First-Principles Study on III-Nitride Polymorphs: AlN/GaN/InN in the Pmn21 Phase



Abstract
1. Introduction
2. Computational Methods
3. Results
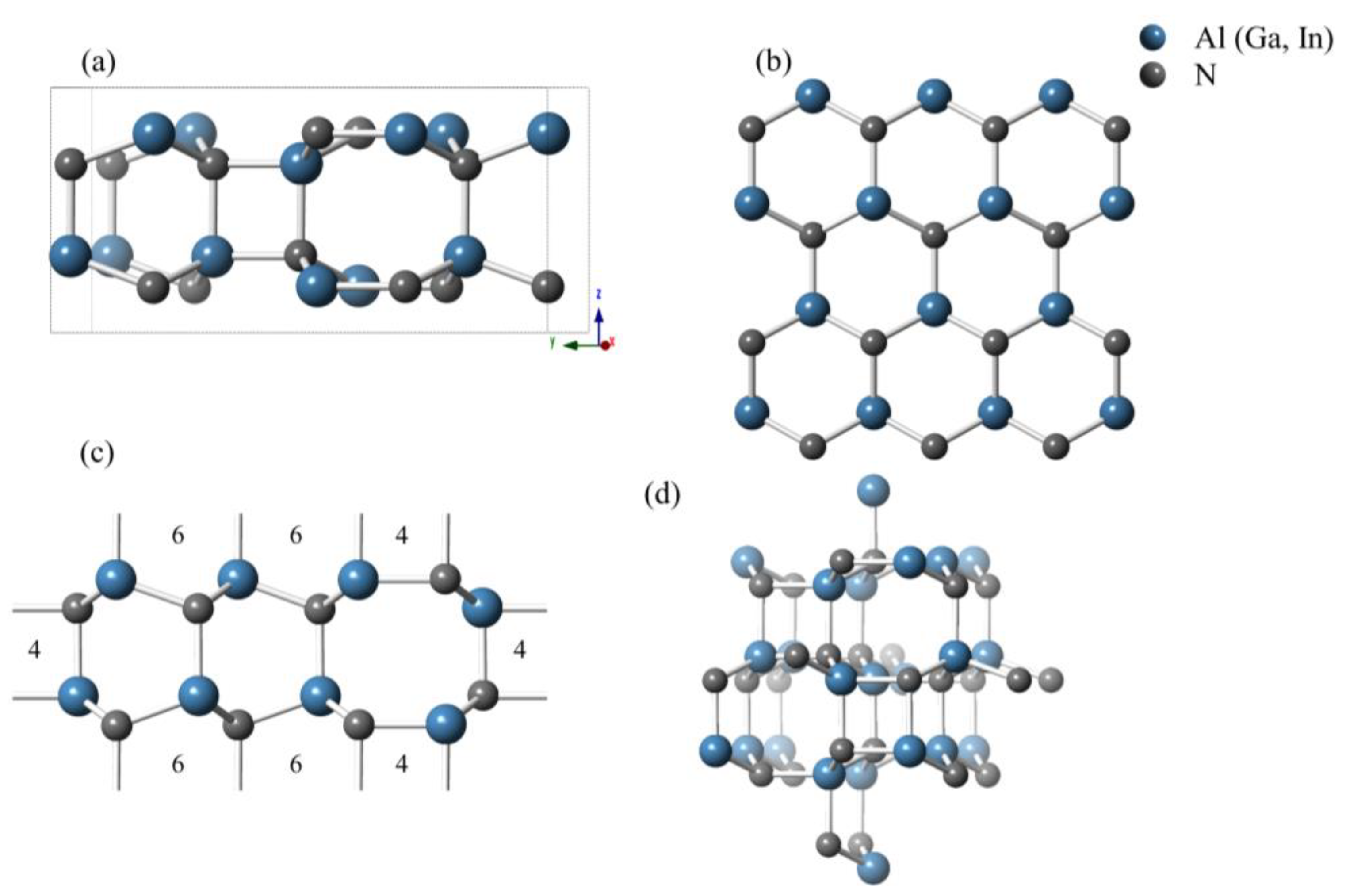
3.1. Structural Properties
3.2. Stability and Mechanical Properties
3.3. Mechanical Anisotropic Properties
3.4. Electrical and Thermal Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, H.-W.; Lu, Y.-J.; Chen, H.-Y.; Lee, H.-M.; Gwo, S. InGaN/GaN nanorod array white light-emitting diode. Appl. Phys. Lett. 2010, 97, 073101. [Google Scholar]
- Serrano, J.; Rubio, A.; Hernández, E.; Muñoz, A.; Mujica, A. Theoretical study of the relative stability of structural phases in group-III nitrides at high pressures. Phys. Rev. B 2000, 62, 16612. [Google Scholar] [CrossRef]
- Ueno, M.; Yoshida, M.; Onodera, A.; Shimomura, O.; Takemura, K. Stability of the wurtzite-type structure under high pressure: GaN and InN. Phys. Rev. B 1994, 49, 14. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.C.; Song, Y.X.; Fan, Q.Y.; Yang, Y.T. Structural, Mechanical, Anisotropic, and Thermal Properties of AlAs in oC12 and hP6 Phases under Pressure. Materials 2018, 11, 740. [Google Scholar] [CrossRef]
- Takahashi, N.; Matsumoto, Y.; Nakamura, T. Investigations of structure and morphology of the AlN nano-pillar crystal films prepared by halide chemical vapor deposition under atmospheric pressure. J. Phys. Chem. Solids 2006, 67, 665–668. [Google Scholar] [CrossRef]
- Miao, J.; Chai, C.; Zhang, W.; Song, Y.; Yang, Y. First-Principles Study on Structural, Mechanical, Anisotropic, Electronic and Thermal Properties of III-Phosphides: XP (X = Al, Ga, or In) in the P6422 Phase. Materials 2020, 13, 686. [Google Scholar] [CrossRef]
- Muranaka, T.; Kikuchi, Y.; Yoshizawa, T.; Shirakawa, N.; Akimitsu, J. Superconductivity in carrier-doped silicon carbide. Sci. Technol. Adv. Mater. 2009, 9, 044204. [Google Scholar] [CrossRef]
- Morkoc, H.; Strite, S.; Gao, G.; Lin, M.; Sverdlov, B.; Burns, M. Large-band-gap SiC, III-V nitride, and II-VI ZnSe-based semiconductor device technologies. J. Appl. Phys. 1994, 76, 1363–1398. [Google Scholar] [CrossRef]
- Zhang, W.; Ristein, J.; Ley, L. Hydrogen-terminated diamond electrodes. II. Redox activity. Phys. Rev. E 2008, 78, 041603. [Google Scholar] [CrossRef]
- Collins, A.T. The optical and electronic properties of semiconducting diamond. Philos. Trans. R. Soc. Lond. Ser. A Phys. Eng. Sci. 1993, 342, 233–244. [Google Scholar]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Weng, K.Q.; Yang, Y.T. Theoretical investigations of Ge1-x Sn (x) alloys (x = 0, 0.333, 0.667, 1) in P4(2)/ncm phase. J. Mater. Sci. 2018, 53, 9611–9626. [Google Scholar] [CrossRef]
- Song, Y.X.; Chai, C.C.; Fan, Q.Y.; Zhang, W.; Yang, Y.T. Physical properties of Si-Ge alloys in C2/m phase: A comprehensive investigation. J. Phys. Condes. Matter 2019, 31, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chai, C.C.; Fan, Q.Y.; Song, Y.X.; Yang, Y.T. PBCF-Graphene: A 2D Sp(2) Hybridized Honeycomb Carbon Allotrope with a Direct Band Gap. ChemNanoMat 2020, 6, 139–147. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. Two novel superhard carbon allotropes with honeycomb structures. J. Appl. Phys. 2019, 126, 145704. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. Penta-C20: A Superhard Direct Band Gap Carbon Allotrope Composed of Carbon Pentagon. Materials 2020, 13, 1926. [Google Scholar] [CrossRef]
- Zhang, W.; Chai, C.; Fan, Q.; Song, Y.; Yang, Y. Six novel carbon and silicon allotropes with their potential application in photovoltaic field. J. Phys. Condens. Matter 2020, 32, 35. [Google Scholar] [CrossRef]
- Fan, Q.; Zhao, Y.; Yu, X.; Song, Y.; Zhang, W.; Yun, S. Physical properties of a novel microporous carbon material. Diam. Relat. Mater. 2020, 106, 107831. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, W.; Song, Y.; Zhang, W.; Yun, S. P63/mmc-Ge and their Si–Ge alloys with a mouldable direct band gap. Semicond. Sci. Technol. 2020, 35, 055012. [Google Scholar] [CrossRef]
- Fan, Q.; Wang, H.; Song, Y.; Zhang, W.; Yun, S. Five carbon allotropes from Squaroglitter structures. Comput. Mater. Sci. 2020, 178, 109634. [Google Scholar] [CrossRef]
- Liu, H.; Fan, Q.-Y.; Yang, F.; Yu, X.-H.; Zhang, W.; Yun, S.-N. tP40 carbon: A novel superhard carbon allotrope. Chin. Phys. B 2020. [Google Scholar] [CrossRef]
- Yang, G.; Chen, B.F. Predicted a novel high-pressure superhard boron nitride phase. J. Alloys Compd. 2014, 598, 54–56. [Google Scholar] [CrossRef]
- Liu, C.; Ma, M.; Yuan, X.; Sun, H.; Ying, P.; Xu, B.; Zhao, Z.; He, J. Metastable phases, phase transformation and properties of AlAs based on first-principle study. Comput. Mater. Sci. 2017, 128, 337–342. [Google Scholar] [CrossRef]
- Xu, L.; Bu, W. Mechanical and thermodynamic properties of AlX (X = N, P, As) compounds. Int. J. Mod. Phys. B 2017, 31, 1750167. [Google Scholar] [CrossRef]
- Zhang, Q.; Zou, Y.; Fan, Q.; Yang, Y. Physical Properties of XN (X = B, Al, Ga, In) in the Pm−3n phase: First-Principles Calc. Materials 2020, 13, 1280. [Google Scholar] [CrossRef]
- Zeng, Y.; Chiu, H.-C.; Rasool, M.; Brodusch, N.; Gauvin, R.; Jiang, D.-T.; Ryan, D.H.; Zaghib, K.; Demopoulos, G.P. Hydrothermal crystallization of Pmn21 Li2FeSiO4 hollow mesocrystals for Li-ion cathode application. Chem. Eng. J. 2019, 359, 1592–1602. [Google Scholar] [CrossRef]
- Pandey, M.; Ramar, V.; Balaya, P.; Kshirsagar, R.J. Infrared Spectroscopy of Li2MnSiO4: A cathode material for Li ion batteries. AIP Conf. Proc. 2015, 1665, 140044. [Google Scholar]
- Fisher, C.A.; Kuganathan, N.; Islam, M.S. Defect chemistry and lithium-ion migration in polymorphs of the cathode material Li2MnSiO4. J. Mater. Chem. A 2013, 1, 4207–4214. [Google Scholar] [CrossRef]
- Yang, R.; Zhu, C.; Wei, Q.; Du, Z. First-principles study of the properties of Pmn2 1-B1–x Al x N. Philos. Mag. 2017, 97, 3008–3026. [Google Scholar] [CrossRef]
- Chittari, B.L. Dynamical stability of Pmn21 phase of NH3BH3: A vdW density functional study. Philos. Mag. Lett. 2014, 94, 278–287. [Google Scholar] [CrossRef]
- Liu, C.; Hu, M.; Luo, K.; Cui, L.; Yu, D.; Zhao, Z.; He, J. Novel high-pressure phases of AlN: A first-principles study. Comput. Mater. Sci. 2016, 117, 496–501. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Density Functional Theory (DFT). Phys. Rev. B 1964, 136, 864. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Ceperley, D.M.; Alder, B.J. Ground state of the electron gas by a stochastic method. Phys. Rev. Lett. 1980, 45, 566. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Baroni, S.; De Gironcoli, S.; Dal Corso, A.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Karch, K.; Bechstedt, F. Ab initio lattice dynamics of BN and AlN: Covalent versus ionic forces. Phys. Rev. B 1997, 56, 7404. [Google Scholar] [CrossRef]
- Edgar, J.H. Properties of Group-III Nitrides; Institution of Electrical Engineers: London, UK, 1997. [Google Scholar]
- Lei, T.; Fanciulli, M.; Molnar, R.; Moustakas, T.; Graham, R.; Scanlon, J. Epitaxial growth of zinc blende and wurtzitic gallium nitride thin films on (001) silicon. Appl. Phys. Lett. 1991, 59, 944–946. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, W.; Yun, S.; Xu, J.; Song, Y. III-Nitride Polymorphs: XN (X = Al, Ga, In) in the Pnma Phase. Chem. A Eur. J. 2018, 24, 17280–17287. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Chai, C.; Wei, Q.; Yang, J.; Zhou, P.; Zhang, D.; Yang, Y. A new phase of GaN. J. Chem. 2016, 2016, 8612892. [Google Scholar] [CrossRef]
- Wright, A. Elastic properties of zinc-blende and wurtzite AlN, GaN, and InN. J. Appl. Phys. 1997, 82, 2833–2839. [Google Scholar] [CrossRef]
- Shimada, K.; Sota, T.; Suzuki, K. First-principles study on electronic and elastic properties of BN, AlN, and GaN. J. Appl. Phys. 1998, 84, 4951–4958. [Google Scholar] [CrossRef]
- Polian, A.; Grimsditch, M.; Grzegory, I. Elastic constants of gallium nitride. J. Appl. Phys. 1996, 79, 3343–3344. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Korozlu, N.; Colakoglu, K.; Deligoz, E.; Aydin, S. The elastic and mechanical properties of MB12 (M = Zr, Hf, Y, Lu) as a function of pressure. J. Alloys Compd. 2013, 546, 157–164. [Google Scholar] [CrossRef]
- Watt, J.P. Hashin-Shtrikman bounds on the effective elastic moduli of polycrystals with monoclinic symmetry. J. Appl. Phys. 1980, 51, 1520–1524. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Lewandowski, J.; Wang, W.; Greer, A. Intrinsic plasticity or brittleness of metallic glasses. Philos. Mag. Lett. 2005, 85, 77–87. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Ting, T. On Anisotropic Elastic Materials for which Y’oung’s Modulus E (n) is Independent of n or the Shear Modulus G (n, m) is Independent of n and m. J. Elast. 2005, 81, 271–292. [Google Scholar] [CrossRef]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef]
- Takahashi, K.; Yoshikawa, A.; Sandhu, A. Wide Bandgap Semiconductors; Springer: Berlin/Heidelberg, Germany, 2007; Volume 239. [Google Scholar]
- Schubert, E.F. Light-Emitting Diodes, 2nd ed.; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Shan, W.; Schmidt, T.; Hauenstein, R.; Song, J.; Goldenberg, B. Pressure-dependent photoluminescence study of wurtzite GaN. Appl. Phys. Lett. 1995, 66, 3492–3494. [Google Scholar] [CrossRef]
- Morkoç, H. Handbook of Nitride Semiconductors and Devices, Materials Properties, Physics and Growth; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 1. [Google Scholar]
- Rinke, P.; Winkelnkemper, M.; Qteish, A.; Bimberg, D.; Neugebauer, J.; Scheffler, M. Consistent set of band parameters for the group-III nitrides AlN, GaN, and InN. Phys. Rev. B 2008, 77, 075202. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | a | b | c | ρ | |
|---|---|---|---|---|---|
| Pmn21-AlN | GGA | 3.130 | 10.832 | 5.099 | 3.150 |
| Pmn21-GaN | GGA | 3.231 | 11.202 | 5.352 | 5.742 |
| Pmn21-InN | GGA | 3.617 | 12.528 | 5.946 | 6.351 |
| wz-AlN | GGA | 3.125 | 5.009 | 3.206 | |
| LDA | 3.066 | 4.905 | 3.408 | ||
| Exp. a | 3.111 | 4.978 | |||
| wz-GaN | GGA | 3.227 | 5.258 | 5.865 | |
| LDA | 3.158 | 5.149 | 6.254 | ||
| Exp. a | 3.192 | 5.166 | |||
| wz-InN | GGA | 3.611 | 5.842 | 6.485 | |
| LDA | 3.531 | 5.714 | 6.936 | ||
| Exp. a | 3.533 | 5.639 | |||
| zb-AlN | GGA | 4.399 | 3.198 | ||
| LDA | 4.309 | 3.404 | |||
| Exp. b | 4.370 | ||||
| zb-GaN | GGA | 4.561 | 5.862 | ||
| LDA | 4.466 | 6.248 | |||
| Exp. c | 4.490 | ||||
| zb-InN | GGA | 5.092 | 6.482 | ||
| LDA | 4.977 | 9.642 |
| Structure | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | B | G | B/G | E | ν | Hv |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pmn21-AlN | 383 | 122 | 97 | 352 | 89 | 294 | 117 | 112 | 115 | 181 | 116 | 1.56 | 288 | 0.235 | 16.3 |
| wz-AlN | 376 | 118 | 88 | 353 | 113 | 129 | 188 | 125 | 1.50 | 307 | 0.227 | 18.0 | |||
| 345 a | 125 | 120 | 395 | 118 | 110 | 18 d | |||||||||
| zb-AlN | 276 | 143 | 181 | 187 | 121 | 1.54 | 299 | 0.233 | 17.0 | ||||||
| 304 b | 160 | 193 | |||||||||||||
| Pmn21-GaN | 333 | 109 | 84 | 299 | 75 | 294 | 87 | 85 | 94 | 162 | 97 | 1.68 | 241 | 0.251 | 12.8 |
| wz-GaN | 322 | 102 | 69 | 361 | 89 | 110 | 165 | 107 | 1.54 | 264 | 0.233 | 15.5 | |||
| 390 c | 145 | 106 | 398 | 105 | 123 | 15.1 d | |||||||||
| zb-GaN | 249 | 127 | 151 | 168 | 105 | 1.60 | 260 | 0.242 | 14.5 | ||||||
| 293 b | 159 | 155 | |||||||||||||
| Pmn21-InN | 207 | 91 | 74 | 185 | 68 | 168 | 42 | 42 | 47 | 113 | 48 | 2.38 | 125 | 0.316 | 3.9 |
| wz-InN | 194 | 90 | 64 | 202 | 48 | 52 | 114 | 54 | 2.12 | 140 | 0.296 | 5.6 | |||
| 223 b | 115 | 92 | 224 | 48 | 5.1 d | ||||||||||
| zb-InN | 154 | 101 | 87 | 119 | 54 | 2.18 | 142 | 0.301 | 5.3 | ||||||
| 187 b | 125 | 86 |
| xDirection | Hole Effective Mass | Electron Effective Mass |
| Pmn21/zinc-blende/Ref | Pmn21/zinc-blende/Ref | |
| AlN | 3.29/1.23/1.02 a | 0.32/0.33/0.23 a |
| GaN | 1.95/0.90/0.80 a | 0.16/0.14/0.14 a |
| InN | 1.14/0.94/1.08 a | 0.08/0.08/0.13 a |
| yDirection | Hole Effective Mass | Electron Effective Mass |
| Pmn21/zinc-blende | Pmn21/zinc-blende/Ref | |
| AlN | 5.31/1.23 | 0.31/0.33/0.23 a |
| GaN | 2.58/0.90 | 0.16/0.14/0.14 a |
| InN | 2.55/0.94 | 0.08/0.08/0.13 a |
| zDirection | Hole Effective Mass | Electron Effective Mass |
| Pmn21/zinc-blende/Ref | Pmn21/zinc-blende/Ref | |
| AlN | 0.27/1.23/1.33 b | 0.30/0.33/0.23 a |
| GaN | 0.15/0.90/0.81 b | 0.15/0.14/0.14 a |
| InN | 0.09/0.94/0.84 b | 0.08/0.08/0.13 a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Chai, C.; Zhang, W.; Song, Y.; Kong, L.; Yang, Y. First-Principles Study on III-Nitride Polymorphs: AlN/GaN/InN in the Pmn21 Phase. Materials 2020, 13, 3212. https://doi.org/10.3390/ma13143212
Zhang Z, Chai C, Zhang W, Song Y, Kong L, Yang Y. First-Principles Study on III-Nitride Polymorphs: AlN/GaN/InN in the Pmn21 Phase. Materials. 2020; 13(14):3212. https://doi.org/10.3390/ma13143212
Chicago/Turabian StyleZhang, Zheren, Changchun Chai, Wei Zhang, Yanxing Song, Linchun Kong, and Yintang Yang. 2020. "First-Principles Study on III-Nitride Polymorphs: AlN/GaN/InN in the Pmn21 Phase" Materials 13, no. 14: 3212. https://doi.org/10.3390/ma13143212
APA StyleZhang, Z., Chai, C., Zhang, W., Song, Y., Kong, L., & Yang, Y. (2020). First-Principles Study on III-Nitride Polymorphs: AlN/GaN/InN in the Pmn21 Phase. Materials, 13(14), 3212. https://doi.org/10.3390/ma13143212