Time Dependent Structure and Property Evolution in Fibres during Continuous Carbon Fibre Manufacturing


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Fourier Transform Infrared Spectroscopy (FTIR)
2.3. X-ray Diffraction Studies (XRD)
2.4. Density Measurements
2.5. Tensile Properties
3. Results
3.1. Influence of Dwell Time on the Progress of Thermal Stabilization in PAN Fibres
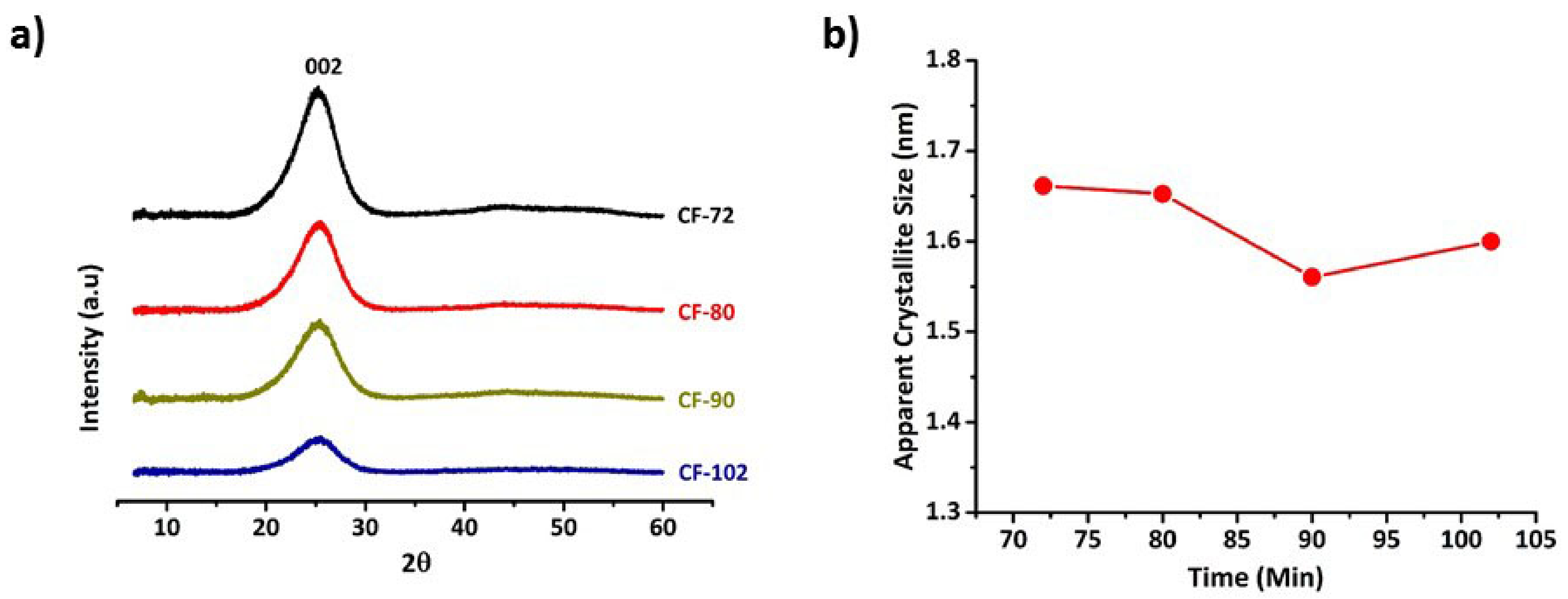
3.2. Microstructure Evolution in Carbon Fibres
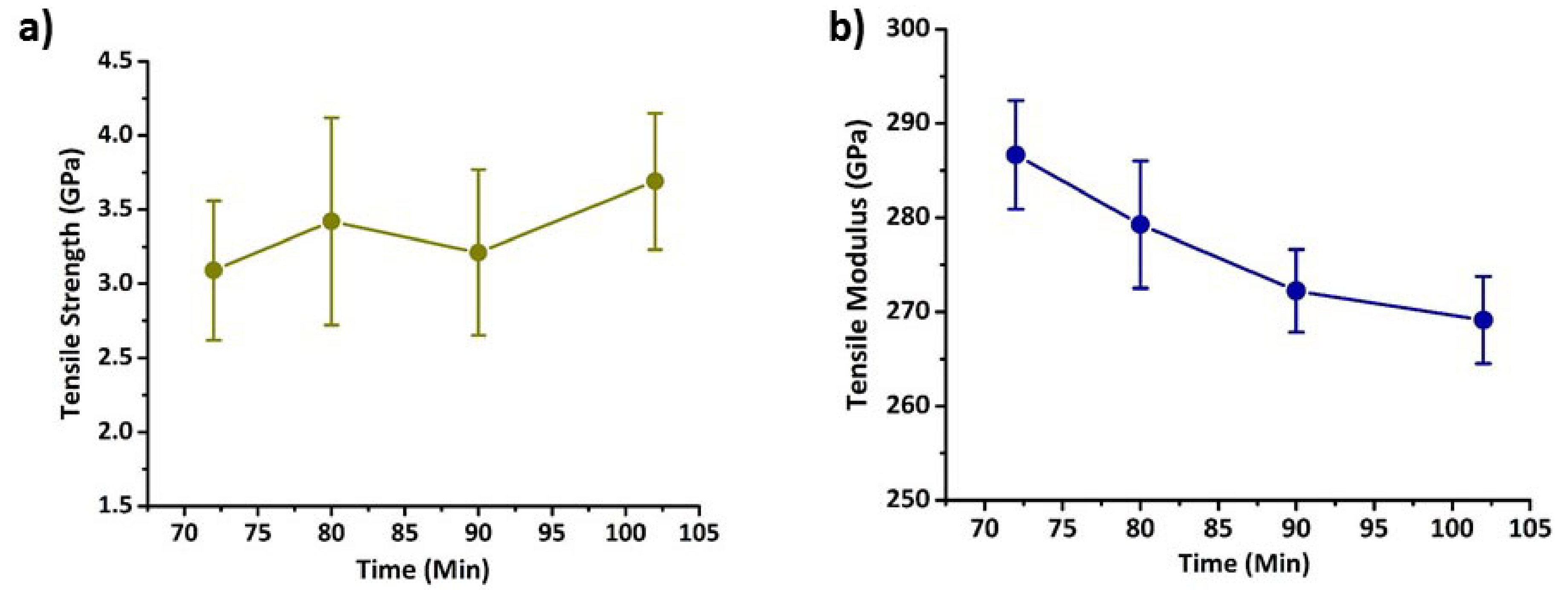
3.3. Properties of Carbon Fibres
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhao, J.; Zhang, J.; Zhou, T.; Liu, X.; Yuan, Q.; Zhang, A. New understanding on the reaction pathways of the polyacrylonitrile copolymer fiber pre-oxidation: Online tracking by two-dimensional correlation ftir spectroscopy. RSC Adv. 2016, 6, 4397–4409. [Google Scholar] [CrossRef]
- Nunna, S.; Naebe, M.; Hameed, N.; Fox, B.L.; Creighton, C. Evolution of radial heterogeneity in polyacrylonitrile fibres during thermal stabilization: An overview. Polym. Degrad. Stab. 2017, 136, 20–30. [Google Scholar] [CrossRef]
- Khayyam, H.; Fakhrhoseini, S.M.; Church, J.S.; Milani, A.S.; Bab-Hadiashar, A.; Jazar, R.N.; Naebe, M. Predictive modelling and optimization of carbon fiber mechanical properties through high temperature furnace. Appl. Therm. Eng. 2017, 125, 1539–1554. [Google Scholar] [CrossRef]
- Rahman, M.A.; Ismail, A.F.; Mustafa, A. The effect of residence time on the physical characteristics of pan-based fibers produced using a solvent-free coagulation process. Mater. Sci. Eng. A 2007, 448, 275–280. [Google Scholar] [CrossRef]
- Yusof, N.; Ismail, A.F. Post spinning and pyrolysis processes of polyacrylonitrile (pan)-based carbon fiber and activated carbon fiber: A review. J. Anal. Appl. Pyrolysis 2012, 93, 1–13. [Google Scholar] [CrossRef]
- Jie, L.; Wangxi, Z. Structural changes during the thermal stabilization of modified and original polyacrylonitrile precursors. J. Appl. Polym. Sci. 2005, 97, 2047–2053. [Google Scholar] [CrossRef]
- Nunna, S.; Creighton, C.; Fox, B.L.; Naebe, M.; Maghe, M.; Tobin, M.J.; Bambery, K.; Vongsvivut, J.; Hameed, N. The effect of thermally induced chemical transformations on the structure and properties of carbon fibre precursors. J. Mater. Chem. A 2017, 5, 7372–7382. [Google Scholar] [CrossRef]
- Chen, J.C.; Harrison, I.R. Modification of polyacrylonitrile (pan) carbon fiber precursor via post-spinning plasticization and stretching in dimethyl formamide (dmf). Carbon 2002, 40, 25–45. [Google Scholar] [CrossRef]
- Andrews, R.; Jacques, D.; Rao, A.M.; Rantell, T.; Derbyshire, F.; Chen, Y.; Chen, J.; Haddon, R.C. Nanotube composite carbon fibers. Appl. Phys. Lett. 1999, 75, 1329–1331. [Google Scholar] [CrossRef]
- Xiang, C.; Behabtu, N.; Liu, Y.; Chae, H.G.; Young, C.C.; Genorio, B.; Tsentalovich, D.E.; Zhang, C.; Kosynkin, D.V.; Lomeda, J.R.; et al. Graphene nanoribbons as an advanced precursor for making carbon fiber. ACS Nano 2013, 7, 1628–1637. [Google Scholar] [CrossRef]
- Newcomb, B.A.; Gulgunje, P.V.; Gupta, K.; Kamath, M.G.; Liu, Y.; Giannuzzi, L.A.; Chae, H.G.; Kumar, S. Processing, structure, and properties of gel spun pan and pan/cnt fibers and gel spun pan based carbon fibers. Polym. Eng. Sci. 2015, 55, 2603–2614. [Google Scholar] [CrossRef]
- Chae, H.G.; Minus, M.L.; Rasheed, A.; Kumar, S. Stabilization and carbonization of gel spun polyacrylonitrile/single wall carbon nanotube composite fibers. Polymer 2007, 48, 3781–3789. [Google Scholar] [CrossRef]
- Maghe, M.; Creighton, C.; Henderson, L.C.; Huson, M.G.; Nunna, S.; Atkiss, S.; Byrne, N.; Fox, B.L. Using ionic liquids to reduce energy consumption for carbon fibre production. J. Mater. Chem. A 2016, 4, 16619–16626. [Google Scholar] [CrossRef]
- Frank, E.; Hermanutz, F.; Buchmeiser, M.R. Carbon fibers: Precursors, manufacturing, and properties. Macromol. Mater. Eng. 2012, 297, 493–501. [Google Scholar] [CrossRef]
- Khayyam, H.; Naebe, M.; Zabihi, O.; Zamani, R.; Atkiss, S.; Fox, B. Dynamic prediction models and optimization of polyacrylonitrile (pan) stabilization processes for production of carbon fiber. IEEE Trans. Ind. Inform. 2015, 11, 887–895. [Google Scholar] [CrossRef]
- Nunna, S.; Naebe, M.; Hameed, N.; Creighton, C.; Naghashian, S.; Jennings, M.J.; Atkiss, S.; Setty, M.; Fox, B.L. Investigation of progress of reactions and evolution of radial heterogeneity in the initial stage of thermal stabilization of pan precursor fibres. Polym. Degrad. Stab. 2016, 125, 105–114. [Google Scholar] [CrossRef]
- Ko, T.-H. Influence of continuous stabilization on the physical properties and microstructure of pan-based carbon fibers. J. Appl. Polym. Sci. 1991, 42, 1949–1957. [Google Scholar] [CrossRef]
- Wang, P.H.; Yue, Z.R.; Li, R.Y.; Liu, J. Aspects on interaction between multistage stabilization of polyacrylonitrile precursor and mechanical properties of carbon fibers. J. Appl. Polym. Sci. 1995, 56, 289–300. [Google Scholar] [CrossRef]
- Wu, G.; Lu, C.; Ling, L.; Hao, A.; He, F. Influence of tension on the oxidative stabilization process of polyacrylonitrile fibers. J. Appl. Polym. Sci. 2005, 96, 1029–1034. [Google Scholar] [CrossRef]
- Wang, P.; Liu, J.; Yue, Z.; Li, R. Thermal oxidative stabilization of polyacrylonitrile precursor fiber—Progression of morphological structure and mechanical properties. Carbon 1992, 30, 113–120. [Google Scholar] [CrossRef]
- Ji, M.; Wang, C.; Bai, Y.; Yu, M.; Wang, Y. Structural evolution of polyacrylonitrile precursor fibers during preoxidation and carbonization. Polym. Bull. 2007, 59, 527–536. [Google Scholar] [CrossRef]
- Jing, M.; Wang, C.-G.; Bai, Y.-J.; Zhu, B.; Wang, Y.-X. Effect of temperatures in the rearmost stabilization zone on structure and properties of pan-based oxidized fibers. Polym. Bull. 2007, 58, 541–551. [Google Scholar] [CrossRef]
- Yu, M.; Wang, C.; Bai, Y.; Wang, Y.; Wang, Q.; Liu, H. Combined effect of processing parameters on thermal stabilization of pan fibers. Polym. Bull. 2006, 57, 525–533. [Google Scholar] [CrossRef]
- Tsai, J.-S. Tension effects on the properties of oxidized polyacrylonitrile and carbon fibers during continuous oxidation. Polym. Eng. Sci. 1995, 35, 1313–1316. [Google Scholar] [CrossRef]
- Lian, F.; Liu, J.; Ma, Z.; Liang, J. Stretching-induced deformation of polyacrylonitrile chains both in quasicrystals and in amorphous regions during the in situ thermal modification of fibers prior to oxidative stabilization. Carbon 2012, 50, 488–499. [Google Scholar] [CrossRef]
- Lee, S. Structural evolution of polyacrylonitrile fibers in stabilization and carbonization. Adv. Chem. Eng. Sci. 2012, 2, 275–282. [Google Scholar] [CrossRef]
- Collins, G.L.; Thomas, N.W.; Williams, G.E. Kinetic relationships between heat generation and nitrile consumption in the reaction of poly(acrylonitrile) in air at 265 °C. Carbon 1988, 26, 671–679. [Google Scholar] [CrossRef]
- Badii, K.; Church, J.S.; Golkarnarenji, G.; Naebe, M.; Khayyam, H. Chemical structure based prediction of pan and oxidized pan fiber density through a non-linear mathematical model. Polym. Degrad. Stab. 2016, 131, 53–61. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A review of heat treatment on polyacrylonitrile fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Arbab, S.; Zeinolebadi, A. A procedure for precise determination of thermal stabilization reactions in carbon fiber precursors. Polym. Degrad. Stab. 2013, 98, 2537–2545. [Google Scholar] [CrossRef]
- Hameed, N.; Sharp, J.; Nunna, S.; Creighton, C.; Magniez, K.; Jyotishkumar, P.; Salim, N.V.; Fox, B. Structural transformation of polyacrylonitrile fibers during stabilization and low temperature carbonization. Polym. Degrad. Stab. 2016, 128, 39–45. [Google Scholar] [CrossRef]
- Arbab, S.; Mirbaha, H.; Zeinolebadi, A.; Nourpanah, P. Indicators for evaluation of progress in thermal stabilization reactions of polyacrylonitrile fibers. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, B.; Sun, L.; Zhang, H. Study on the thermal oxidative stabilization reactions and the formed structures in polyacrylonitrile during thermal treatment. Polym. Degrad. Stab. 2017, 140, 104–113. [Google Scholar] [CrossRef]
- Karacan, I.; Erdoğan, G. A study on structural characterization of thermal stabilization stage of polyacrylonitrile fibers prior to carbonization. Fibers Polym 2012, 13, 329–338. [Google Scholar] [CrossRef]
- Takaku, A.; Hashimoto, T.; Miyoshi, T. Tensile properties of carbon fibers from acrylic fibers stabilized under isothermal conditions. J. Appl. Polym. Sci. 1985, 30, 1565–1571. [Google Scholar] [CrossRef]
- Li, X.; Ji, X.; He, C. Evolution of the morphological and structural properties of plasticized spinning polyacrylonitrile fibers during the stabilization process. RSC Adv. 2015, 5, 81399–81406. [Google Scholar] [CrossRef]
- Xiao, S.; Cao, W.; Wang, B.; Xu, L.; Chen, B. Mechanism and kinetics of oxidation during the thermal stabilization of polyacrylonitrile fibers. J. Appl. Polym. Sci. 2013, 127, 3198–3203. [Google Scholar] [CrossRef]
- Liu, F.; Wang, H.; Xue, L.; Fan, L.; Zhu, Z. Effect of microstructure on the mechanical properties of pan-based carbon fibers during high-temperature graphitization. J. Mater. Sci. 2008, 43, 4316–4322. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Tensile Strength (GPa) | Tensile Modulus (GPa) | Elongation at Break (%) | Linear Density (dtex) | Diameter (μm) |
|---|---|---|---|---|---|
| PAN | 0.95 ± 0.06 | 16.95 ± 0.63 | 11.69 ± 0.61 | 0.76 ± 0.09 | 9.06 ± 0.54 |
| Parameters | Stabilization | LT | HT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Zone-1 | Zone-2 | Zone-3 | Zone-4 | Zone-1 | Zone-2 | Zone-3 | Zone-1 | Zone-2 | ||
| Temperature (°C) | 228 | 236 | 248 | 258 | ~500 | ~800 | ~1000 | ~1100 | ~1500 | |
| Tension (cN) | ~2000 | ~2100 | ~2000 | ~2100 | ~1100 | ~1700 | ||||
| Line Speed (m/h) | Dwell Time (min) | |||||||||
| CF-72 | 20 | 18 | 18 | 18 | 18 | 5.4 | 3.6 | |||
| CF-80 | 18 | 20 | 20 | 20 | 20 | 6 | 4 | |||
| CF-90 | 16 | 22.5 | 22.5 | 22.5 | 22.5 | 6.75 | 4.5 | |||
| CF-102 | 14 | 25.7 | 25.7 | 25.7 | 25.7 | 7.7 | 5.14 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunna, S.; Maghe, M.; Rana, R.; Varley, R.J.; Knorr, D.B., Jr.; Sands, J.M.; Creighton, C.; Henderson, L.C.; Naebe, M. Time Dependent Structure and Property Evolution in Fibres during Continuous Carbon Fibre Manufacturing. Materials 2019, 12, 1069. https://doi.org/10.3390/ma12071069
Nunna S, Maghe M, Rana R, Varley RJ, Knorr DB Jr., Sands JM, Creighton C, Henderson LC, Naebe M. Time Dependent Structure and Property Evolution in Fibres during Continuous Carbon Fibre Manufacturing. Materials. 2019; 12(7):1069. https://doi.org/10.3390/ma12071069
Chicago/Turabian StyleNunna, Srinivas, Maxime Maghe, Rohit Rana, Russell J. Varley, Daniel B. Knorr, Jr., James M. Sands, Claudia Creighton, Luke C. Henderson, and Minoo Naebe. 2019. "Time Dependent Structure and Property Evolution in Fibres during Continuous Carbon Fibre Manufacturing" Materials 12, no. 7: 1069. https://doi.org/10.3390/ma12071069
APA StyleNunna, S., Maghe, M., Rana, R., Varley, R. J., Knorr, D. B., Jr., Sands, J. M., Creighton, C., Henderson, L. C., & Naebe, M. (2019). Time Dependent Structure and Property Evolution in Fibres during Continuous Carbon Fibre Manufacturing. Materials, 12(7), 1069. https://doi.org/10.3390/ma12071069