Application of the Microwave Technique in Continuous Flow Processing of Organophosphorus Chemical Reactions




Abstract
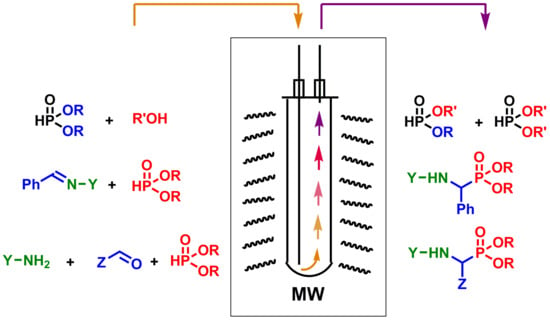
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Batch vs. Continuous Flow Systems
1.2. Batch Microwave Chemistry
1.3. Continuous Flow Microwave Attempts
1.4. Microwaves in Batch Organophosphorus Syntheses
2. Microwave-Assisted Continuous Flow Applications
2.1. Development of the Continuous Flow Microwave Device
2.2. Elaboration of the Continuous Flow Transesterification of Dialkyl Phosphites
2.3. Continuous Flow Synthesis of α-Aminophosphonates by the aza-Pudovik Reaction
2.4. The Synthesis of α-Aminophosphonates by Continuous Flow Kabachnik–Fields Reaction
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Wiley-VCH: Weinheim, Germany, 2000; ISBN 978-3-527-30385-4.
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s guide to flow chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef] [PubMed]
- Glasnov, T. Continuous-Flow Chemistry in the Research Laboratory; Springer International Publishing: Basel, Switzerland, 2016; ISBN 978-3-319-32194-3. [Google Scholar]
- Vaccaro, L. Sustainable Flow Chemistry: Methods and Applications; Wiley: Weinheim, Germany, 2017; ISBN 978-3-527-338528. [Google Scholar]
- Movsisyan, M.; Delbeke, E.I.P.; Berton, J.K.E.T.; Battiloccihio, C.; Ley, S.V.; Stevens, C.V. Taming hazardous chemistry by continuous flow technology. Chem. Soc. Rev. 2016, 45, 4892–4928. [Google Scholar] [CrossRef] [PubMed]
- Bakeev, K.A. Process Analytical Technology: Spectroscopic Tools and Implemented Strategies for the Chemical and Pharmaceutical Industries; Wiley-VCH: Chichester, UK, 2010; ISBN 978-0-470-72207-7. [Google Scholar]
- May, S.A. Flow Chemistry, Continuous Processing, and Continuous Manufacturing: A Pharmaceutical Perspective. J. Flow Chem. 2017, 7, 137–145. [Google Scholar] [CrossRef]
- Strying, P.; Parracho, A.I.R. From discovery to production: Scale-out of continuous flow meso reactors. Beilstein J. Org. Chem. 2009, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Pashkova, A.; Greiner, L. Towards Small-Scale Continuous Chemical Production: Technology Gaps and Challenges. Chem. Ing. Tech. 2011, 83, 1337–1342. [Google Scholar] [CrossRef]
- Cravotto, G.; Carnaroglio, D. Microwave Chemistry; De Gruyter: Berlin, Germany, 2017; ISBN 978-3-11-047993-5. [Google Scholar]
- Horikoshi, S.; Schiffmann, R.F.; Fukushima, J.; Serpone, N. Microwave Chemical and Materials Processing; Springer: Singapore, 2018; ISBN 978-981-10-6465-4. [Google Scholar]
- Bálint, E.; Keglevich, G. The Spread of the Application of the Microwave Technique in Organic Synthesis. In Milestones in Microwave Chemistry; Keglevich, G., Ed.; Springer: Cham, Switzerland, 2016; pp. 1–10. [Google Scholar]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Kappe, C.O.; Stadler, A.; Dallinger, D. Microwaves in Organic and Medicinal Chemistry, 2nd ed.; Wiley: Weinheim, Germany, 2012; Volume 52, ISBN 978-3-527-33185-7. [Google Scholar]
- Zhao, X.; Zhang, Z.; Wang, L.; Xi, K.; Cao, Q.; Wang, D.; Yang, Y.; Du, Y. Excellent microwave absorption property of Graphene-coated Fe nanocomposites. Sci. Rep. 2013, 3, 3421. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Yang, J.; Pu, B.; Chen, H.; Li, Y.; Wang, Z.; Niu, X. Excellent microwave absorption of lead halide perovskites with high stability. J. Mater. Chem. C 2018, 6, 4201–4207. [Google Scholar] [CrossRef]
- Gong, J.; Yang, F.; Shao, Q.; He, X.; Zhang, X.; Liu, S.; Tang, L.; Deng, Y. Microwave absorption performance of methylimidazolium ionic liquids: Towards novel ultra-wideband metamaterial absorbers. RSC Adv. 2017, 7, 41980–41988. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Bálint, E.; Keglevich, G. Microwave-Assisted Syntheses in Organic Chemistry. In Milestones in Microwave Chemistry; Keglevich, G., Ed.; Springer: Cham, Switzerland, 2016; pp. 11–45. [Google Scholar]
- Kitchen, H.J.; Vallance, S.R.; Kennedy, J.L.; Tapia-Ruiz, N.; Carassiti, L.; Harrison, A.; Whittaker, A.G.; Drysdale, T.D.; Kingman, S.W.; Gregory, D.H. Modern Microwave Methods in Solid-State Inorganic Materials Chemistry: From Fundamentals to Manufacturing. Chem. Rev. 2014, 114, 1170–1206. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Sallay, P.; Greiner, I. Continuous flow microwave reactors. Hung. Chem. J. 2008, 63, 278–283. [Google Scholar]
- de la Hoz, A.; Díaz-Ortiz, A. Nonconventional Techniques in Sustainable Flow Chemistry. In Sustainable Flow Chemistry: Methods and Applications; Vaccaro, L., Ed.; Wiley: Weinheim, Germany, 2017; pp. 219–248. [Google Scholar]
- Estel, L.; Poux, M.; Benamara, N.; Polaert, I. Continuous flow-microwave reactor: Where are we? Chem. Eng. Process. 2016, 113, 56–64. [Google Scholar] [CrossRef]
- Wang, C.S. Processing parameters of continuous microwave heating of ethylene-propylene terpolymer. Rubber Chem. Technol. 1984, 57, 134–144. [Google Scholar] [CrossRef]
- Gunasekaran, S. Grain drying using continuous and pulsed microwave energy. Dry. Technol. 1990, 8, 1039–1047. [Google Scholar] [CrossRef]
- Strauss, C.R. A Strategic, ‘Green’ Approach to Organic Chemistry with Microwave Assistance and Predictive Yield Optimization as Core, Enabling Technologies. Aust. J. Chem. 1999, 52, 83–96. [Google Scholar] [CrossRef]
- Baxendale, I.; Hayward, J.; Ley, S. Microwave reactions under continuous flow conditions. Comb. Chem. High Throughput Screen. 2007, 10, 802–836. [Google Scholar] [CrossRef] [PubMed]
- Öhrngren, P.; Fardost, A.; Russo, F.; Schanche, J.-S.; Fagrell, M.; Larhed, M. Evaluation of a nonresonant microwave applicator for continuous-flow chemistry applications. Org. Proc. Res. Dev. 2012, 16, 1053–1063. [Google Scholar] [CrossRef]
- Rydfjord, J.; Svensson, F.; Fagrell, M.; Savmarker, J.; Thulin, M.; Larhed, M. Temperature measurements with two different IR sensors in a continuous-flow microwave heated system. Beilstein J. Org. Chem. 2013, 9, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorsi, L.; Proverbio, E. Influence of process parameters in microwave continuous synthesis of zeolite LTA. Microporous Mesoporous Mater. 2008, 112, 481–493. [Google Scholar] [CrossRef]
- Khadilkar, B.M.; Madyar, V.R. Scaling up of dihydropyridine ester synthesis by using aqueous hydrotrope solutions in a continuous microwave reactor. Org. Process Res. Dev. 2001, 5, 452–455. [Google Scholar] [CrossRef]
- Pillai, U.R.; Sahle-Demessie, E.; Varma, R.S. Hydrodechlorination of chlorinated benzenes in a continuous microwave reactor. Green Chem. 2004, 6, 295–298. [Google Scholar] [CrossRef]
- Correa, R.; Gonzalez, G.; Dougar, V. Emulsion polymerization in a microwave reactor. Polymer 1998, 39, 1471–1474. [Google Scholar] [CrossRef]
- Cáceres, A.; Jaimes, M.; Chávez, G.; Bravo, B.; Ysambertt, F.; Márquez, N. Continuous system with microwave irradiation to obtain alkyl benzoates. Talanta 2005, 68, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Vámosi, P.; Matsuo, K.; Masuda, T.; Sato, K.; Narumi, T.; Takeda, K.; Mase, N. (2018) Rapid Optimization of Reaction Conditions Based on Comprehensive Reaction Analysis Using a Continuous Flow Microwave Reactor. Chem. Rec. 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Bonnet, C.; Estel, L.; Ledoux, A.; Mazari, B.; Louis, A. Study of the thermal repartition in a microwave reactor: Application to the nitrobenzene hydrogenation. Chem. Eng. Proc. 2004, 43, 1435–1440. [Google Scholar] [CrossRef]
- Bo, L.; Quan, X.; Chen, S.; Zhao, H.; Zhao, Y. Degradation of p-nitrophenol in aqueous solution by microwave assisted oxidation process through a granular activated carbon fixed bed. Water Res. 2006, 40, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Bagley, M.C.; Jenkins, R.L.; Lubinu, M.C.; Mason, C.; Wood, R. A simple continuous flow microwave reactor. J. Org. Chem. 2005, 70, 7003–7006. [Google Scholar] [CrossRef] [PubMed]
- Kabza, K.G.; Chapados, B.R.; Getswicki, J.; McGrath, J.L. Microwave-induced esterification using heterogeneous acid catalyst in a low dielectric constant medium. J. Org. Chem. 2000, 65, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Haswell, S.J.; Fletcher, P.D.I. Efficiency, monitoring and control of microwave heating within a continuous flow capillary reactor. Sens. Actuator B Chem. 2005, 105, 516–520. [Google Scholar] [CrossRef]
- Shore, G.; Morin, S.; Organ, M.G. Catalysis in capillaries by Pd thin films using microwave-assisted continuous-flow organic synthesis (MACOS). Angew. Chem. Int. Ed. 2006, 45, 2761–2766. [Google Scholar] [CrossRef] [PubMed]
- Comer, E.; Organ, M.G. A microreactor for microwave-assisted capillary (continuous flow) organic synthesis (MACOS). J. Am. Chem. Soc. 2005, 127, 8160–8167. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Kaval, N.; Tomar, S.; Van der Eycken, E.; Parmar, V.S. Transition metal-catalyzed carbon–carbon bond formation Suzuki, Heck, and Sonogashira reactions using microwave and microtechnology. Org. Process Res. Dev. 2008, 12, 468–474. [Google Scholar] [CrossRef]
- Glasnov, T.N.; Kappe, C.O. Microwave-assisted synthesis under continuous-flow conditions. Macromol. Rapid Commun. 2007, 28, 395–410. [Google Scholar] [CrossRef]
- Ullah, F.; Samarakoon, T.; Rolfe, A.; Kurtz, R.D.; Hanson, P.R.; Organ, M.G. Scaling out by microwave-assisted, continuous flow organic synthesis (MACOS): Multi-gram synthesis of bromo- and fluoro-benzofused sultams benzthiaoxazepine-1,1-dioxides. Chem. Eur. J. 2010, 16, 10959–10962. [Google Scholar] [CrossRef] [PubMed]
- Dressen, M.H.C.L.; van de Kruijs, B.H.P.; Meuldijk, J.; Vekemans, J.A.J.M.; Hulshof, L.A. Flow processing of microwave-assisted (heterogeneous) organic reactions. Org. Process Res. Dev. 2010, 14, 351–361. [Google Scholar] [CrossRef]
- Bergamelli, F.; Iannelli, M.; Marafie, J.A.; Moseley, J.D. A commercial continuous flow microwave reactor evaluated for scale-up. Org. Process Res. Dev. 2010, 14, 926–930. [Google Scholar] [CrossRef]
- Bagley, M.C.; Fusillo, V.; Jenkins, R.L.; Lubinu, M.C.; Mason, C. Continuous flow processing from microreactors to mesoscale: The Bohlmann-Rahtz cyclodehydration reaction. Org. Biomol. Chem. 2010, 8, 2245–2251. [Google Scholar] [CrossRef] [PubMed]
- Moseley, J.D.; Lawton, S.J. Initial results from a commercial continuous flow microwave reactor for scale-up. Chem. Today 2007, 25, 16–19. [Google Scholar]
- Benaskar, F.; Hessel, V.; Krtschil, U.; Löb, P.; Stark, A. Intensification of the capillary-based Kolbe-Schmitt synthesis from resorcinol by reactive ionic liquids, microwave heating or a combination thereof. Org. Process Res. Dev. 2009, 13, 970–982. [Google Scholar] [CrossRef]
- Leadbeater, N.E.; Barnard, T.M.; Stencel, L.M. Batch and continuous-flow preparation of biodiesel derived from butanol and facilitated by microwave heating. Energy Fuels 2008, 22, 2005–2008. [Google Scholar] [CrossRef]
- Smith, C.J.; Iglesias-Siguenza, F.J.; Baxendale, I.R.; Ley, S.V. Flow and batch mode focused microwave synthesis of 5-amino-4-cyanopyrazoles and their further conversion to 4-aminopyrazolopyrimidines. Org. Biomol. Chem. 2007, 5, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Organ, M.G.; Hanson, P.R.; Rolfe, A.; Samarakoon, T.B.; Ullah, F. Accessing stereochemically rich sultams via microwave-assisted, continuous flow organic synthesis (MACOS) scale-out. J. Flow Chem. 2011, 1, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Morschhäuser, R.; Krull, M.; Kayser, C.; Boberski, C.; Bierbaum, R.; Püschner, P.A.; Glasnov, T.N.; Kappe, C.O. Microwave-assisted continuous flow synthesis on industrial scale. Green Process Synth. 2012, 1, 281–290. [Google Scholar] [CrossRef]
- Guenin, E.; Meziane, D. Microwave Assisted Phosphorus Organic Chemistry: A Review. Curr. Org. Chem. 2011, 15, 3465–3485. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Ludányi, K.; Drahos, L.; Keglevich, G. Novel Synthesis of Phosphinates by the Microwave-Assisted Esterification of Phosphinic Acids. Synth. Commun. 2009, 39, 2392–2404. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E.; Kiss, N.Z.; Jablonkai, E.; Hegedűs, L.; Grün, A.; Greiner, I. Microwave-Assisted Esterification of Phosphinic Acids. Curr. Org. Chem. 2011, 15, 1802–1810. [Google Scholar] [CrossRef]
- Keglevich, G.; Kiss, N.Z.; Mucsi, Z.; Körtvélyesi, T. Insights into a surprising reaction: The microwave-assisted direct esterification of phosphinic acids. Org. Biomol. Chem. 2012, 10, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Kiss, N.Z.; Böttger, É.; Drahos, L.; Keglevich, G. Microwave-assisted direct esterification of cyclic phosphinic acids. Heteroat. Chem. 2013, 24, 283–288. [Google Scholar] [CrossRef]
- Mucsi, Z.; Kiss, N.Z.; Keglevich, G. A quantum chemical study on the mechanism and energetics of the direct esterification, thioesterification and amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide. RSC Adv. 2014, 4, 11948–11954. [Google Scholar] [CrossRef]
- Kiss, N.Z.; Keglevich, G. Microwave-assisted direct esterification of cyclic phosphinic acids in the presence of ionic liquids. Tetrahedron Lett. 2016, 57, 971–974. [Google Scholar] [CrossRef]
- Bálint, E.; Jablonkai, E.; Bálint, M.; Keglevich, G. Alkylating esterification of 1-hydroxy-3-phospholene oxides under solventless MW conditions. Heteroat. Chem. 2010, 21, 211–214. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A.; Bölcskei, A.; Drahos, L.; Kraszni, M.; Balogh, G.T. Synthesis and proton dissociation properties of arylphosphonates: A microwave-assisted catalytic arbuzov reaction with aryl bromides. Heteroat. Chem. 2012, 23, 574–582. [Google Scholar] [CrossRef]
- Dargó, G.; Bölcskei, A.; Grün, A.; Béni, S.; Szántó, Z.; Lopata, A.; Keglevich, G.; Balogh, G.T. Proton dissociation properties of arylphosphonates: Determination of accurate Hammett equation parameters. J. Pharm. Biomed. Anal. 2017, 143, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Greiner, I.; Grün, A.; Ludányi, K.; Keglevich, G. Solid–liquid two-phase alkylation of tetraethyl methylenebisphosphonate under microwave irradiation. Heteroat. Chem. 2011, 22, 11–14. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A.; Blastik, Z.; Greiner, I. Solid-liquid phase alkylation of P=O-functionalized CH acidic compounds utilizing phase transfer catalysis and microwave irradiation. Heteroat. Chem. 2011, 22, 174–179. [Google Scholar] [CrossRef]
- Grün, A.; Blastik, Z.; Drahos, L.; Keglevich, G. Microwave-assisted alkylation of diethyl ethoxycarbonylmethylphosphonate under solventless conditions. Heteroat. Chem. 2012, 23, 241–246. [Google Scholar] [CrossRef]
- Keglevich, G.; Grün, A. Microwave Irradiation as a Substitute for Phase Transfer Catalyst in CAlkylation Reactions. Curr. Green Chem. 2015, 2, 254–263. [Google Scholar] [CrossRef]
- Grün, A.; Bálint, E.; Keglevich, G. Solid-Liquid Phase C-Alkylation of Active Methylene Containing Compounds under Microwave Conditions. Catalysts 2015, 5, 634–652. [Google Scholar] [CrossRef]
- Grün, A.; Blastik, Z.; Drahos, L.; Keglevich, G. Dialkylation of diethyl ethoxycarbonylmethylphosphonate under microwave and solventless conditions. Heteroat. Chem. 2014, 25, 107–113. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrényi, A. Eco-Friendly Accomplishment of the Extended Kabachnik–Fields Reaction; a Solvent- and Catalyst-Free Microwave-Assisted Synthesis of α-Aminophosphonates and α-Aminophosphine Oxides. Lett. Org. Chem. 2008, 5, 616–622. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields Reaction: Mechanism and Synthetic Use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef] [PubMed]
- Tajti, Á.; Bálint, E.; Keglevich, G. Synthesis of ethyl octyl α-aminophosphonate derivatives. Curr. Org. Synth. 2016, 13, 638–645. [Google Scholar] [CrossRef]
- Bálint, E.; Tóth, R.E.; Keglevich, G. Synthesis of alkyl α-aminomethyl-phenylphosphinates and N,N-bis(alkoxyphenylphosphinylmethyl)amines by the microwave-assisted Kabachnik–Fields reaction. Heteroat. Chem. 2016, 27, 323–335. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Kalocsai, D.; Mátravölgyi, B.; Karaghiosoff, K.; Czugler, M.; Keglevich, G. Synthesis and utilization of optically active α-aminophosphonate derivatives by Kabachnik-Fields reaction. Tetrahedron 2017, 73, 5659–5667. [Google Scholar] [CrossRef]
- Bálint, E.; Tripolszky, A.; Jablonkai, E.; Karaghiosoff, K.; Czugler, M.; Mucsi, Z.; Kollár, L.; Pongrácz, P.; Keglevich, G. Synthesis and use of α-aminophosphine oxides and N,N-bis(phosphinoylmethyl)amines—A study on the related ring platinum complexes. J. Organomet. Chem. 2016, 801, 111–121. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrényi, A.; Szöllősy, Á.; Drahos, L. Synthesis of bis(phosphonatomethyl)-,bis(phosphinatomethyl)-, and bis(phosphinoxidomethyl)amines, as well as related ring bis(phosphine) platinum complexes. Synth. Commun. 2011, 41, 2265–2272. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Pintér, G.; Szöllősy, Á.; Holczbauer, T.; Czugler, M.; Drahos, L.; Körtvélyesi, T.; Keglevich, G. Synthesis and utilization of the bis(>P(O)CH2)amine derivatives obtained by the double Kabachnik–Fields reaction with cyclohexylamine; Quantum chemical and X-ray study of the related bidentate chelate platinum complexes. Curr. Org. Chem. 2012, 16, 547–554. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Pongrácz, P.; Kollár, L.; Drahos, L.; Holczbauer, T.; Czugler, M.; Keglevich, G. N-Benzyl and N-aryl bis(phospha-Mannich adducts): Synthesis and catalytic activity of the related bidentate chelate platinum complexes in hydroformylation. J. Organomet. Chem. 2012, 717, 75–82. [Google Scholar] [CrossRef]
- Li, Y.H.; Das, S.; Zhou, S.L.; Junge, K.; Beller, M. General and Selective Copper-Catalyzed Reduction of Tertiary and Secondary Phosphine Oxides: Convenient Synthesis of Phosphines. J. Am. Chem. Soc. 2012, 134, 9727–9732. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Keglevich, G. The reduction of tertiary phosphine oxides by silanes. Curr. Org. Chem. 2017, 21, 569–585. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, T.; Csatlós, F. The deoxygenation of phosphine oxides under green chemical conditions. Heteroat. Chem. 2015, 26, 199–205. [Google Scholar] [CrossRef]
- Kovács, T.; Urbanics, A.; Csatlós, F.; Binder, J.; Falk, A.; Uhlig, F.; Keglevich, G. A Study on the Deoxygenation of Phosphine Oxides by Different Silane Derivatives. Curr. Org. Synth. 2016, 13, 148–153. [Google Scholar] [CrossRef]
- Kovács, T.; Urbanics, A.; Csatlós, F.; Keglevich, G. A study on the deoxygenation of trialkyl-, dialkyl-phenyl- and alkyl-diphenyl phosphine oxides by hydrosilanes. Heteroat. Chem. 2017, 28, e21376. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Yamada, N.; Ohshiro, Y.; Agawa, T. Palladium-catalyzed New Carbon-Phosphorus Bond Formation. Bull. Chem. Soc. Jpn. 1982, 55, 909–913. [Google Scholar] [CrossRef]
- Hirao, T.; Masunaga, T.; Ohshiro, Y.; Agawa, T. A Novel Synthesis of Dialkyl Arenephosphonates. Synthesis 1981, 1981, 56–57. [Google Scholar] [CrossRef]
- Jablonkai, E.; Keglevich, G. P-ligand-free, microwave-assisted variation of the Hirao reaction under solvent-free conditions; The P-C coupling reaction of >p(O)H species and bromoarenes. Tetrahedron Lett. 2013, 54, 4185–4188. [Google Scholar] [CrossRef]
- Keglevich, G.; Jablonkai, E.; Balázs, L.B. A “green” variation of the Hirao reaction: The P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides with bromoarenes using a P-ligand-free Pd(OAc)2catalyst under microwave and solvent-free conditions. RSC Adv. 2014, 4, 22808–22816. [Google Scholar] [CrossRef]
- Keglevich, G.; Henyecz, R.; Mucsi, Z.; Kiss, N.Z. The Palladium Acetate-Catalyzed Microwave-Assisted Hirao Reaction without an Added Phosphorus Ligand as a “Green” Protocol: A Quantum Chemical Study on the Mechanism. Adv. Synth. Catal. 2017, 359, 4322–4331. [Google Scholar] [CrossRef] [PubMed]
- Ergan, B.T.; Bayaromoglu, M. Poly (l-lactic acid) synthesis using continuous microwave irradiation–simultaneous cooling method. Chem. Eng. Commun. 2018, 205, 1665–1677. [Google Scholar] [CrossRef]
- Chen, S.-T.; Chiou, S.-H.; Wang, K.-T. Preparative scale organic synthesis using a kitchen microwave oven. J. Chem. Soc. Chem. Commun. 1990, 807–809. [Google Scholar] [CrossRef]
- Pipus, G.; Plazl, I.; Koloini, T. Esterification of benzoic acid in microwave tubular flow reactor. Chem. Eng. J. 2000, 76, 239–245. [Google Scholar] [CrossRef]
- Cablewski, T.; Faux, A.F.; Strauss, C.R. Development and Application of a Continuous Microwave Reactor for Organic Synthesis. J. Org. Chem. 1994, 59, 3408–3412. [Google Scholar] [CrossRef]
- Krull, M.; Moschhaeuser, R. Continuous Method for Producing Esters of Aromatic Carboxylic Acids. U.S. Patent 0088918, 12 April 2012. [Google Scholar]
- Tajti, Á.; Tóth, N.; Bálint, E.; Keglevich, G. Esterification of benzoic acid in a continuous flow microwave reactor. J. Flow Chem. 2018, 8, 11–19. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Drahos, L.; Ilia, G.; Keglevich, G. Alcoholysis of dialkyl phosphites under Microwave conditions. Curr. Org. Chem. 2013, 17, 555–562. [Google Scholar] [CrossRef]
- Bálint, E.; Tripolszky, A.; Tajti, Á. Synthesis of α-aminophosphonates by the Kabachnik–Fields reaction. In Organophosphorus Chemistry; Keglevich, G., Ed.; De Gruyter: Berlin, Germany, 2018; pp. 108–147. [Google Scholar]
- Keglevich, G.; Bálint, E.; Tajti, Á.; Mátravölgyi, B.; Balogh, G.T.; Bálint, M.; Ilia, G. Microwave-assisted alcoholysis of dialkyl phosphites by ethylene glycol and ethanolamine. Pure Appl. Chem. 2014, 86, 1723–1728. [Google Scholar] [CrossRef]
- Tajti, Á.; Keglevich, G.; Bálint, E. Microwave-assisted alcoholysis of dialkyl H-phosphonates by diols and amino alcohols. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 769–775. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Yuan, Y.-Y.; Du, J.-Z.; Yang, X.-Z.; Wang, J. Recent Progress in Polyphosphoesters: From Controlled Synthesis to Biomedical Applications. Macromol. Biosci. 2009, 9, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Troev, K.D. Polyphosphoesters: Chemistry and Application; Elsevier: Oxford, UK, 2012. [Google Scholar]
- Bálint, E.; Tajti, Á.; Tóth, N.; Keglevich, G. Continuous Flow Alcoholysis of Dialkyl H-Phosphonates with Aliphatic Alcohols. Molecules 2018, 23, 1618. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Tajti, Á.; Ladányi-Pára, K.; Tóth, N.; Mátravölgyi, B.; Keglevich, G. Continuous flow synthesis of α-aryl-α-aminophosphonates. Pure Appl. Chem. 2019, 91, 67–76. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Ádám, A.; Csontos, I.; Karaghiosoff, K.; Czugler, M.; Ábrányi-Balogh, P.; Keglevich, G. The synthesis of α-aryl-α-aminophosphonates and α-aryl-α-aminophosphine oxides by the microwave-assisted Pudovik reaction. Beilstein J. Org. Chem. 2017, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bálint, E.; Tajti, Á.; Keglevich, G. Application of the Microwave Technique in Continuous Flow Processing of Organophosphorus Chemical Reactions. Materials 2019, 12, 788. https://doi.org/10.3390/ma12050788
Bálint E, Tajti Á, Keglevich G. Application of the Microwave Technique in Continuous Flow Processing of Organophosphorus Chemical Reactions. Materials. 2019; 12(5):788. https://doi.org/10.3390/ma12050788
Chicago/Turabian StyleBálint, Erika, Ádám Tajti, and György Keglevich. 2019. "Application of the Microwave Technique in Continuous Flow Processing of Organophosphorus Chemical Reactions" Materials 12, no. 5: 788. https://doi.org/10.3390/ma12050788
APA StyleBálint, E., Tajti, Á., & Keglevich, G. (2019). Application of the Microwave Technique in Continuous Flow Processing of Organophosphorus Chemical Reactions. Materials, 12(5), 788. https://doi.org/10.3390/ma12050788