Author Contributions
The authors contributions are as follows: conceptualization, M.M.; methodology, M.M., A.M. (Alexandre Maupu) and Y.K.; formal analysis, M.M., A.M. (Alexandre Maupu, A.M. (Adrien Metafiot)) and Y.K.; investigation, A.M. (Alexandre Maupu), A.M. (Adrien Metafiot) and Y.K.; data curation, A.M. (Alexandre Maupu), A.M. (Adrien Metafiot), Y.K. and M.M.; writing—original draft preparation, A.M. (Alexandre Maupu) and Y.K.; writing—review and editing, M.M. supervision, M.M.; project administration, M.M.; funding acquisition, M.M.
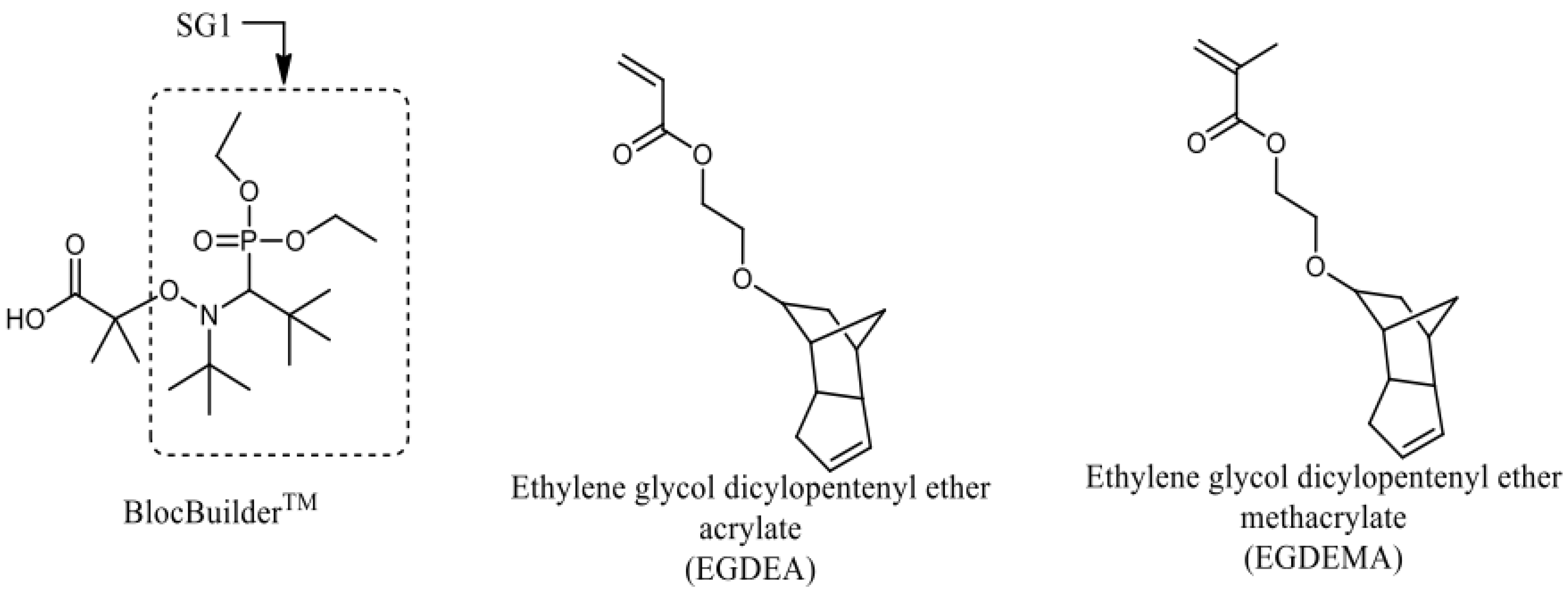
Figure 1.
Structures of BlocBuilder, SG1 free nitroxide and ethylene glycol dicyclopentenyl ether (meth)acrylate (EGDEA and EGDEMA).
Figure 1.
Structures of BlocBuilder, SG1 free nitroxide and ethylene glycol dicyclopentenyl ether (meth)acrylate (EGDEA and EGDEMA).
Figure 2.
1H NMR of a sample of the reaction mixture for the homopolymerization of EGDEA (Expt. ID: E-T90, temperature = 90 °C, tpolymerization = 150 min, Mn = 3.0 kg mol−1, Ð = 1.54).
Figure 2.
1H NMR of a sample of the reaction mixture for the homopolymerization of EGDEA (Expt. ID: E-T90, temperature = 90 °C, tpolymerization = 150 min, Mn = 3.0 kg mol−1, Ð = 1.54).
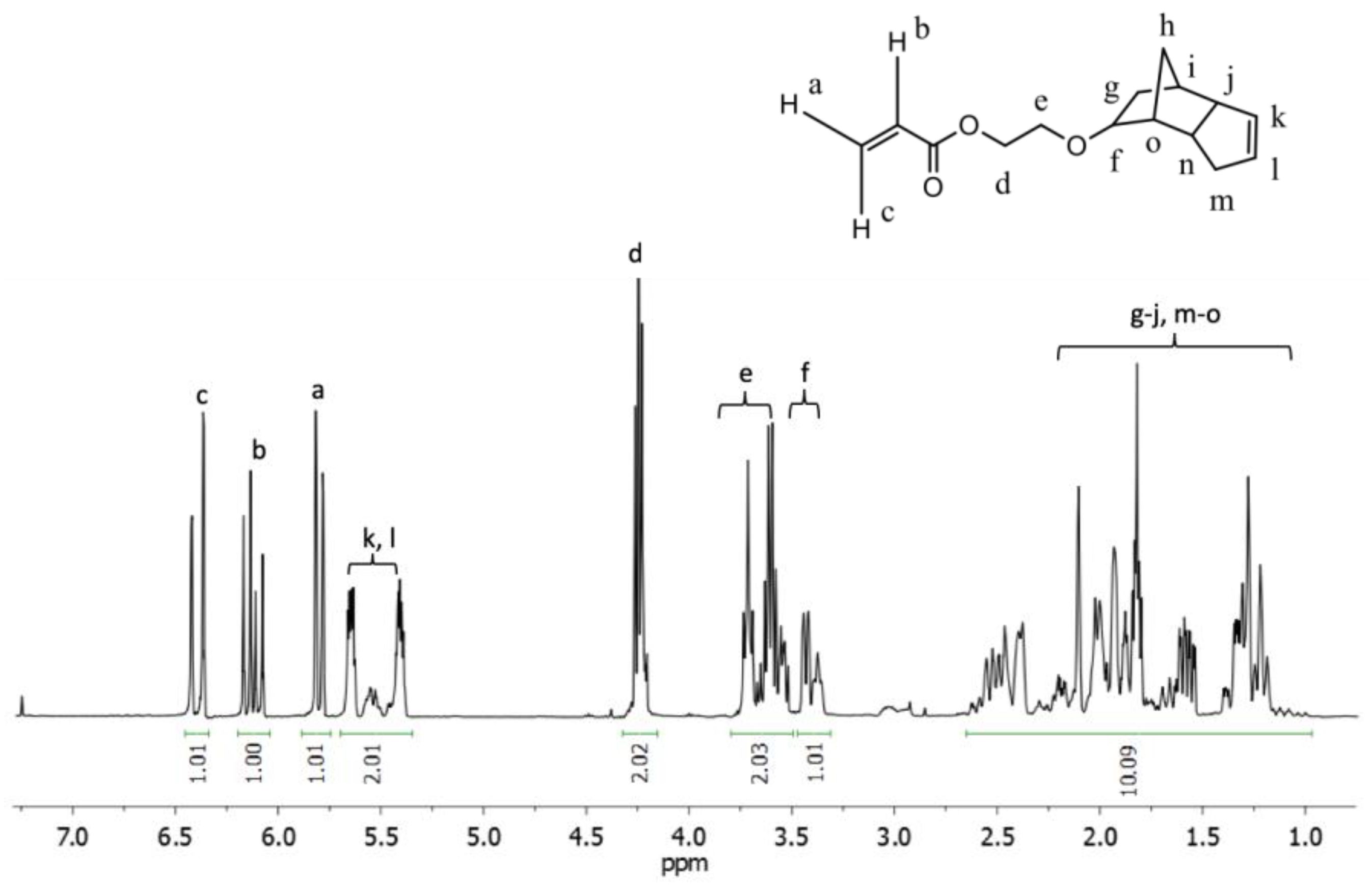
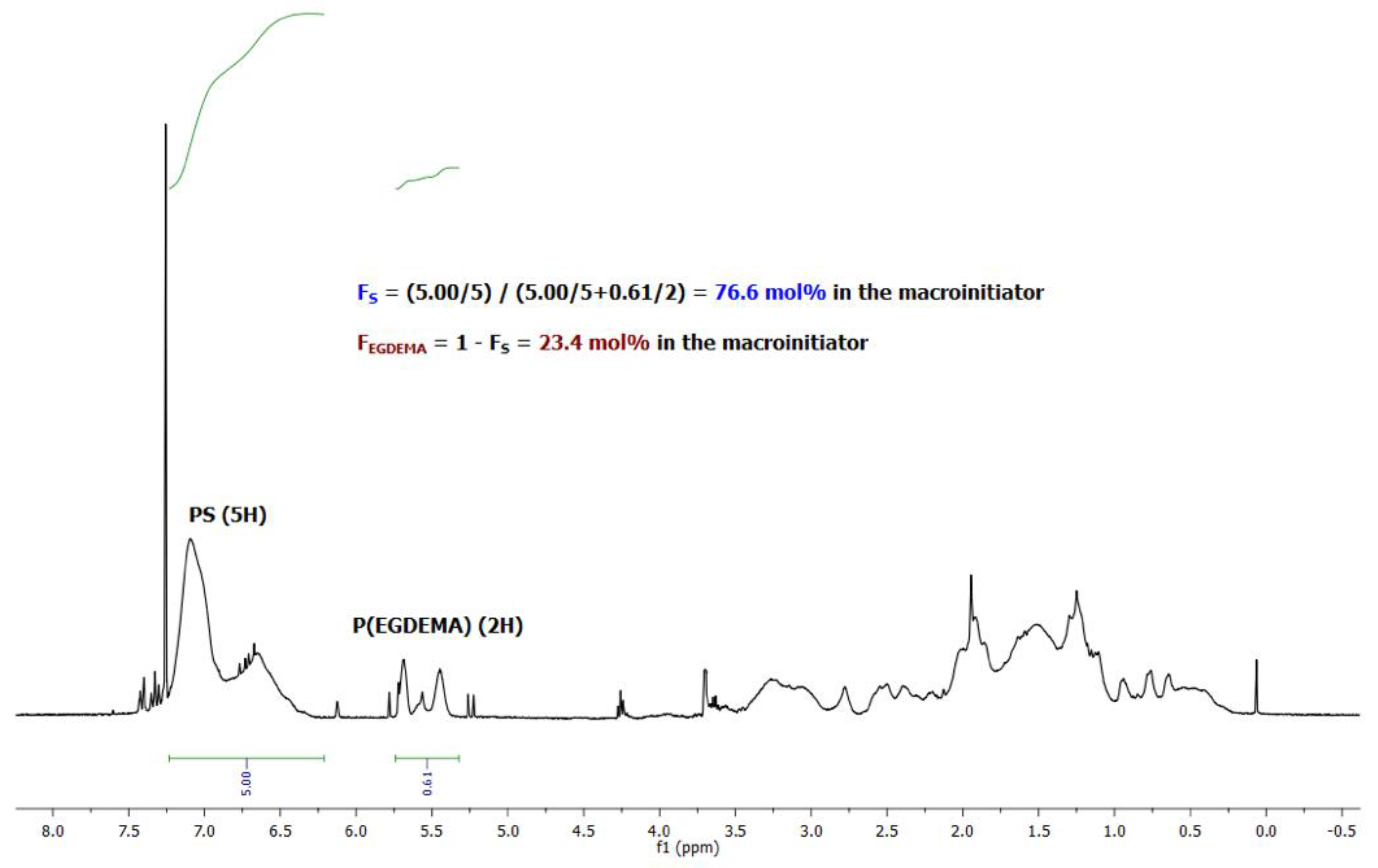
Figure 3.
1H NMR of an EGDEMA/S copolymer (EGDEMA/S-N-80-90) with Mn = 5.6 kg mol−1, Ð = 1.27, FS = 0.77. Note that the cyclopentenyl peaks at about 5.5 ppm were used to indicate EGDEMA composition while the aromatic protons at 6.5–7.0 ppm were used to indicate S composition.
Figure 3.
1H NMR of an EGDEMA/S copolymer (EGDEMA/S-N-80-90) with Mn = 5.6 kg mol−1, Ð = 1.27, FS = 0.77. Note that the cyclopentenyl peaks at about 5.5 ppm were used to indicate EGDEMA composition while the aromatic protons at 6.5–7.0 ppm were used to indicate S composition.
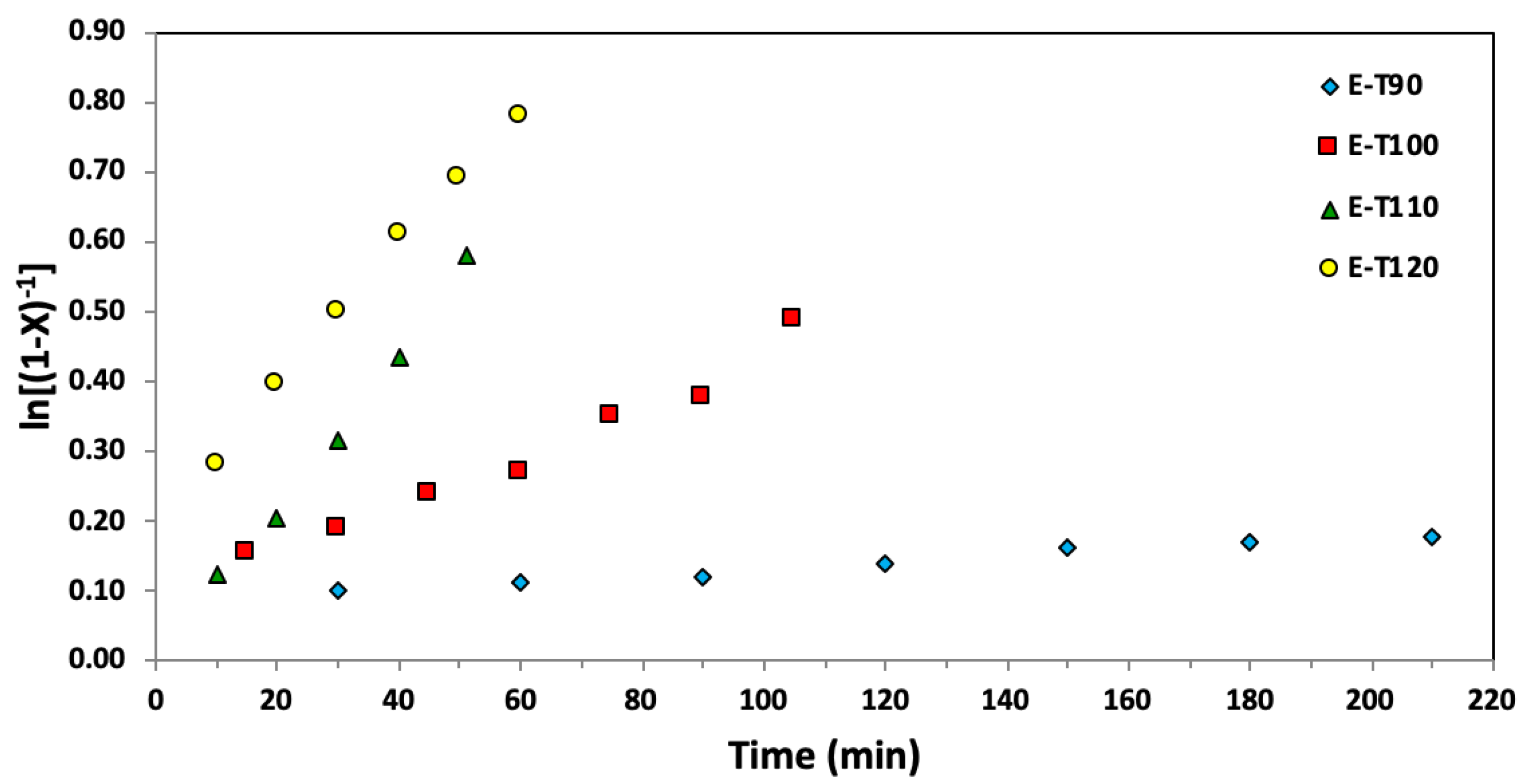
Figure 4.
Semi-logarithmic kinetic plots of ln[(1−X)−1], where X represents the overall monomer conversion, versus time for ethylene glycol dicyclopentenyl ether acrylate (EGDEA) homopolymerizations at 90–120 °C via nitroxide mediated polymerization with BlocBuilderTM and 5 mol% additional SG1 free nitroxide relative to BlocBuilderTM.
Figure 4.
Semi-logarithmic kinetic plots of ln[(1−X)−1], where X represents the overall monomer conversion, versus time for ethylene glycol dicyclopentenyl ether acrylate (EGDEA) homopolymerizations at 90–120 °C via nitroxide mediated polymerization with BlocBuilderTM and 5 mol% additional SG1 free nitroxide relative to BlocBuilderTM.
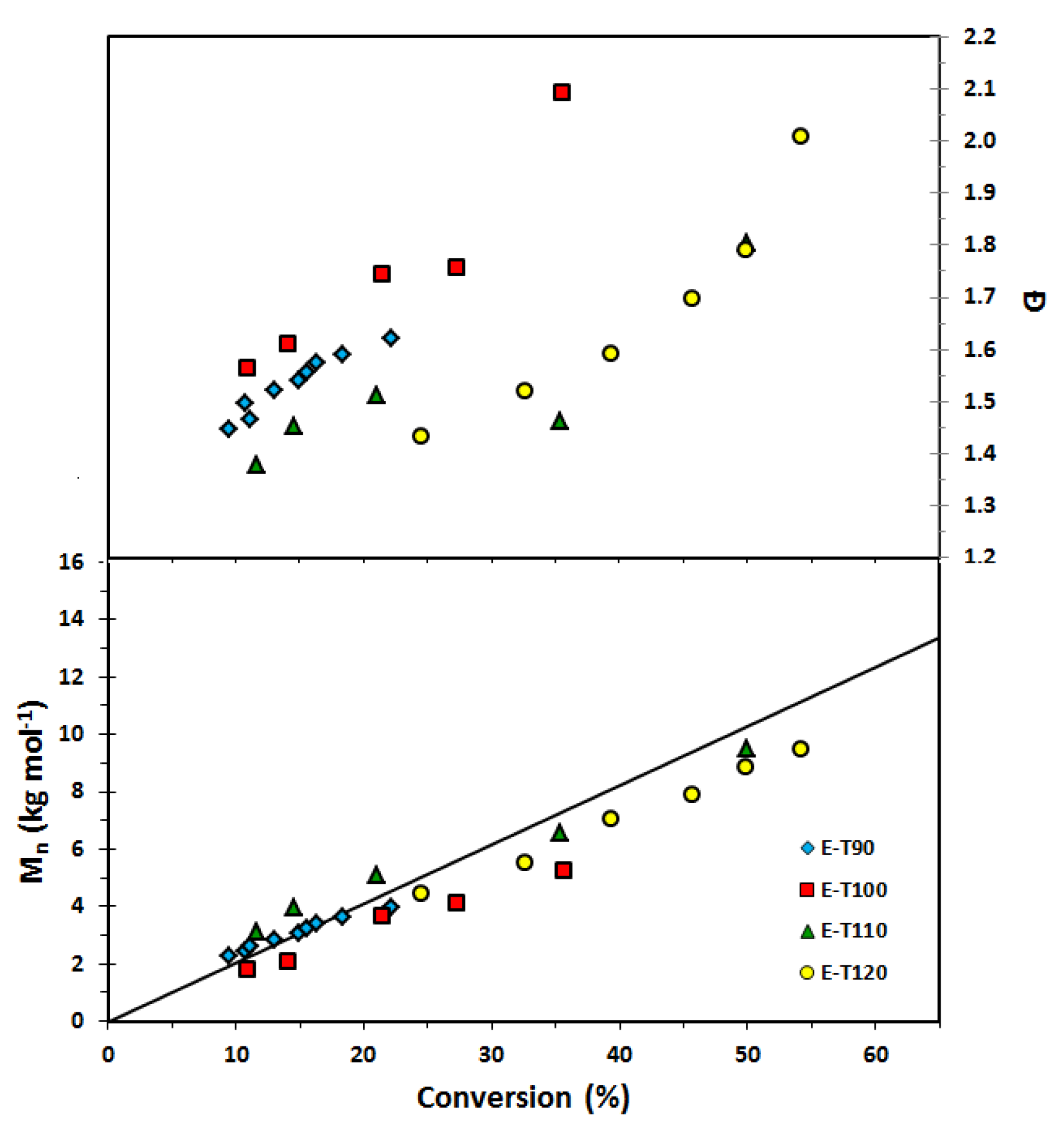
Figure 5.
Number average molecular weight (Mn) and dispersity (Ð) of homopolymers of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) with r = 0.05 (added free SG1 nitroxide relative to BlocBuilder initially) at various temperatures (E-T90 = EGDEA homopolymerization at 90 °C; E-T100 = EGDEA homopolymerization at 100 °C; E-T110 = EGDEA homopolymerization at 110 °C; E-T120 = EGDEA homopolymerization at 120 °C). The straight solid line indicates the predicted Mn versus conversion if the polymerization was living (Mn, theoretical at 100% conversion ≈ 20 kg mol−1 for this set of polymerizations).
Figure 5.
Number average molecular weight (Mn) and dispersity (Ð) of homopolymers of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) with r = 0.05 (added free SG1 nitroxide relative to BlocBuilder initially) at various temperatures (E-T90 = EGDEA homopolymerization at 90 °C; E-T100 = EGDEA homopolymerization at 100 °C; E-T110 = EGDEA homopolymerization at 110 °C; E-T120 = EGDEA homopolymerization at 120 °C). The straight solid line indicates the predicted Mn versus conversion if the polymerization was living (Mn, theoretical at 100% conversion ≈ 20 kg mol−1 for this set of polymerizations).
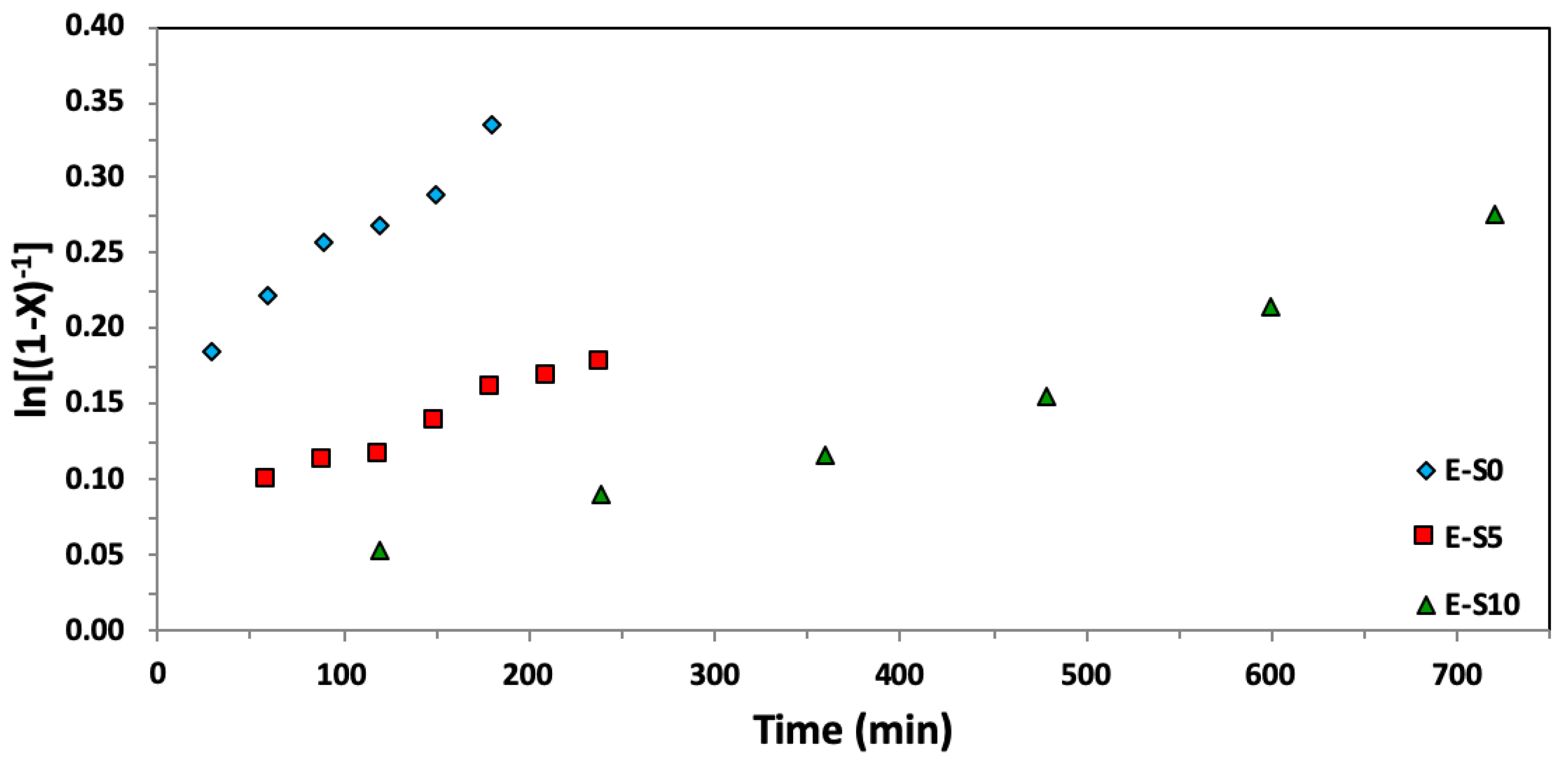
Figure 6.
Semi-logarithmic kinetic plots of ln[(1−X)−1], where X represents the overall monomer conversion, versus time for ethylene glycol dicyclopentenyl ether acrylate (EGDEA) homopolymerizations at 90 °C via Nitroxide-Mediated Polymerization with BlocBuilderTM and r = 0–10% excess SG1 free nitroxide relative to BlocBuilderTM.
Figure 6.
Semi-logarithmic kinetic plots of ln[(1−X)−1], where X represents the overall monomer conversion, versus time for ethylene glycol dicyclopentenyl ether acrylate (EGDEA) homopolymerizations at 90 °C via Nitroxide-Mediated Polymerization with BlocBuilderTM and r = 0–10% excess SG1 free nitroxide relative to BlocBuilderTM.
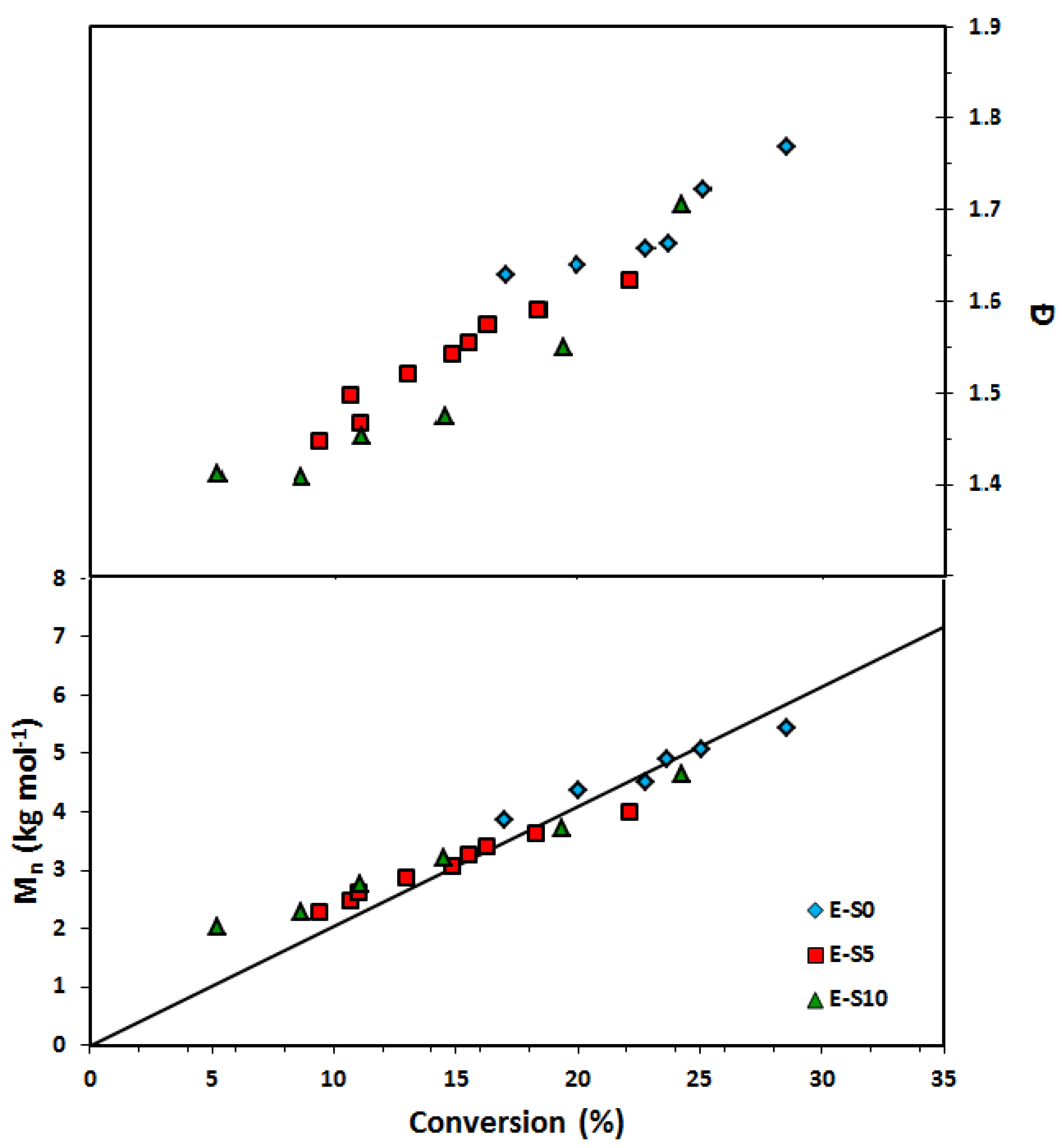
Figure 7.
Number average molecular weight (Mn) and dispersity (Ð) of homopolymers of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) measured by GPC calibrated with poly(methyl methacrylate) standards (symbols: E-0 (r = 0): blue diamonds, E-5: red squares (r = 0.05), E-10: green triangles (r = 0.10)).
Figure 7.
Number average molecular weight (Mn) and dispersity (Ð) of homopolymers of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) measured by GPC calibrated with poly(methyl methacrylate) standards (symbols: E-0 (r = 0): blue diamonds, E-5: red squares (r = 0.05), E-10: green triangles (r = 0.10)).
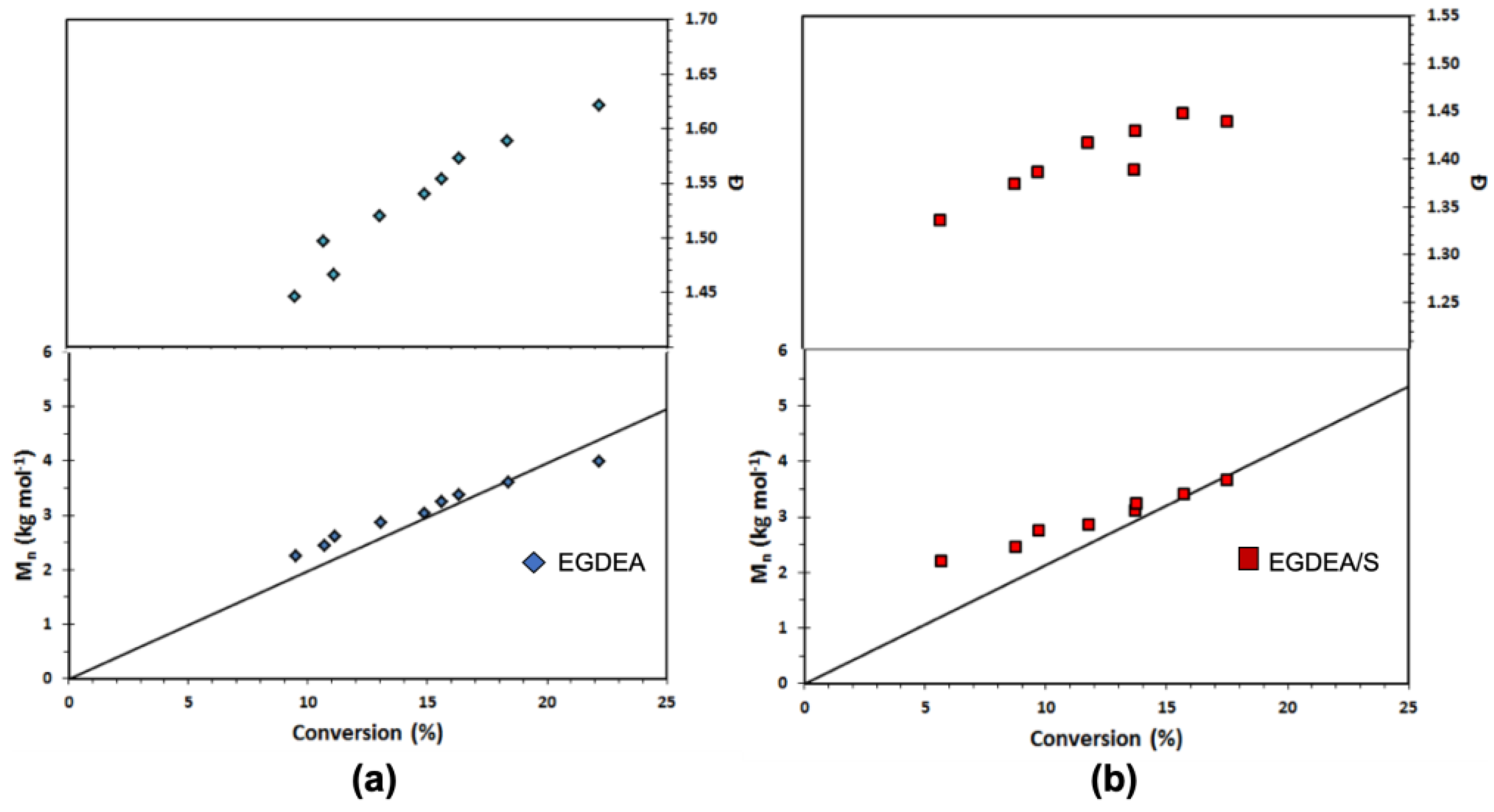
Figure 8.
Number-average molecular weight (
Mn) and dispersity (
Ð) versus conversion of (
a) EGDEA homopolymerization and (
b) EGDEA/S copolymerization with
fS,0 = 0.05 in the initial monomer composition at 90 °C. This data is referred to expt. IDs EGDEA and EGDEA/S in
Table 7.
Figure 8.
Number-average molecular weight (
Mn) and dispersity (
Ð) versus conversion of (
a) EGDEA homopolymerization and (
b) EGDEA/S copolymerization with
fS,0 = 0.05 in the initial monomer composition at 90 °C. This data is referred to expt. IDs EGDEA and EGDEA/S in
Table 7.
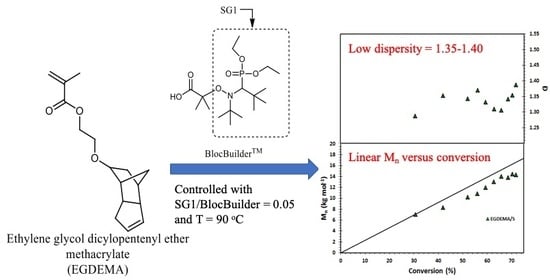
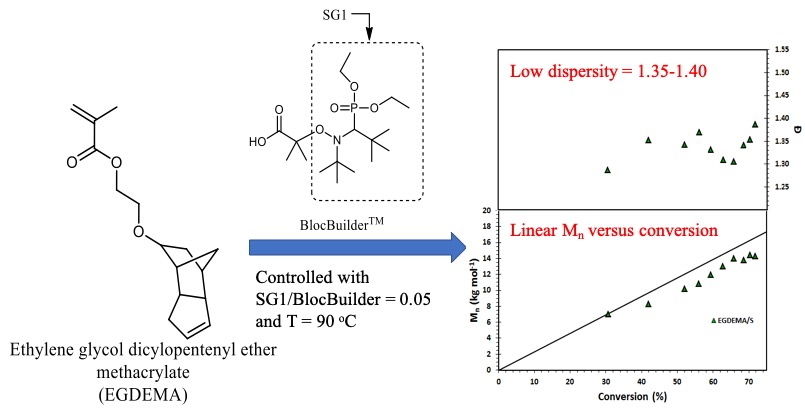
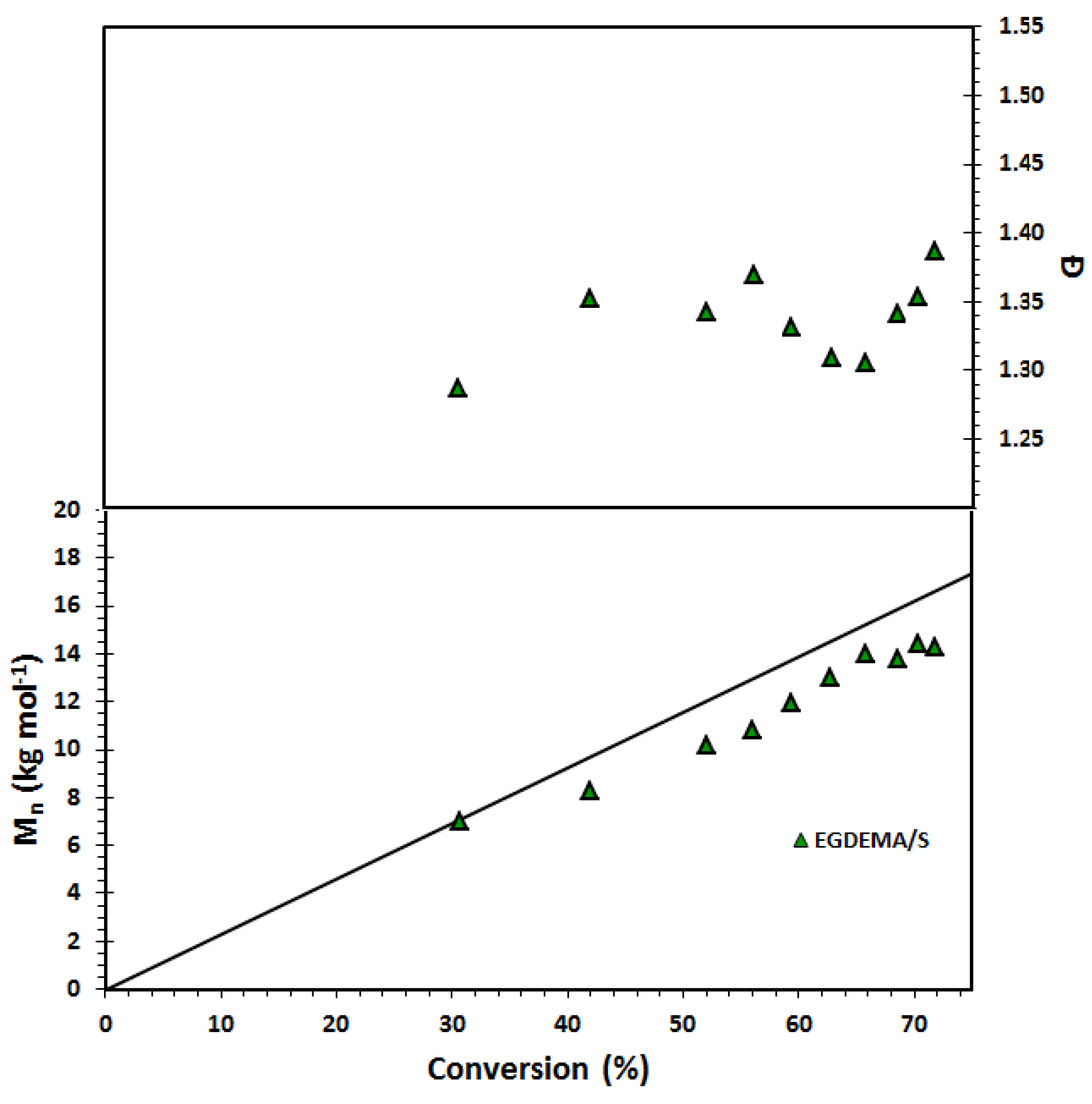
Figure 9.
Number-average molecular weight (
Mn) and dispersity (
Ð) of statistical copolymer of ethylene glycol dicyclopentenyl ether methacrylate (EGDEMA) and styrene (S) measured by GPC calibrated with poly(methyl methacrylate) standards (Expt. ID. EGDEMA/S in
Table 7). This polymerization was done at 90 °C in 50 wt.% dioxane solution.
Figure 9.
Number-average molecular weight (
Mn) and dispersity (
Ð) of statistical copolymer of ethylene glycol dicyclopentenyl ether methacrylate (EGDEMA) and styrene (S) measured by GPC calibrated with poly(methyl methacrylate) standards (Expt. ID. EGDEMA/S in
Table 7). This polymerization was done at 90 °C in 50 wt.% dioxane solution.
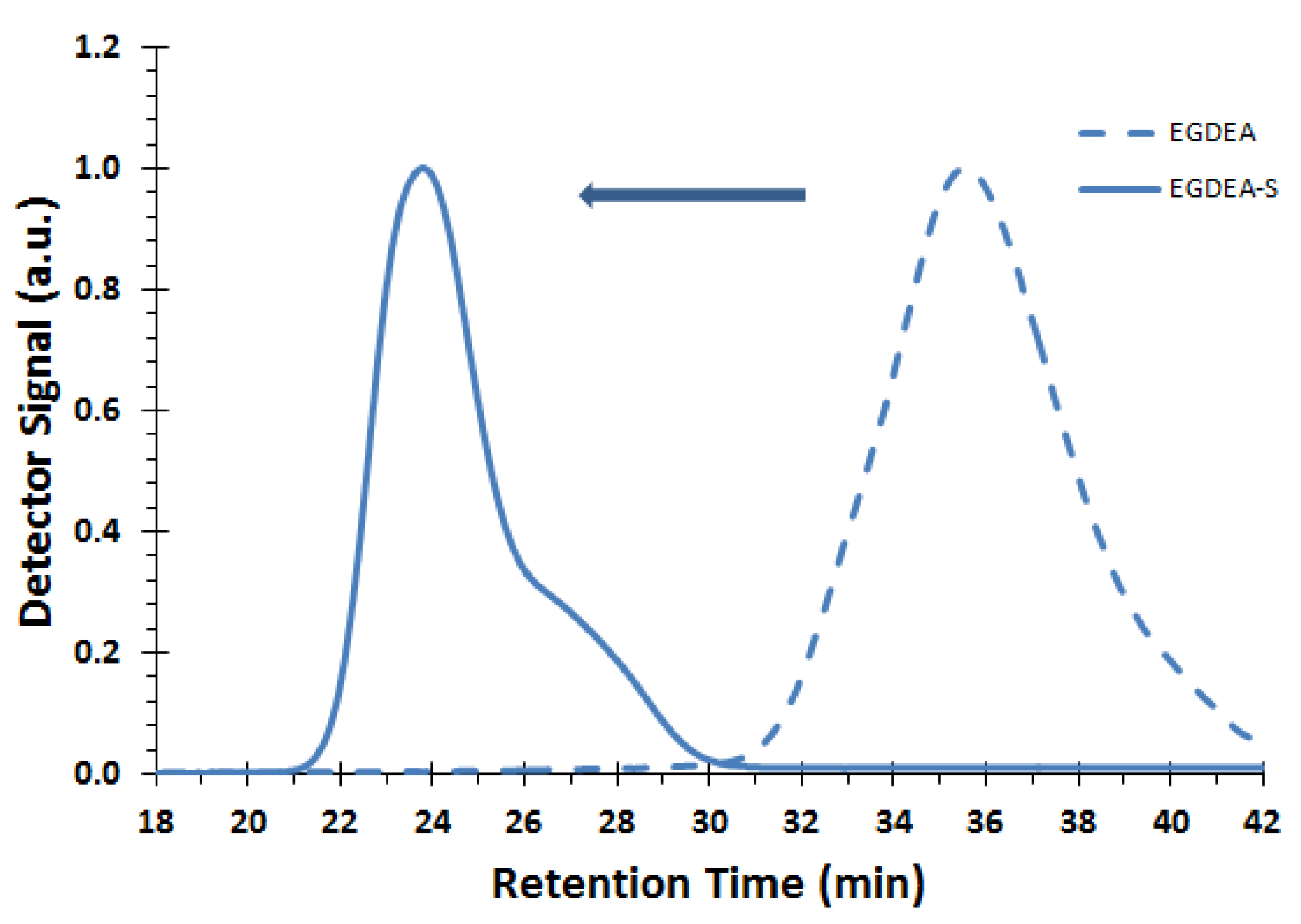
Figure 10.
GPC traces of corresponding macroinitiator EGDEA (dashed line, sample E-T90, Mn = 4.0 kg mol−1, Ð = 1.62) and chain extended block copolymer EGDEA-S (solid line, Mn = 58.6 kg mol−1, Ð = 1.97, FEGDEA = 0.08). Chromatogram of chain-extended block copolymer showed no significant change after fractionation.
Figure 10.
GPC traces of corresponding macroinitiator EGDEA (dashed line, sample E-T90, Mn = 4.0 kg mol−1, Ð = 1.62) and chain extended block copolymer EGDEA-S (solid line, Mn = 58.6 kg mol−1, Ð = 1.97, FEGDEA = 0.08). Chromatogram of chain-extended block copolymer showed no significant change after fractionation.
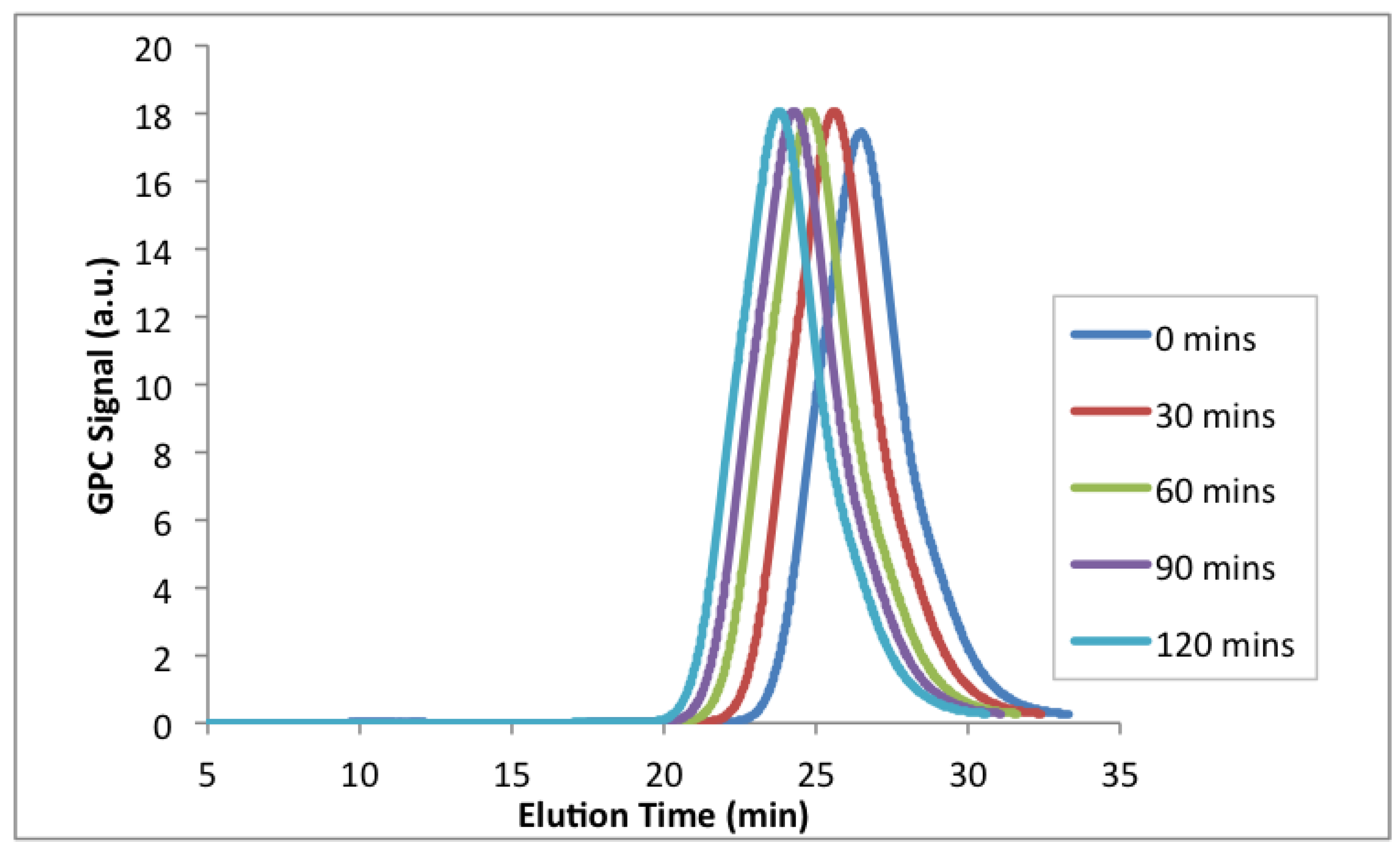
Figure 11.
GPC chromatograms of the chain extension with styrene at 100 °C from a poly(EGDEMA-
stat-S) macroinitiator (see
Table 8 for properties of the macroinitiator with ID = EGDEMA/S-15-90).
Figure 11.
GPC chromatograms of the chain extension with styrene at 100 °C from a poly(EGDEMA-
stat-S) macroinitiator (see
Table 8 for properties of the macroinitiator with ID = EGDEMA/S-15-90).
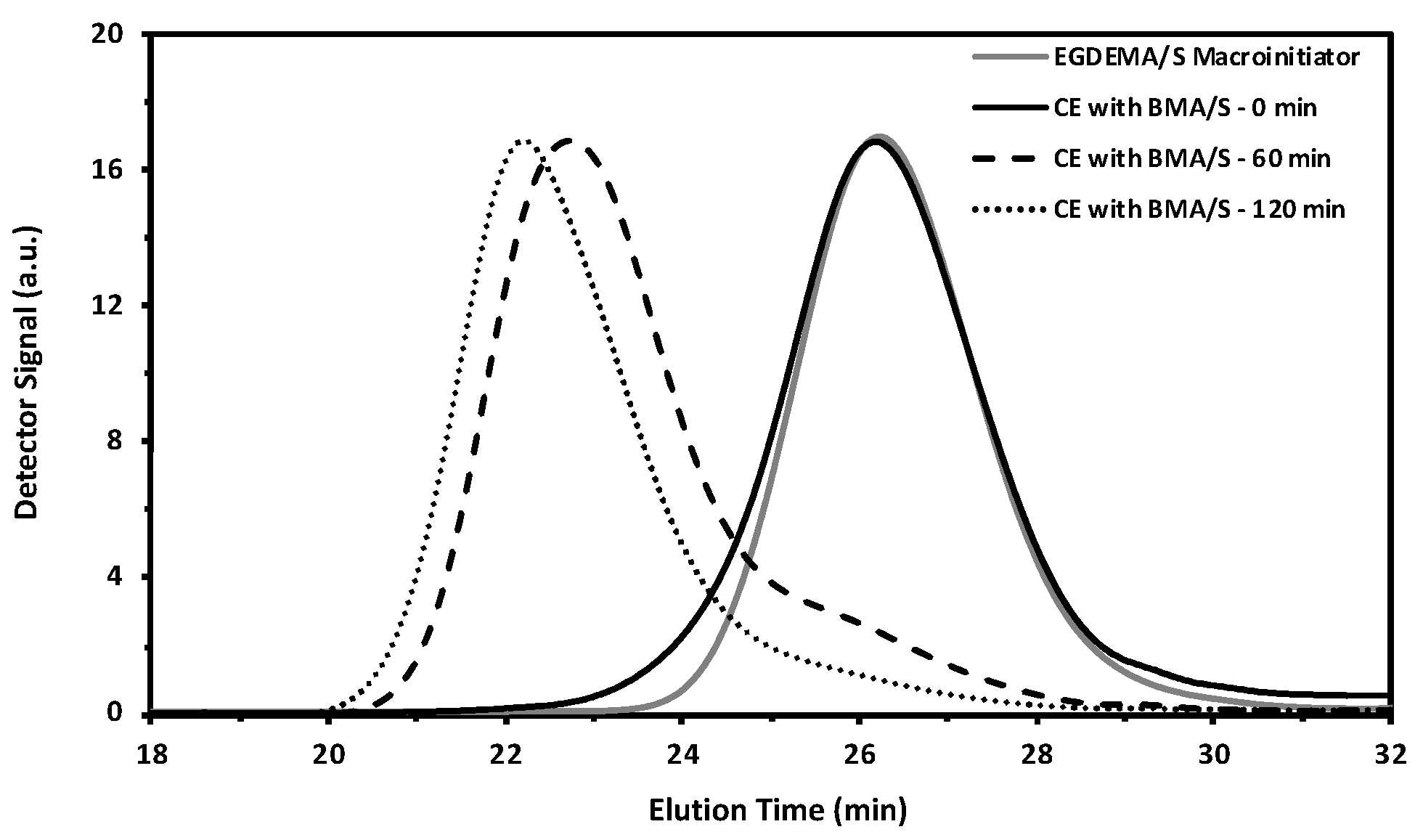
Figure 12.
GPC traces from an EGDEMA/S macroinitiator (EGDEMA/S-N-80-90, Mn = 5.6 kg mol−1, Đ = 1.27, FEGDEMA = 0.23) to the chain extended species with BMA/S at various polymerizations to form the poly(EGDEMA/S-block-BMA/S) block copolymer (Mn = 32.3 kg mol−1, Đ = 1.69, FBMA= 0.64, FEGDEMA = 0.05).
Figure 12.
GPC traces from an EGDEMA/S macroinitiator (EGDEMA/S-N-80-90, Mn = 5.6 kg mol−1, Đ = 1.27, FEGDEMA = 0.23) to the chain extended species with BMA/S at various polymerizations to form the poly(EGDEMA/S-block-BMA/S) block copolymer (Mn = 32.3 kg mol−1, Đ = 1.69, FBMA= 0.64, FEGDEMA = 0.05).
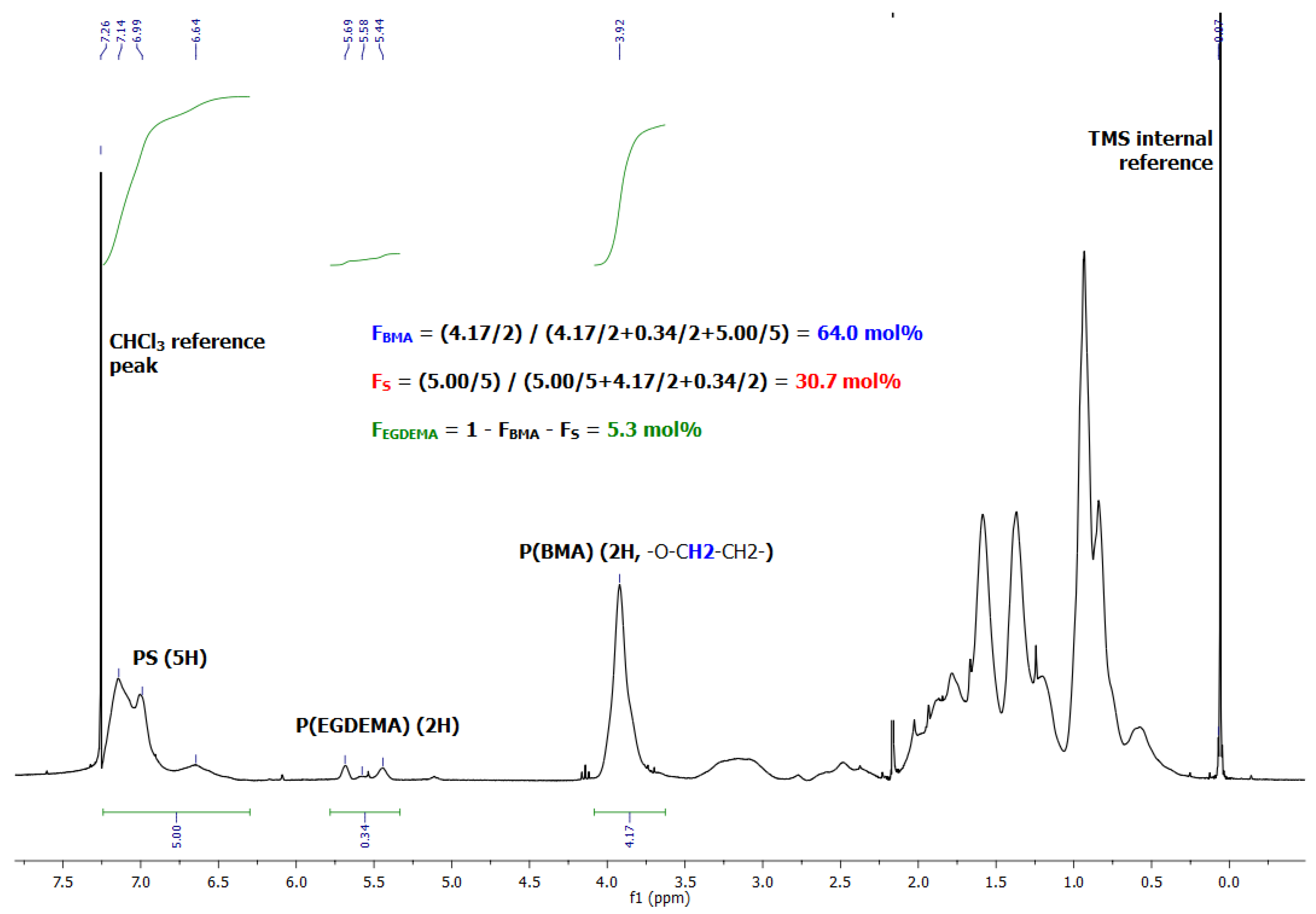
Figure 13.
1H NMR spectrum in CDCl3 of poly(EGDEMA/S-block-BMA/S) showing the integrations to provide the overall compositions of the block copolymer.
Figure 13.
1H NMR spectrum in CDCl3 of poly(EGDEMA/S-block-BMA/S) showing the integrations to provide the overall compositions of the block copolymer.
Table 1.
Experimental conditions for homopolymerization of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) in 1,4-dioxane at 90–120 °C and various levels of free SG1 nitroxide via Nitroxide-Mediated Polymerization with BlocBuilder.
Table 1.
Experimental conditions for homopolymerization of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) in 1,4-dioxane at 90–120 °C and various levels of free SG1 nitroxide via Nitroxide-Mediated Polymerization with BlocBuilder.
| Expt. ID a | Temperature (°C) | [BlocBuilder]0 (mol L−1) | [SG1]0 (mol L−1) | r b | [EGDEA]0 (mol L−1) | [1,4-dioxane]0 (mol L−1) | Mn,targetc (kg mol−1) |
|---|
| E-T90 (E-S5) | 90 | 0.026 | 0.001 | 0.05 | 2.01 | 5.68 | 21.2 |
| E-T100 | 100 | 0.024 | 0.001 | 0.06 | 1.98 | 5.76 | 20.4 |
| E-T110 | 110 | 0.024 | 0.001 | 0.06 | 2.01 | 5.69 | 20.8 |
| E-T120 | 120 | 0.024 | 0.001 | 0.06 | 2.01 | 5.68 | 21.1 |
| E-S0 | 90 | 0.024 | 0.000 | 0 | 2.02 | 5.67 | 21.1 |
| E-S10 | 90 | 0.025 | 0.003 | 0.10 | 2.11 | 5.93 | 21.3 |
Table 2.
Experimental conditions for ethylene glycol dicyclopentenyl ether acrylate/styrene (EGDEA/S) and ethylene glycol dicyclopentenyl ether methacrylate/styrene (EGDEMA/S) mixtures polymerized at 90 °C in 50 wt.% 1,4-dioxane solution with BlocBuilderTM and additional SG1 free nitroxide.
Table 2.
Experimental conditions for ethylene glycol dicyclopentenyl ether acrylate/styrene (EGDEA/S) and ethylene glycol dicyclopentenyl ether methacrylate/styrene (EGDEMA/S) mixtures polymerized at 90 °C in 50 wt.% 1,4-dioxane solution with BlocBuilderTM and additional SG1 free nitroxide.
| Expt. ID a | [BlocBuilder]0 (mol L−1) | [SG1]0 (mol L−1) | r b | [Styrene]0 (mol L−1) | [X]0 c (mol L−1) | [1,4-dioxane]0 (mol L−1) | Mn,targetd (kg mol−1) |
|---|
| EGDEA/S | 0.024 | 0.001 | 0.051 | 0.11 | 1.99 | 5.61 | 21.5 |
| EGDEMA/S | 0.023 | 0.001 | 0.051 | 0.21 | 1.91 | 5.41 | 23.1 |
| EGDEA (E-T90) | 0.026 | 0.001 | 0.053 | 0 | 2.01 | 5.68 | 21.2 |
Table 3.
Experimental conditions for ethylene glycol dicyclopentenyl ether methacrylate/styrene (EGDEMA/S) mixtures polymerized in 50 wt.% 1,4-dioxane solution with NHS-BlocBuilder.
Table 3.
Experimental conditions for ethylene glycol dicyclopentenyl ether methacrylate/styrene (EGDEMA/S) mixtures polymerized in 50 wt.% 1,4-dioxane solution with NHS-BlocBuilder.
| Experiment ID | T (°C) | [NHS-BB]0 (mol L−1) | [EGDEMA]0 (mol L−1) | [Styrene]0 (mol L−1) | fS,0 | Reaction Time (min) |
|---|
| EGDEMA/S–N-5-90 | 90 | 0.022 | 1.93 | 0.10 | 0.05 | 90 |
| EGDEMA/S–N-10-90 | 90 | 0.022 | 1.89 | 0.21 | 0.10 | 90 |
| EGDEMA/S–N-15-90 | 90 | 0.022 | 1.84 | 0.33 | 0.15 | 120 |
| EGDEMA/S-N-5-100 | 100 | 0.022 | 1.95 | 0.10 | 0.05 | 90 |
| EGDEMA/S-N–10-100 | 100 | 0.022 | 1.90 | 0.21 | 0.10 | 90 |
| EGDEMA/S–N-15-100 | 100 | 0.022 | 1.85 | 0.33 | 0.15 | 90 |
| EGDEMA/S-N-80-90 | 90 | 0.020 | 0.71 | 2.86 | 0.80 | 150 |
Table 4.
Experimental conditions for chain extension experiment with styrene from a poly(ethylene glycol dicyclopentenyl ether acrylate) macroinitiator at 90 °C.
Table 4.
Experimental conditions for chain extension experiment with styrene from a poly(ethylene glycol dicyclopentenyl ether acrylate) macroinitiator at 90 °C.
| Expt. ID a | [Macroinitiator]0 b (mol L−1) | [Styrene]0 (mol L−1) | [SG1]0 (mol L−1) | [1,4-dioxane]0 (mol L−1) | Mn,targetc (kg mol−1) |
|---|
| EGDEA-S | 0.012 | 4.79 | 0.0006 | 5.68 | 45.7 |
Table 5.
Summary of polymerizations and molecular properties for the homopolymerizations of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) via nitroxide-mediated polymerization with BlocBuilderTM and 5 mol% excess free SG1 nitroxide relative to BlocBuilderTM in 1,4-dioxane at 90–120 °C.
Table 5.
Summary of polymerizations and molecular properties for the homopolymerizations of ethylene glycol dicyclopentenyl ether acrylate (EGDEA) via nitroxide-mediated polymerization with BlocBuilderTM and 5 mol% excess free SG1 nitroxide relative to BlocBuilderTM in 1,4-dioxane at 90–120 °C.
| Expt. ID a | Temperature (°C) | r b | Polymerization Time (min) | Conversion c (%) | Mn,target (kg mol−1) | Mnd (kg mol−1) | Ðd |
|---|
| E-T90 (E-S5) | 90 | 0.05 | 270 | 22 | 19.8 | 4.0 | 1.62 |
| E-T100 | 100 | 0.06 | 100 | 36 | 20.7 | 5.2 | 2.09 |
| E-T110 | 110 | 0.06 | 50 | 51 | 20.8 | 9.5 | 1.80 |
| E-T120 | 120 | 0.06 | 60 | 54 | 21.1 | 9.4 | 2.01 |
| E-S0 | 90 | 0.00 | 180 | 29 | 21.1 | 5.4 | 1.76 |
| E-S10 | 90 | 0.10 | 720 | 24 | 20.7 | 4.6 | 1.70 |
Table 6.
Summary of kpK values for the EGDEA homopolymerization via NMP with BlocBuilderTM in the presence of 5 mol % excess SG1 free nitroxide relative to BlocBuilderTM.
Table 6.
Summary of kpK values for the EGDEA homopolymerization via NMP with BlocBuilderTM in the presence of 5 mol % excess SG1 free nitroxide relative to BlocBuilderTM.
| Temperature (°C) | kpK (10−5 s−1) |
|---|
| 90 | 1.0 ± 0.1 |
| 100 | 6.8 ± 0.7 |
| 110 | 18.8 ± 1.0 |
| 120 | 16.7 ± 0.5 |
Table 7.
Summary of polymerizations and molecular properties for the statistical copolymerizations ethylene glycol dicyclopentenyl ether acrylate (EGDEA) and ethylene glycol dicyclopentenyl ether methacrylate (EGDEMA) with styrene and homopolymerization of EGDEA via Nitroxide-Mediated Polymerization with BlocBuilderTM in 1,4-dioxane at 90 °C.
Table 7.
Summary of polymerizations and molecular properties for the statistical copolymerizations ethylene glycol dicyclopentenyl ether acrylate (EGDEA) and ethylene glycol dicyclopentenyl ether methacrylate (EGDEMA) with styrene and homopolymerization of EGDEA via Nitroxide-Mediated Polymerization with BlocBuilderTM in 1,4-dioxane at 90 °C.
| Expt. ID a | T (°C) | fS,0b | Polymerization Time (min) | X c | FSb | <kp><K> (10−6 s−1) | Mn,target (kg mol−1) | Mn d (kg mol−1) | Ð e |
|---|
| EGDEA/S | 90 | 0.05 | 320 | 0.17 | 0.14 | 9.5 ± 0.9 | 21.5 | 3.6 | 1.44 |
| EGDEMA/S | 90 | 0.10 | 130 | 0.72 | 0.13 | 7.0 ± 0.5 | 23.1 | 14.3 | 1.38 |
| EGDEA | 90 | 0 | 270 | 0.22 | 0 | 145 ± 4 | 19.8 | 4.0 | 1.62 |
Table 8.
Summary of EGDEMA-rich polymerizations controlled with styrene (S) at 90 and 100 °C using NHS-BlocBuilder in 50 wt.% 1,4 dioxane solutions.
Table 8.
Summary of EGDEMA-rich polymerizations controlled with styrene (S) at 90 and 100 °C using NHS-BlocBuilder in 50 wt.% 1,4 dioxane solutions.
| Experiment ID a | Temperature (°C) | t (min) | fS,0b | FSb | X c | Mn (kg mol−1)d | Ð d |
|---|
| EGDEMA/S–N-5-90 | 90 | 90 | 0.05 | 0.08 | 0.64 | 13.2 | 1.64 |
| EGDEMA/S-N–10-90 | 90 | 90 | 0.10 | 0.14 | 0.65 | 15.8 | 1.56 |
| EGDEMA/S-N–15-90 | 90 | 120 | 0.15 | 0.20 | 0.71 | 18.3 | 1.67 |
| EGDEMA/S-N–5-100 | 100 | 90 | 0.05 | 0.07 | 0.69 | 17.3 | 1.73 |
| EGDEMA/S–N-10-100 | 100 | 90 | 0.10 | 0.13 | 0.71 | 21.9 | 1.76 |
| EGDEMA/S–N-15-100 | 100 | 90 | 0.15 | 0.17 | 0.67 | 16.4 | 1.74 |
| EGDEMA/S-N-80-90 | 90 | 150 | 0.80 | 0.77 | 0.30 | 5.6 | 1.27 |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}