
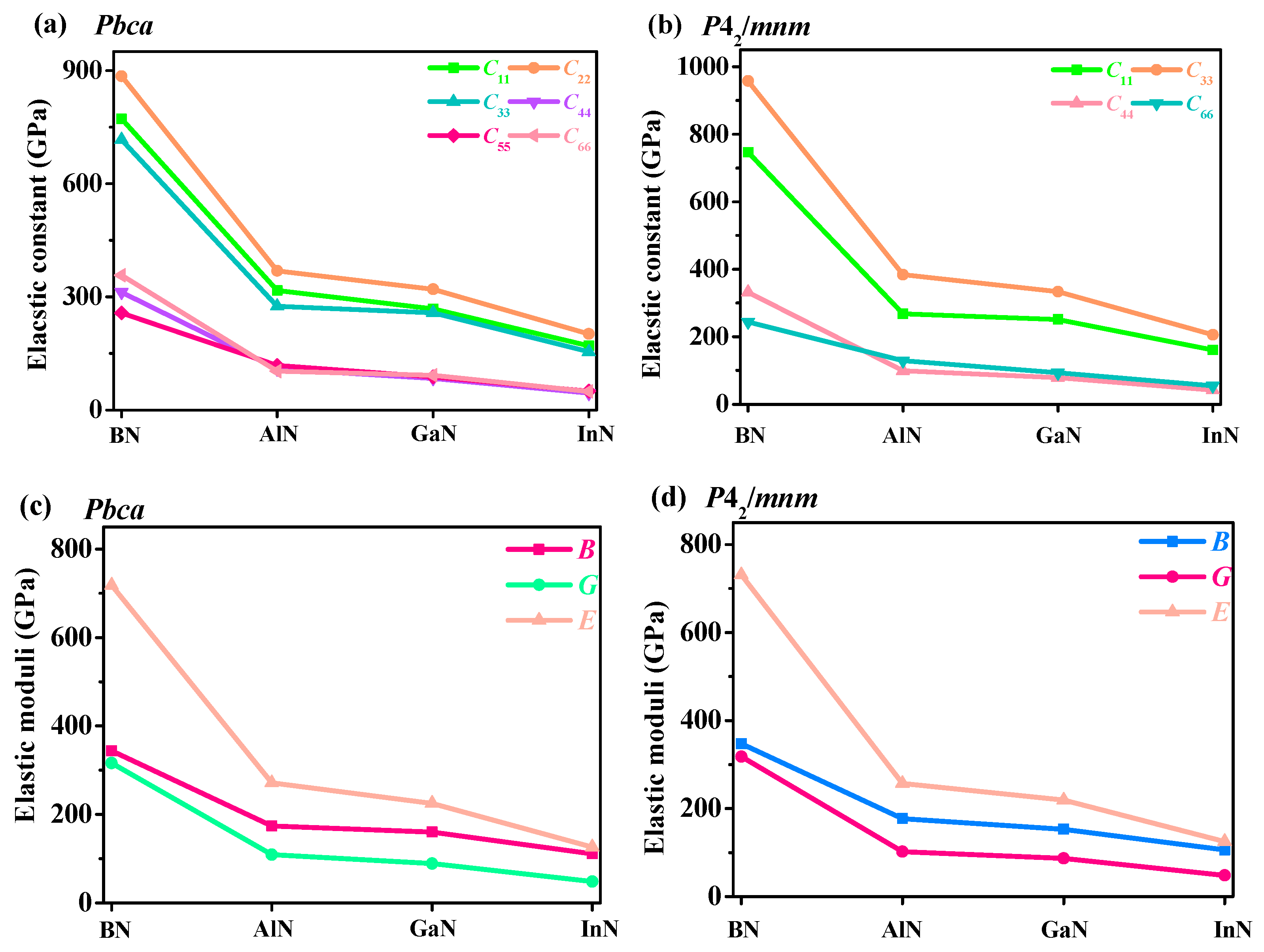
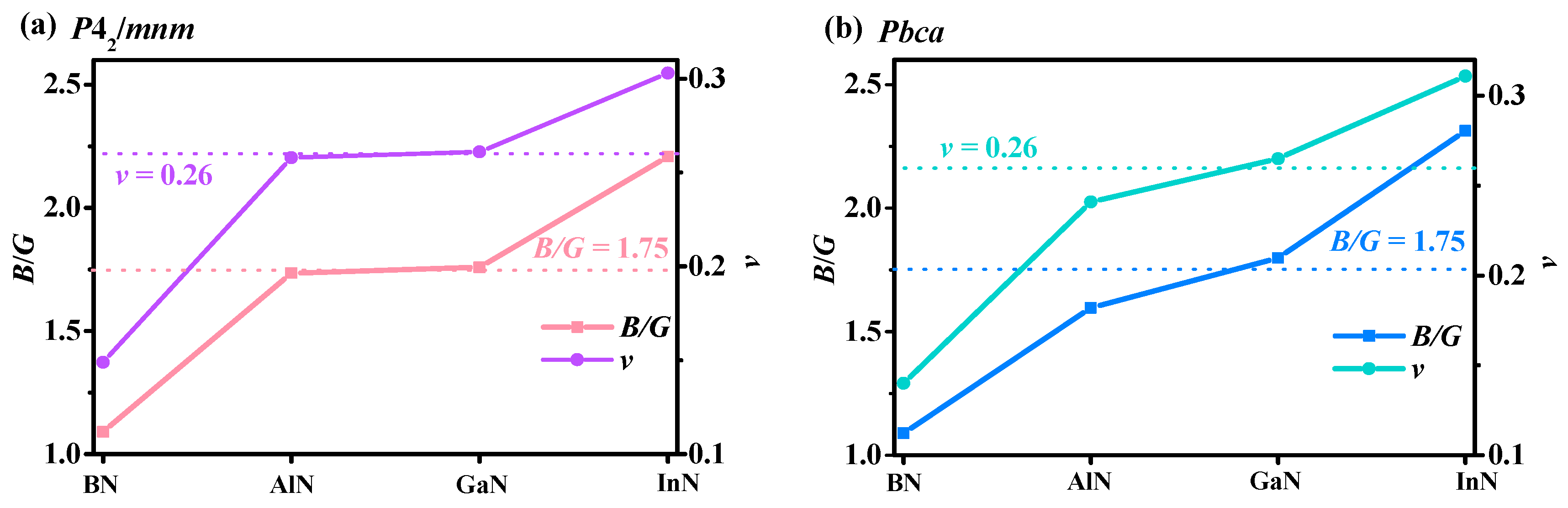
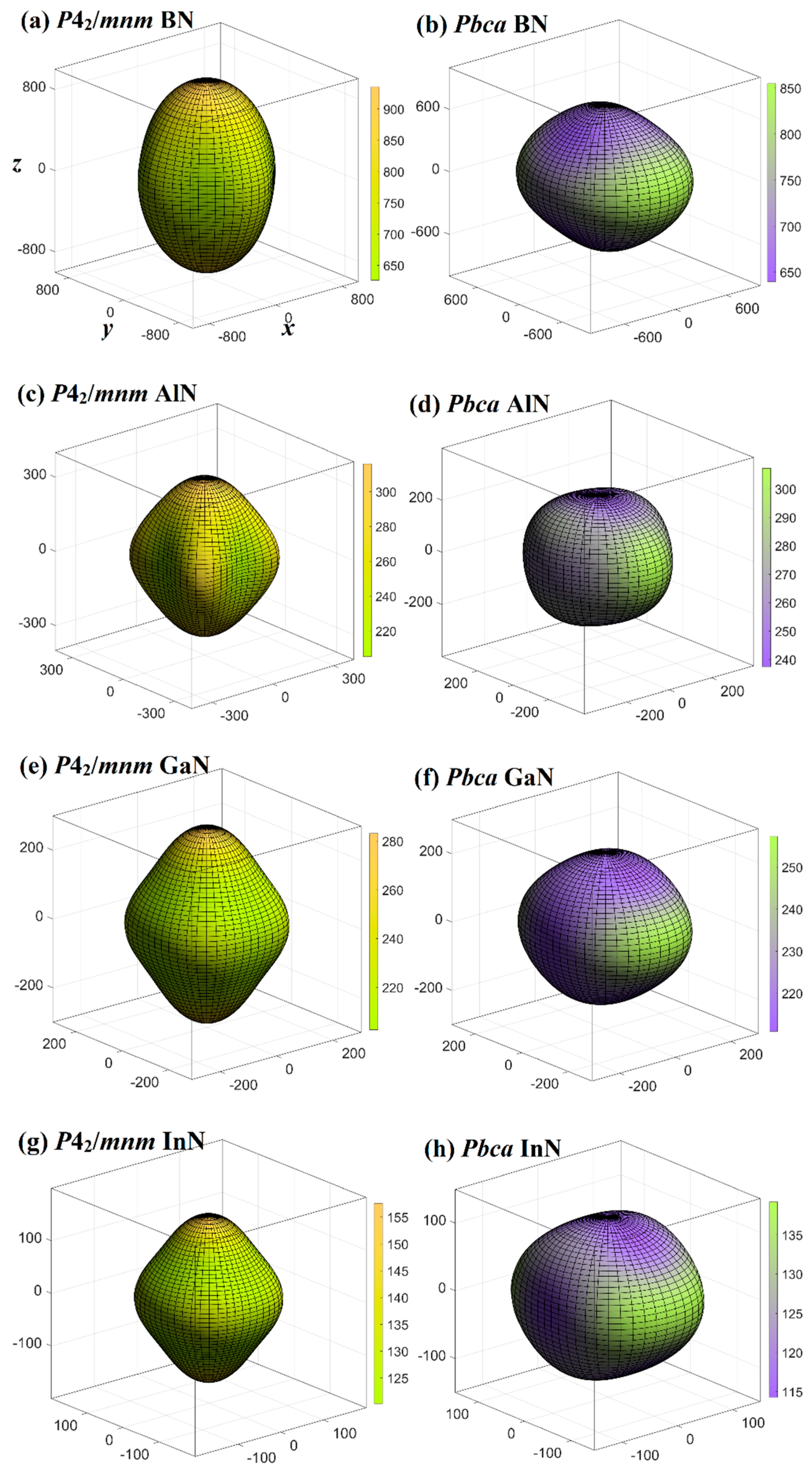
3.3. Mechanical and Anisotropy Properties
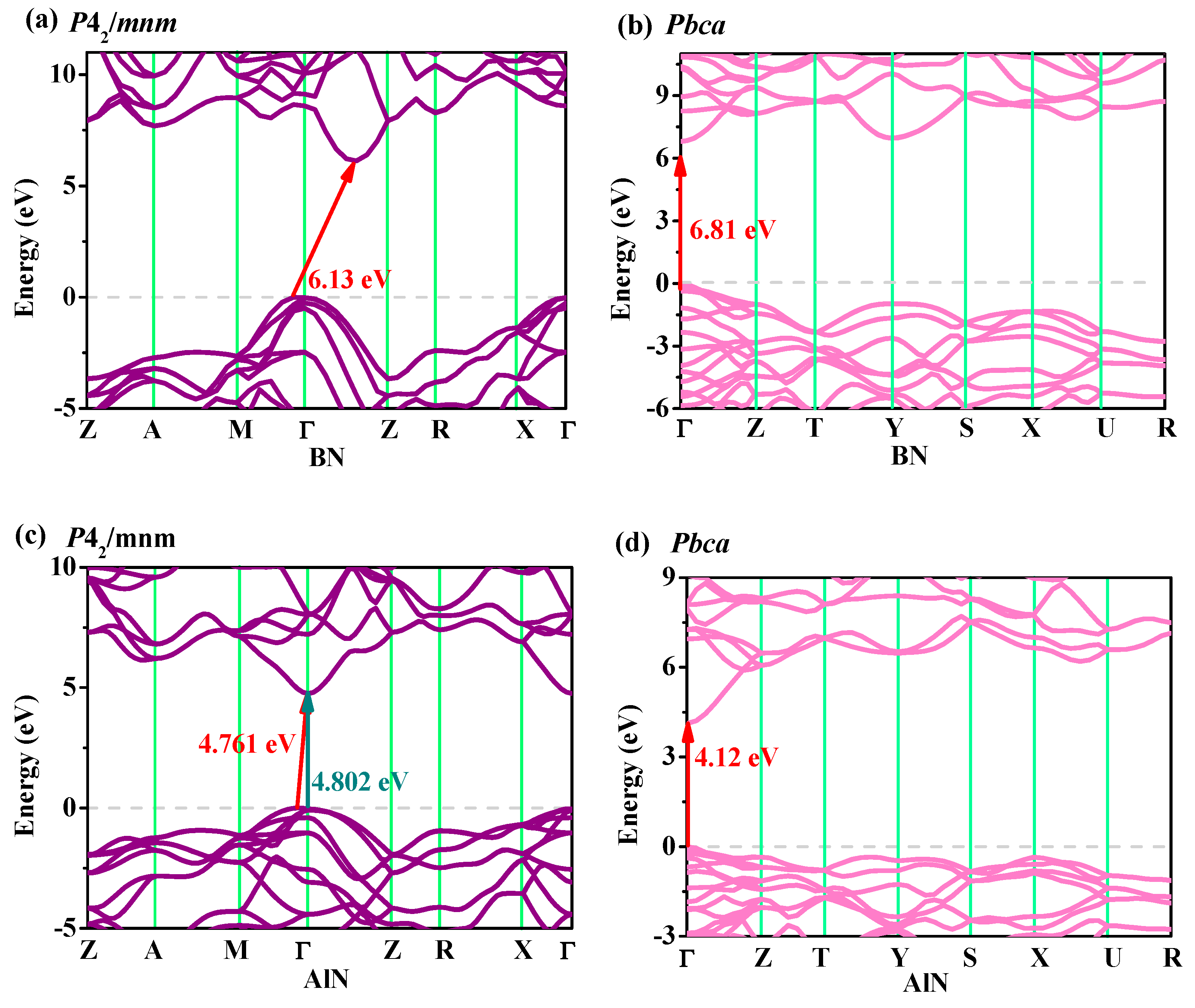
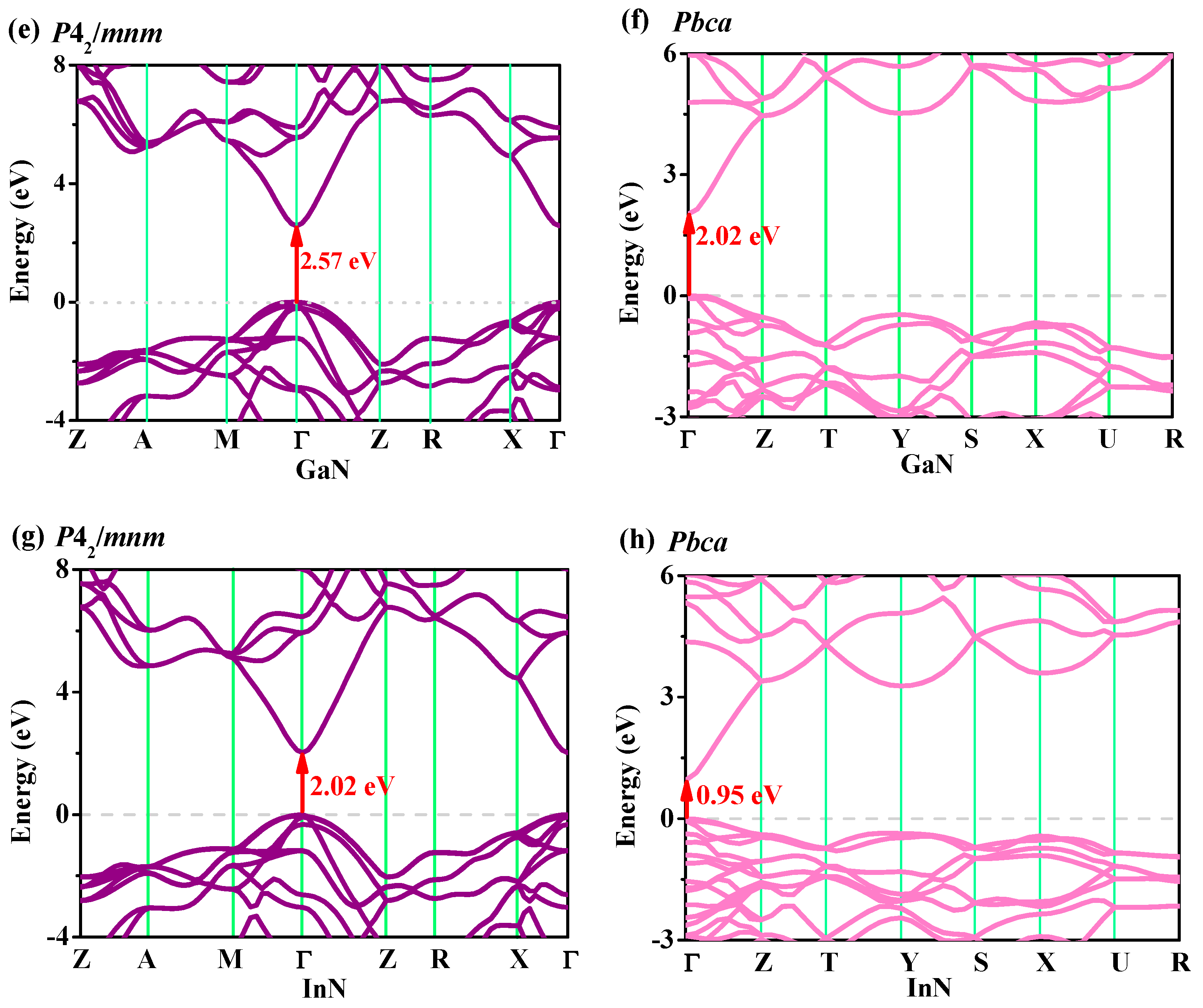
3.4. Electronic Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fan, Q.Y.; Wang, H.Q.; Song, Y.X.; Zhang, W.; Yun, S.N. Five carbon allotropes from Squaroglitter structures. Comput. Mater. Sci. 2020, 178, 109634. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Song, Y.X.; Yang, Y. Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon. Materials 2020, 13, 1926. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Xing, M.J. Prediction of a novel carbon allotrope from first-principle calculations: A potential superhard material in monoclinic symmetry. Mater. Chem. Phys. 2020, 242, 122480. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Zhao, Y.B.; Yu, X.H.; Song, Y.X.; Zhang, W.; Yun, S.N. Physical properties of a novel microporous carbon material. Diam. Relat. Mater. 2020, 106, 107831. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Song, Y.X.; Yang, Y.T. PBCF-Graphene: A 2D Sp2 Hybridized Honeycomb Carbon Allotrope with a Direct Band Gap. ChemNanoMat 2020, 6, 139–147. [Google Scholar] [CrossRef]
- Yin, H.C.; Shi, X.Z.; He, C.Y.; Martinez-Canales, M.; Li, J.; Pickard, C.J.; Tang, C.; Ouyang, T.; Zhang, C.X.; Zhong, J.X. Stone-Wales graphene: A two-dimensional carbon semimetal with magic stability. Phys. Rev. B 2019, 99, 041405(R). [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Song, Y.X.; Yang, Y.T. Six novel carbon and silicon allotropes with their potential application in photovoltaic field. J. Phys. Condens. Matter 2020, 32, 355701. [Google Scholar] [CrossRef]
- He, C.Y.; Shi, X.Z.; Clark, S.J.; Li, J.; Pickard, C.J.; Ouyang, T.; Zhang, C.X.; Tang, C.; Zhong, J.X. Complex Low Energy Tetrahedral Polymorphs of Group IV Elements from First Principles. Phys. Rev. Lett. 2018, 121, 175701. [Google Scholar] [CrossRef]
- Gong, Z.H.; Shi, X.Z.; Li, J.; Li, S.F.; He, C.Y.; Ouyang, T.; Zhang, C.X.; Tang, C.; Zhong, J.X. Theoretical prediction of low-energy Stone-Wales graphene with an intrinsic type-III Dirac cone. Phys. Rev. B 2020, 101, 155427. [Google Scholar] [CrossRef]
- Qiao, L.P.; Jin, Z. Two B-C-O Compounds: Structural, Mechanical Anisotropy and Electronic Properties under Pressure. Materials 2017, 10, 1413. [Google Scholar] [CrossRef]
- Li, X.Z.; Xing, M.J. Novel carbon-rich nitride C3N: A superhard phase in monoclinic symmetry. Comput. Mater. Sci. 2019, 158, 170–177. [Google Scholar] [CrossRef]
- Qiao, L.P.; Jin, Z.; Yan, G.Y.; Li, P.; Hang, L.M.; Li, L. Density-functional-studying of oP8–, tI16–, and tP4–B2CO physical properties under pressure. J. Solid State Chem. 2019, 270, 642–650. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Zhou, P.K.; Zhang, J.Q.; Yang, Y.T. Si96: A New Silicon Allotrope with Interesting Physical Properties. Materials 2016, 9, 284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Zhou, P.; Li, J.; He, C.Y.; Zhong, J.X. Si-Cmma: A silicon thin film with excellent stability and Dirac nodal loop. Phys. Rev. B 2019, 100, 115425. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Niu, R.; Zhang, W.Z.; Zhang, W.; Ding, Y.C.; Yun, S.N. t-Si64: A Novel Silicon Allotrope. ChemPhysChem 2019, 20, 128–133. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Zhang, W.Z.; Song, Y.X.; Zhang, W.; Yun, S.N. P63/mmc-Ge and their Si–Ge alloys with a mouldable direct band gap. Semicond. Sci. Technol. 2020, 35, 055012. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, Q.D. Optical, Electronic Properties and Anisotropy in Mechanical Properties of “X” Type Carbon Allotropes. Materials 2020, 13, 2079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Weng, K.Q.; Yang, Y.T. Theoretical investigations of Ge1-xSnx alloys (x = 0, 0.333, 0.667, 1) in P42/ncm phase. J. Mater. Sci. 2018, 53, 9611–9626. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Yang, R.L.; Zhang, W.; Yun, S.N. Elastic anisotropy and thermal conductivity of silicon allotropes. Results Phys. 2019, 15, 102580. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Wong, K.Q.; Liu, Y.Q.; Yang, Y.T. Theoretical investigations of group IV alloys in the Lonsdaleite phase. J. Mater. Sci. 2018, 53, 2785–2801. [Google Scholar] [CrossRef]
- Dai, J.; Wu, X.J.; Yang, J.L.; Zeng, X.C. Porous Boron Nitride with Tunable Pore Size. J. Phys. Chem. Lett. 2014, 5, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.J.; Chai, C.C.; Zhang, W.; Song, Y.X.; Yang, Y.T. First-Principles Study on Structural, Mechanical, Anisotropic, Electronic and Thermal Properties of III-Phosphides: XP (X = Al, Ga, or In) in the P6422 Phase. Materials 2020, 13, 686. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, W.; Huai, P. Novel 3D metallic boron nitride containing only sp2 bonds. J. Phys. D Appl. Phys. 2017, 50, 385302. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, J.H.; Zhou, P.K.; Zhang, D.Y.; Yang, Y.T. A New Phase of GaN. J. Chem. 2016, 2016, 8612892. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Chai, C.C.; Wei, Q.; Yang, Y.T. Thermodynamic, elastic, elastic anisotropy and minimum thermal conductivity of β-GaN under high temperature. Chin. J. Phys. 2017, 55, 400–411. [Google Scholar] [CrossRef]
- Zhang, Q.D.; Zou, Y.C.; Fan, Q.Y.; Yang, Y.T. Physical Properties of XN (X = B, Al, Ga, In) in the Pm-3n phase: First-Principles Calculations. Materials 2020, 13, 1280. [Google Scholar] [CrossRef]
- Xiong, M.; Luo, K.; Pan, Y.L.; Liu, L.Y.; Gao, G.Y.; Yu, D.L.; He, J.L.; Xu, B.; Zhao, Z.S. Hard three-dimensional BN framework with one-dimensional metallicity. J. Alloys Compd. 2018, 731, 364–368. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Han, Z.; Liu, X.H.; Yu, X.H.; Wang, D.Y.; Tian, Y. Pnma-BN: Another Boron Nitride Polymorph with Interesting Physical Properties. Nanomaterials 2017, 7, 3. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Zhang, W.Z.; Yun, S.N.; Xu, J.; Song, Y.X. III-Nitride Polymorphs: XN (X=Al, Ga, In) in the Pnma Phase. Chem.-A Eur. J. 2018, 24, 17280–17287. [Google Scholar] [CrossRef]
- Yang, R.K.; Zhu, C.S.; Wei, Q.; Du, Z. A first-principles study of the properties of four predicted novel phases of AlN. J. Phys. Chem. Solids 2017, 104, 68–78. [Google Scholar] [CrossRef]
- Fan, Q.; Wei, Q.; Yan, H.; Zhang, M.; Zhang, Z.; Zhang, J.; Zhang, D. Elastic and electronic properties of Pbca-BN: First-principles calculations. Comput. Mater. Sci. 2014, 85, 80–87. [Google Scholar] [CrossRef]
- Yang, R.K.; Zhu, C.S.; Wei, Q.; Zhang, D.Y. First-principles study on phases of AlP. Solid State Commun. 2017, 267, 23–28. [Google Scholar] [CrossRef]
- Xu, L.F.; Bu, W. Mechanical and thermodynamic properties of AlX (X = N, P, As) compounds. Int. J. Mod. Phys. B 2017, 31, 1750167. [Google Scholar] [CrossRef]
- Louhibi-Fasla, S.; Achour, H.; Kefif, K.; Ghalem, Y. First-principles study of high-pressure phases of AlN. Phys. Proced. 2014, 55, 324–328. [Google Scholar] [CrossRef]
- Li, Z.; Gao, F. Structure, bonding, vibration and ideal strength of primitive-centered tetragonal boron nitride. Phys. Chem. Chem. Phys. 2012, 14, 869–876. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892R–7895R. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the Quasi-Newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M.J. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Petrescu, M.L. Boron nitride theoretical hardness compared to carbon polymorphs. Diam. Relat. Mater. 2004, 13, 1848–1853. [Google Scholar] [CrossRef]
- Sherwin, M.E.; Drummond, T.J. Predicted elastic constants and critical layer thicknesses for cubic phase AlN, GaN, and InN on β-SiC. J. Appl. Phys. 1991, 69, 8423–8425. [Google Scholar] [CrossRef]
- Bundy, F.P.; Wentorf, R.H., Jr. Direct transformation of hexagonal boron nitride to denser forms. J. Chem. Phys. 1963, 38, 1144–1149. [Google Scholar] [CrossRef]
- Bungaro, C.; Rapcewicz, K.; Bernholc, J. Ab initio phonon dispersions of wurtzite AlN, GaN, and InN. Phys. Rev. B 2000, 61, 6720–6725. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Grimsditch, M.; Zouboulis, E.S.; Polian, A. Elastic constants of boron nitride. J. Appl. Phys. 1994, 76, 832–834. [Google Scholar] [CrossRef]
- Wright, A.F. Elastic properties of zinc-blende and wurtzite AlN, GaN, and InN. J. Appl. Phys. 1997, 82, 2833–2839. [Google Scholar] [CrossRef]
- Shimada, K.; Sota, T.; Suzuki, K. First-principles study on electronic and elastic properties of BN, AlN, and GaN. J. Appl. Phys. 1998, 84, 4951–4958. [Google Scholar] [CrossRef]
- Wu, Z.H.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Paudel, T.R.; Lambrecht, W.R.L. Computational study of phonon modes in short-period AlN/GaN superlattices. Phys. Rev. B 2009, 80, 104202. [Google Scholar] [CrossRef]
- Davydov, V.Y.; Roginskii, E.M.; Smirnov, A.N.; Kitaev, Y.E.; Yagovkina, M.A.; Kyutt, R.N.; Rozhavskaya, M.M.; Zavarin, E.E.; Lundin, W.V.; Smirnov, M.B. Lattice dynamics of short-period AlN/GaN superlattices: Theory and experiment. Phys. Status Solidi A 2013, 210, 484–487. [Google Scholar] [CrossRef]
- Davydov, V.Y.; Roginskii, E.M.; Kitaev, Y.E.; Smirnov, A.N.; Eliseyev, I.A.; Nechaev, D.V.; Jmerik, V.N.; Zavarin, E.E.; Lundin, W.V. Phonons in short-period (GaN)m(AlN)n superlattices: Ab initio calculations and group-theoretical analysis of modes and their genesis. J. Phys. Conf. Ser. 2019, 1400, 066016. [Google Scholar] [CrossRef]
- Voigt, W. Lehrburch der Kristallphysik; Teubner, B.G., Ed.; Johnson Reprint Corp: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. J. Appl. Math. Mech. 1929, 9, 49–58. (In German) [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Phys. Soc. Lond. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Hu, W.C.; Liu, Y.; Li, D.J.; Zeng, X.Q.; Xu, C.S. First-principles study of structural and electronic properties of C14-type Laves phase Al2Zr and Al2Hf. Comput. Mater. Sci. 2014, 83, 27–34. [Google Scholar] [CrossRef]
- Duan, Y.H.; Sun, Y.; Peng, M.J.; Zhou, S.G. Anisotropic elastic properties of the Ca–Pb compounds. J. Alloys Compd. 2014, 595, 14–21. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Wang, P.; Yan, F.; Shi, C.L.; Tian, Y. Physical properties of B4N4-I and B4N4-II: First-principles study. Chin. Phys. B 2019, 28, 036101. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Zuo, J.; Tang, C.Z.; Wang, P.; Shi, C.L. Physical properties of a novel phase of boron nitride and its potential applications. Mater. Chem. Phys. 2020, 252, 123245. [Google Scholar] [CrossRef]
- Fan, Q.Y.; Xu, J.; Zhang, W.Z.; Song, Y.X.; Yun, S.N. Physical properties of group 14 semiconductor alloys in orthorhombic phase. J. Appl. Phys. 2019, 126, 045709. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Zuo, J.; Wang, P.; Shi, C. Physical properties of Ima2-BN under pressure: First principles calculations. Chin. J. Phys. 2019, 59, 317–324. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Methods | a | b | c | c/a | ρ | V |
|---|---|---|---|---|---|---|---|
| Pbca BN | GGA | 5.110 | 4.434 | 4.399 | 3.308 | 12.459 | |
| Pbca AlN | GGA | 6.183 | 5.428 | 5.291 | 3.067 | 22.192 | |
| Pbca GaN | GGA | 6.428 | 5.602 | 5.518 | 5.599 | 24.837 | |
| Pbca InN | GGA | 7.172 | 6.262 | 6.150 | 6.196 | 34.525 | |
| P42/mnm BN | GGA | 4.421 | 2.548 | 3.309 | 12.203 | ||
| P42/mnm AlN | GGA | 5.320 | 3.116 | 3.087 | 22.049 | ||
| P42/mnm GaN | GGA | 5.547 | 3.221 | 5.612 | 24.779 | ||
| P42/mnm InN | GGA | 6.184 | 3.608 | 6.202 | 34.493 | ||
| c-BN | GGA | 3.623 | 11.884 | ||||
| LDA | 3.576 | 11.436 | |||||
| Exp. a | 3.620 | ||||||
| c-AlN | GGA | 4.396 | 3.206 | 21.229 | |||
| c-GaN | GGA | 4.557 | 5.878 | 23.658 | |||
| LDA | 4.523 | 23.132 | |||||
| Exp. b | 4.520 | 23.332 | |||||
| c-InN | GGA | 5.088 | 6.496 | 32.932 | |||
| Pm-3n BN | GGA c | 4.438 | 2.829 | 14.569 | |||
| Pm-3n AlN | GGA c | 5.366 | 2.643 | 25.751 | |||
| Pm-3n GaN | GGA c | 5.584 | 4.793 | 29.015 | |||
| Pm-3n InN | GGA c | 6.237 | 5.291 | 40.428 | |||
| Pnma BN | GGA d | 4.890 | 2.589 | 4.284 | 3.040 | 13.557 | |
| Pnma AlN | GGA e | 5.775 | 3.198 | 5.123 | 2.828 | 24.068 | |
| Pnma GaN | GGA e | 6.076 | 3.302 | 5.421 | 5.114 | 27.190 | |
| Pnma InN | GGA e | 6.777 | 3.699 | 6.051 | 5.642 | 37.918 | |
| wurtzite BN | GGA | 2.539 | 4.200 | 1.654 | |||
| Exp. f | 2.550 | 4.199 | 1.647 | ||||
| wurtzite AlN | GGA | 3.103 | 5.010 | 1.615 | |||
| Exp. g | 3.110 | 4.980 | 1.601 | ||||
| wurtzite GaN | GGA | 3.186 | 5.223 | 1.639 | |||
| Exp. g | 3.190 | 5.200 | 1.630 | ||||
| wurtzite InN | GGA | 3.547 | 5.726 | 1.614 | |||
| Exp. g | 3.540 | 5.710 | 1.613 |
| C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | B/G | E | v | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pbca BN | 772 | 135 | 139 | 885 | 92 | 716 | 312 | 257 | 357 | 344 | 316 | 1.09 | 718 | 0.140 |
| Pbca AlN | 317 | 122 | 92 | 369 | 94 | 275 | 109 | 118 | 103 | 174 | 109 | 1.60 | 271 | 0.241 |
| Pbca GaN | 268 | 117 | 92 | 320 | 91 | 258 | 84 | 88 | 92 | 160 | 89 | 1.80 | 225 | 0.265 |
| Pbca InN | 170 | 95 | 72 | 202 | 75 | 154 | 45 | 50 | 49 | 111 | 48 | 2.31 | 126 | 0.311 |
| P42/mnm BN | 747 | 144 | 99 | 747 | 958 | 332 | 244 | 347 | 318 | 1.09 | 731 | 0.149 | ||
| P42/mnm AlN | 268 | 116 | 114 | 268 | 384 | 99 | 129 | 177 | 102 | 1.74 | 257 | 0.258 | ||
| P42/mnm GaN | 251 | 90 | 93 | 251 | 334 | 79 | 93 | 153 | 87 | 1.76 | 219 | 0.261 | ||
| P42/mnm InN | 161 | 59 | 80 | 161 | 206 | 42 | 55 | 106 | 48 | 2.21 | 125 | 0.303 | ||
| c-BN | 805 | 187 | 468 | 393 | 396 | 0.99 | 889 | 0.123 | ||||||
| 820a | 190 | 480 | 400 | |||||||||||
| c-AlN | 280 | 146 | 184 | 190 | 123 | 1.54 | 304 | 0.234 | ||||||
| 294b | 160 | 189 | ||||||||||||
| c-GaN | 247 | 127 | 150 | 167 | 104 | 1.61 | 258 | 0.242 | ||||||
| 263c | 145 | 156 | 184 | 106 | 1.74 | 267 | 0.258 | |||||||
| 285d | 161 | 149 | 202e | 105e | ||||||||||
| c-InN | 152 | 95 | 86 | 114 | 55 | 2.07 | 142 | 0.292 | ||||||
| Pm-3n BN | 700 f | 85 | 209 | 290 | 244 | 1.19 | 572 | 0.171 | ||||||
| Pm-3n AlN | 335 f | 59 | 58 | 151 | 83 | 1.82 | 210 | 0.268 | ||||||
| Pm-3n GaN | 238 f | 61 | 58 | 120 | 69 | 1.74 | 174 | 0.259 | ||||||
| Pm-3n InN | 173 f | 55 | 36 | 95 | 44 | 2.16 | 114 | 0.299 | ||||||
| Pnma BN | 392 g | 99 | 256 | 770 | 116 | 675 | 299 | 272 | 187 | 298 | 227 | 1.31 | 543 | 0.196 |
| Pnma AlN | 160 h | 68 | 133 | 332 | 137 | 282 | 92 | 118 | 82 | 149 | 80 | 1.86 | 204 | 0.272 |
| Pnma GaN | 152 h | 59 | 116 | 278 | 119 | 267 | 68 | 93 | 67 | 133 | 69 | 1.93 | 176 | 0.279 |
| Pnma InN | 109 h | 48 | 81 | 173 | 92 | 165 | 34 | 52 | 38 | 94 | 38 | 2.47 | 100 | 0.322 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Ai, X.; Zhou, J.; Yu, X.; Zhang, W.; Yun, S. Novel III-V Nitride Polymorphs in the P42/mnm and Pbca Phases. Materials 2020, 13, 3743. https://doi.org/10.3390/ma13173743
Fan Q, Ai X, Zhou J, Yu X, Zhang W, Yun S. Novel III-V Nitride Polymorphs in the P42/mnm and Pbca Phases. Materials. 2020; 13(17):3743. https://doi.org/10.3390/ma13173743
Chicago/Turabian StyleFan, Qingyang, Xin Ai, Junni Zhou, Xinhai Yu, Wei Zhang, and Sining Yun. 2020. "Novel III-V Nitride Polymorphs in the P42/mnm and Pbca Phases" Materials 13, no. 17: 3743. https://doi.org/10.3390/ma13173743
APA StyleFan, Q., Ai, X., Zhou, J., Yu, X., Zhang, W., & Yun, S. (2020). Novel III-V Nitride Polymorphs in the P42/mnm and Pbca Phases. Materials, 13(17), 3743. https://doi.org/10.3390/ma13173743