Structural Characterisation and Chemical Stability of Commercial Fibrous Carbons in Molten Lithium Salts

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
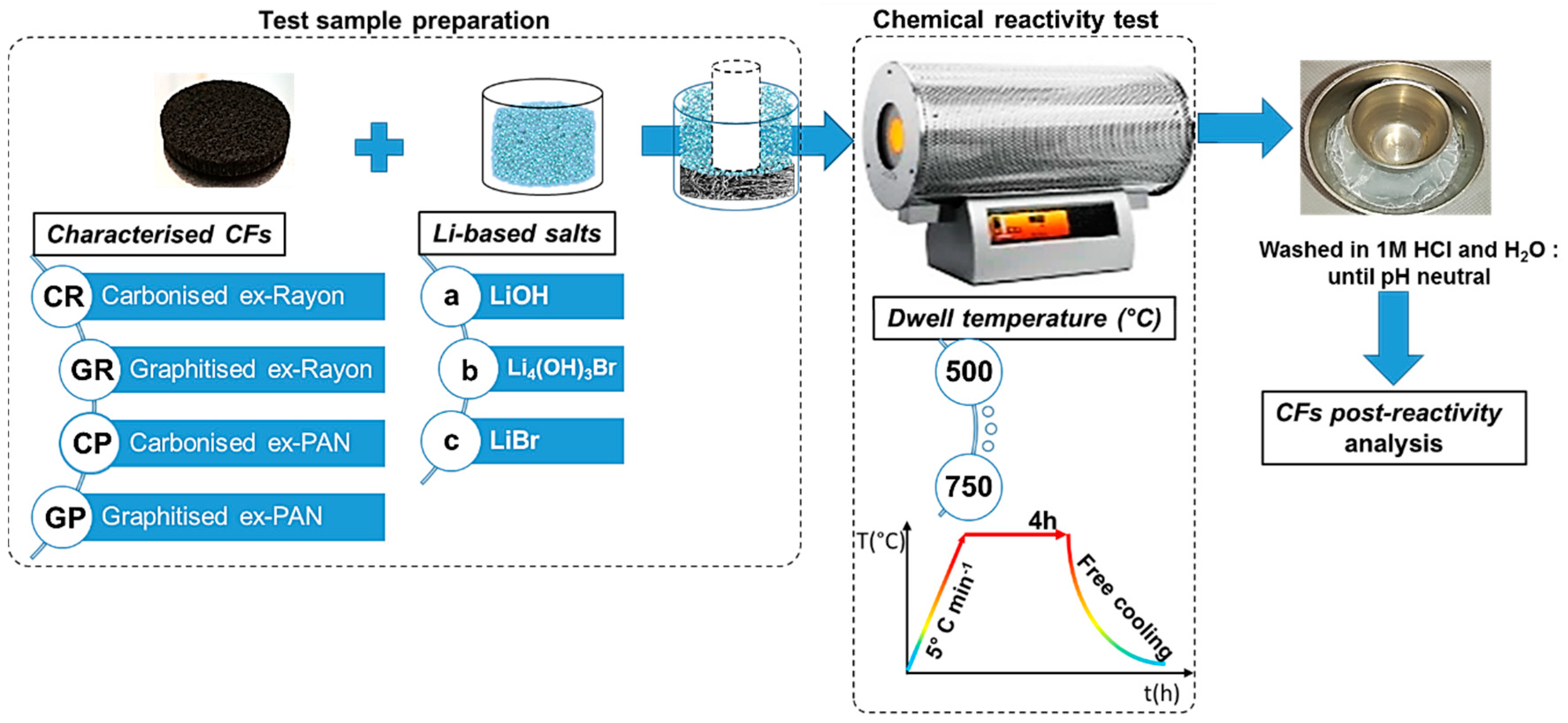
2.2. Chemical Reactivity Tests
2.3. Characterisation
2.3.1. Elemental Analysis
2.3.2. X-ray Diffraction (XRD)
2.3.3. Raman Spectroscopy
2.3.4. Scanning Electron Microscopy
2.3.5. Textural Properties of CFs
3. Results and Discussion
3.1. CFs Elemental Composition
3.2. CFs Nanostructure
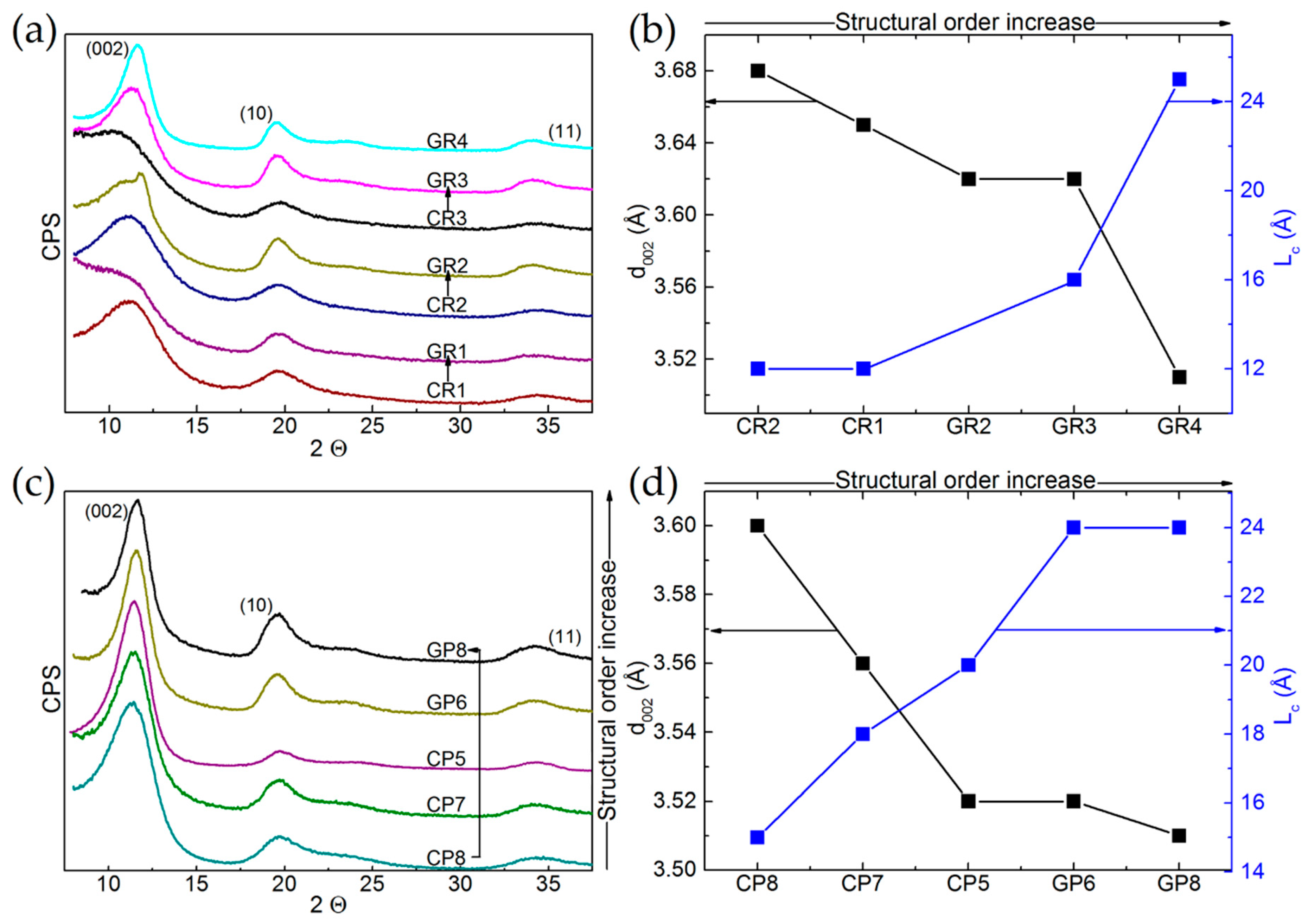
3.2.1. XRD Analysis
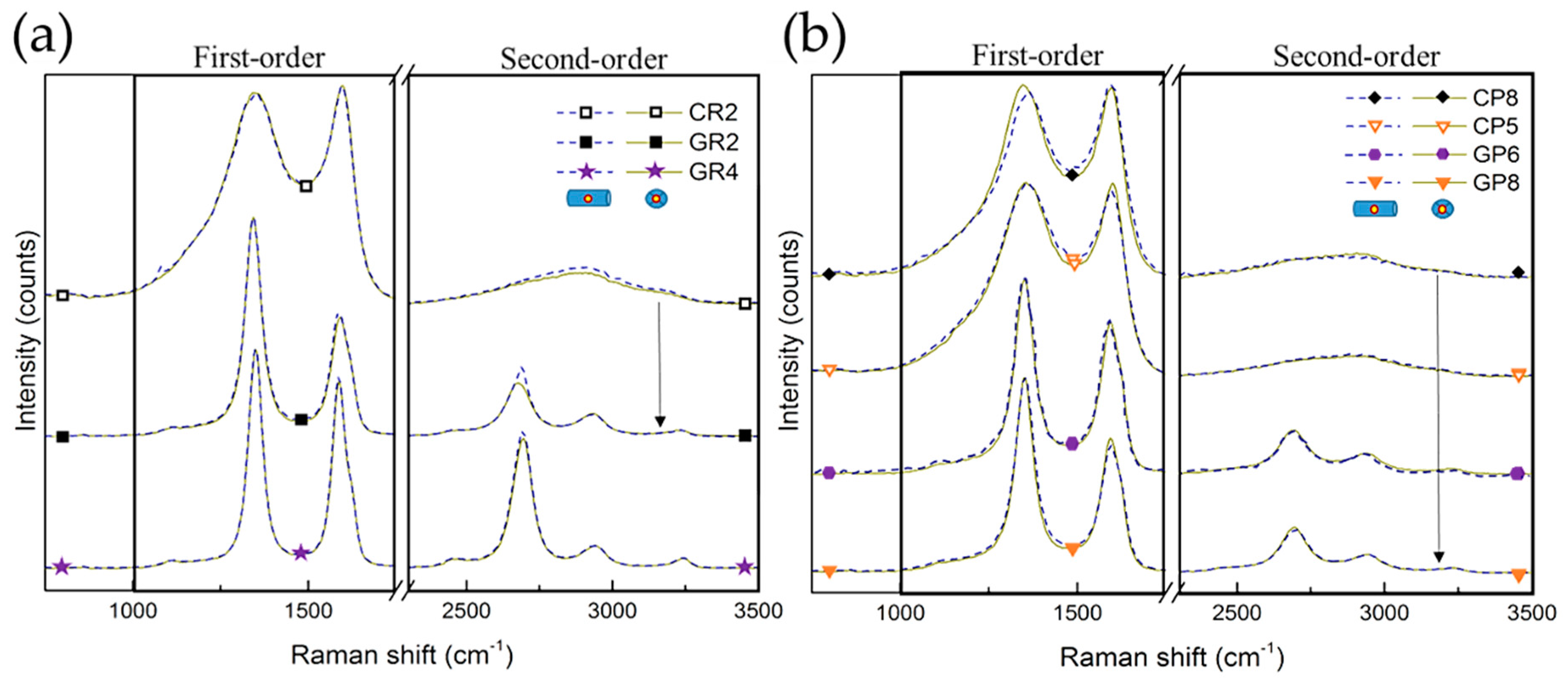
3.2.2. Raman Spectroscopy Analysis
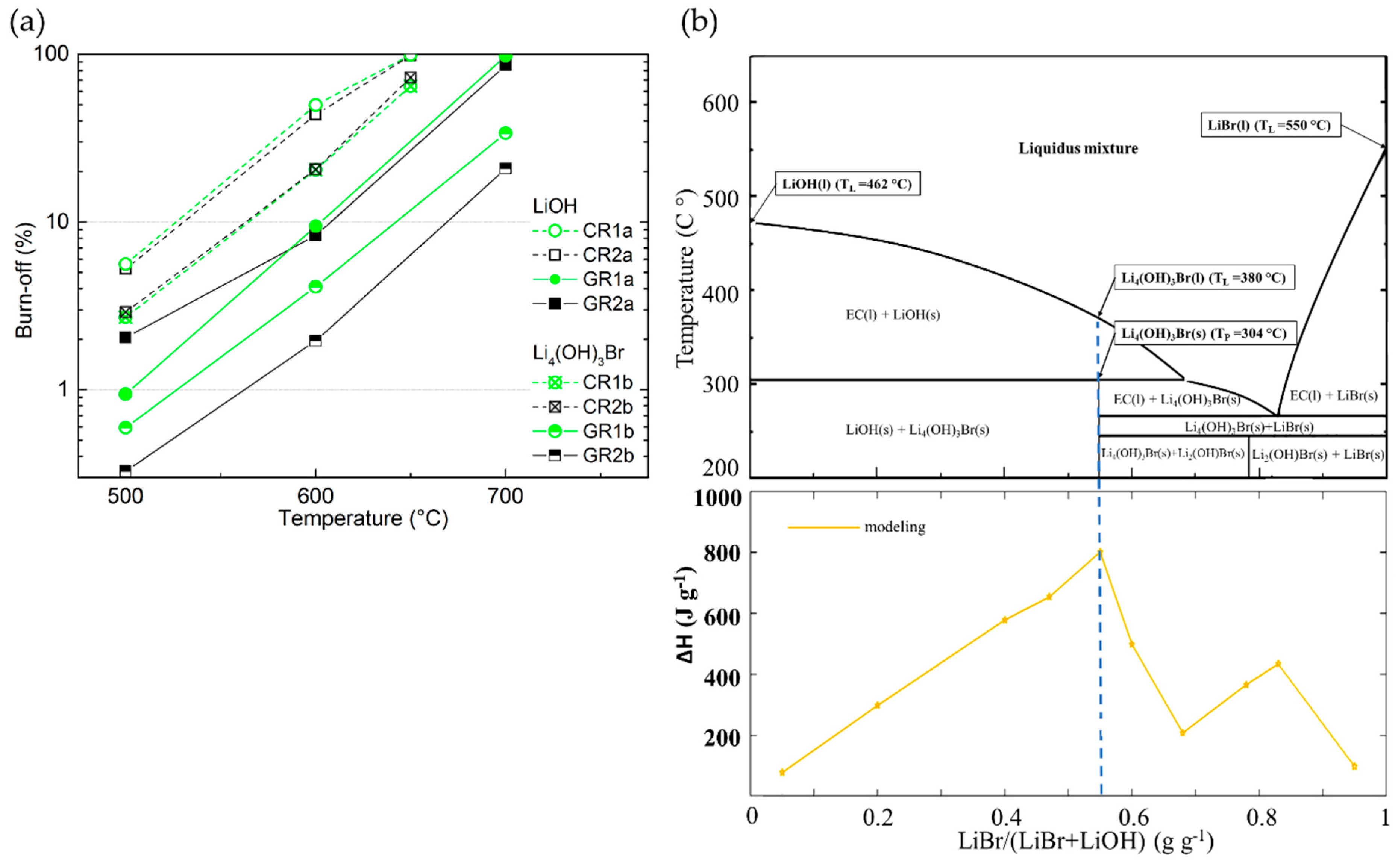
3.3. High-Temperature CFs Chemical Stability in Molten Li-Based Salts
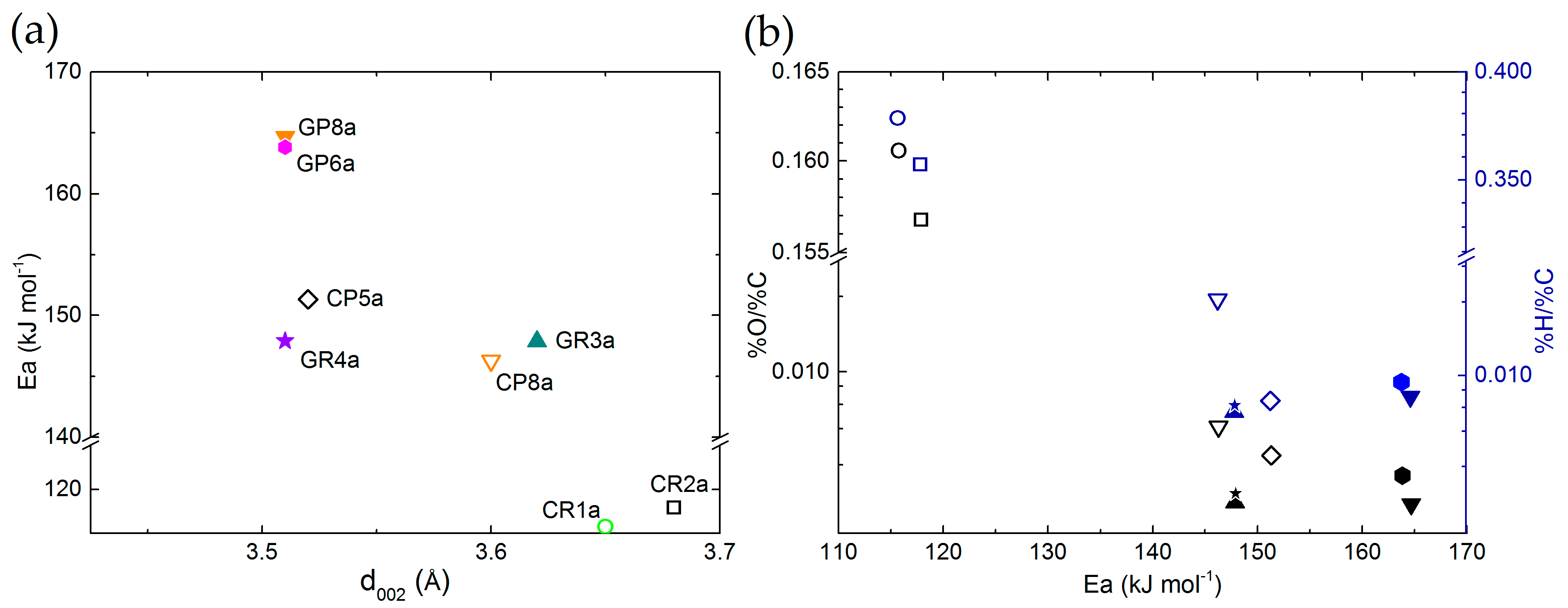
3.3.1. Chemical Stability of CFs in LiOH with Regard to Their Structural Properties
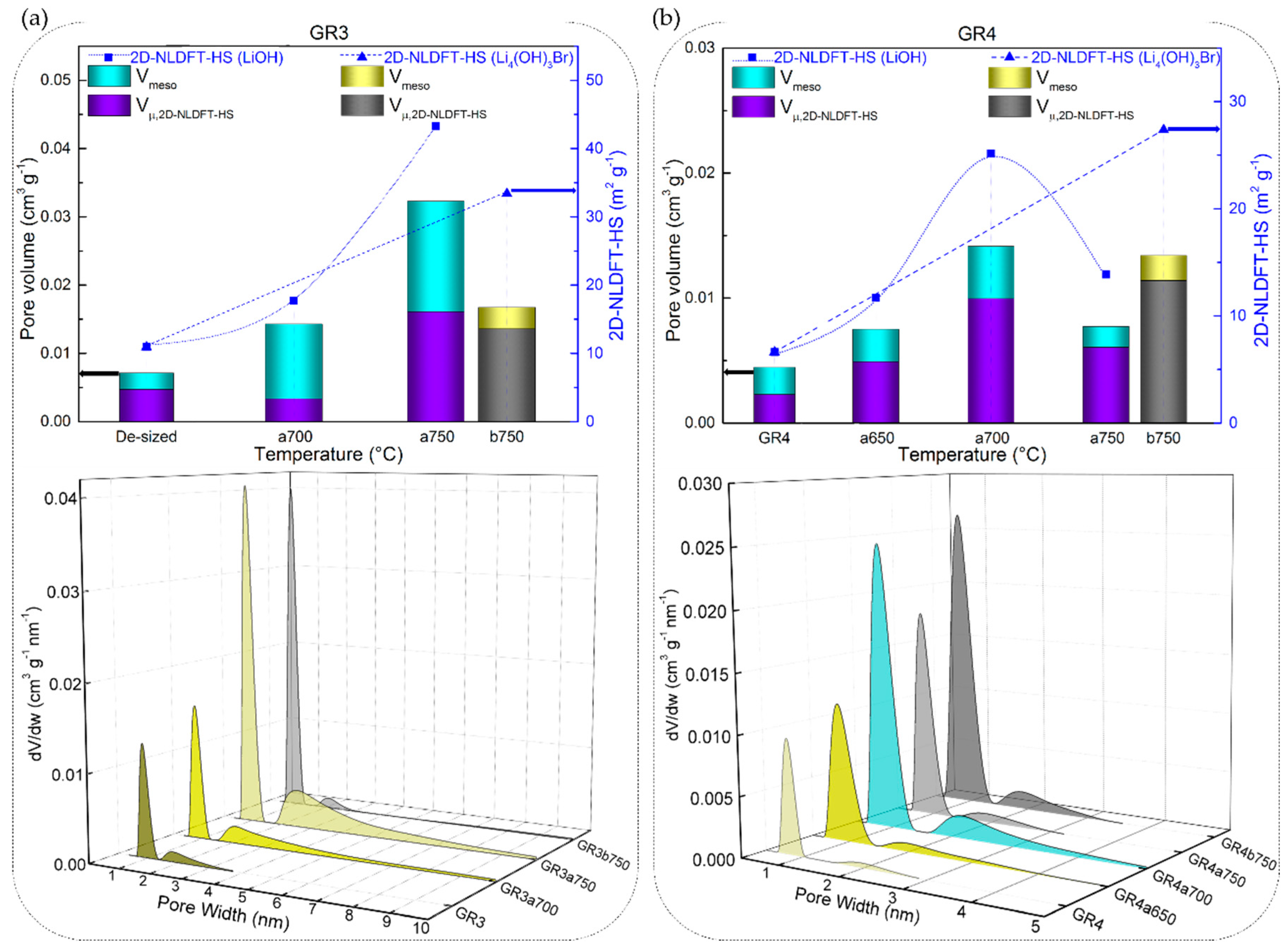
3.3.2. Chemical Stability of CFs in LiOH with Regard to Their Textural Properties
3.3.3. Chemical Stability of CFs in LiOH with Regard to Their Morphology
3.3.4. Chemical Stability of CFs in LiBr or Li4(OH)3Br Compared to that in LiOH
3.3.5. Estimation of CFs Chemical Stability at the TES Application Temperature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nejat, P.; Jomehzadeh, F.; Taheri, M.M.; Gohari, M.; Majid, M.Z.A. A global review of energy consumption, CO2 emissions and policy in the residential sector (with an overview of the top ten CO2 emitting countries). Renew. Sustain. Energy Rev. 2015, 43, 843–862. [Google Scholar] [CrossRef]
- Dincer, I.; Acar, C. Chapter 1.1—Potential Energy Solutions for Better Sustainability. In Exergetic, Energetic and Environmental Dimensions; Dincer, I., Colpan, C.O., Kizilkan, O., Eds.; Academic Press: New York, NY, USA, 2018; pp. 3–37. ISBN 978-0-12-813734-5. [Google Scholar]
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strategy Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Global Market Outlook for Solar Power 2018–2022; SolarPower: Brussels, Belgium, 2018.
- Calderón, A.; Barreneche, C.; Hernández-Valle, K.; Galindo, E.; Segarra, M.; Fernández, A.I. Where is Thermal Energy Storage (TES) research going?—A bibliometric analysis. Sol. Energy 2019. [Google Scholar] [CrossRef]
- Palomba, V.; Frazzica, A. Recent advancements in sorption technology for solar thermal energy storage applications. Sol. Energy 2018. [Google Scholar] [CrossRef]
- Pelay, U.; Luo, L.; Fan, Y.; Stitou, D.; Rood, M. Thermal energy storage systems for concentrated solar power plants. Renew. Sustain. Energy Rev. 2017, 79, 82–100. [Google Scholar] [CrossRef]
- Ibrahim, N.I.; Al-Sulaiman, F.A.; Rahman, S.; Yilbas, B.S.; Sahin, A.Z. Heat transfer enhancement of phase change materials for thermal energy storage applications: A critical review. Renew. Sustain. Energy Rev. 2017, 74, 26–50. [Google Scholar] [CrossRef]
- Liu, M.; Steven Tay, N.H.; Bell, S.; Belusko, M.; Jacob, R.; Will, G.; Saman, W.; Bruno, F. Review on concentrating solar power plants and new developments in high temperature thermal energy storage technologies. Renew. Sustain. Energy Rev. 2016, 53, 1411–1432. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, S. Medium-and high-temperature latent heat thermal energy storage: Material database, system review, and corrosivity assessment. Int. J. Energy Res. 2019, 43, 621–661. [Google Scholar] [CrossRef]
- Pandey, A.K.; Hossain, M.S.; Tyagi, V.V.; Abd Rahim, N.; Selvaraj, J.A.L.; Sari, A. Novel approaches and recent developments on potential applications of phase change materials in solar energy. Renew. Sustain. Energy Rev. 2018, 82, 281–323. [Google Scholar] [CrossRef]
- Achchaq, F.; Barrio, E.P.D. A proposition of peritectic structures as candidates for thermal energy storage. Energy Procedia 2017, 139, 346–351. [Google Scholar] [CrossRef]
- Achchaq, F.; Palomo del Barrio, E.; Renauld, A.; Ben-Khemis, S. Characterization of Li2K(OH)3 as material for thermal energy storage at high temperature. In Proceedings of the 13th International Conference on Energy Storage, Beijing, China, 19–21 May 2015; Volume 2, pp. 1–8. [Google Scholar]
- Badenhorst, H. A review of the application of carbon materials in solar thermal energy storage. Sol. Energy 2019, 192, 35–68. [Google Scholar] [CrossRef]
- Nomura, T.; Tabuchi, K.; Zhu, C.; Sheng, N.; Wang, S.; Akiyama, T. High thermal conductivity phase change composite with percolating carbon fiber network. Appl. Energy 2015, 154, 678–685. [Google Scholar] [CrossRef]
- Jiang, Z.; Ouyang, T.; Yang, Y.; Chen, L.; Fan, X.; Chen, Y.; Li, W.; Fei, Y. Thermal conductivity enhancement of phase change materials with form-stable carbon bonded carbon fiber network. Mater. Des. 2018, 143, 177–184. [Google Scholar] [CrossRef]
- Fukai, J.; Kanou, M.; Kodama, Y.; Miyatake, O. Thermal conductivity enhancement of energy storage media using carbon fibers. Energy Convers. Manag. 2000, 41, 1543–1556. [Google Scholar] [CrossRef]
- Hamada, Y.; Otsu, W.; Fukai, J.; Morozumi, Y.; Miyatake, O. Anisotropic heat transfer in composites based on high-thermal conductive carbon fibers. Energy 2005, 30, 221–233. [Google Scholar] [CrossRef]
- Frusteri, F.; Leonardi, V.; Vasta, S.; Restuccia, G. Thermal conductivity measurement of a PCM based storage system containing carbon fibers. Appl. Therm. Eng. 2005, 25, 1623–1633. [Google Scholar] [CrossRef]
- Karaipekli, A.; Sarı, A.; Kaygusuz, K. Thermal conductivity improvement of stearic acid using expanded graphite and carbon fiber for energy storage applications. Renew. Energy 2007, 32, 2201–2210. [Google Scholar] [CrossRef]
- Wang, J.; Xie, H.; Xin, Z.; Li, Y.; Yin, C. Investigation on thermal properties of heat storage composites containing carbon fibers. J. Appl. Phys. 2011, 110, 094302. [Google Scholar] [CrossRef]
- Samimi, F.; Babapoor, A.; Azizi, M.; Karimi, G. Thermal management of a Li-ion battery using carbon fiber-PCM composites. Appl. Therm. Eng. 2015, 82, 281–290. [Google Scholar] [CrossRef]
- Huang, X.; Alva, G.; Liu, L.; Fang, G. Microstructure and thermal properties of cetyl alcohol/high density polyethylene composite phase change materials with carbon fiber as shape-stabilized thermal storage materials. Appl. Energy 2017, 200, 19–27. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, Z.; Guo, Q.; Wu, G. Preparation and thermal properties of short carbon fibers/erythritol phase change materials. Energy Convers. Manag. 2017, 136, 220–228. [Google Scholar] [CrossRef]
- Ye, F.; Ge, Z.; Ding, Y.; Yang, J. Multi-walled carbon nanotubes added to Na2CO3/MgO composites for thermal energy storage. Particuology 2014, 15, 56–60. [Google Scholar] [CrossRef]
- Singh, D.; Kim, T.; Zhao, W.; Yu, W.; France, D.M. Development of graphite foam infiltrated with MgCl2 for a latent heat based thermal energy storage (LHTES) system. Renew. Energy 2016, 94, 660–667. [Google Scholar] [CrossRef]
- Guillot, S.; Faik, A.; Rakhmatullin, A.; Lambert, J.; Veron, E.; Echegut, P.; Bessada, C.; Calvet, N.; Py, X. Corrosion effects between molten salts and thermal storage material for concentrated solar power plants. Appl. Energy 2012, 94, 174–181. [Google Scholar] [CrossRef]
- Calvet, N.; Gomez, J.C.; Faik, A.; Roddatis, V.V.; Meffre, A.; Glatzmaier, G.C.; Doppiu, S.; Py, X. Compatibility of a post-industrial ceramic with nitrate molten salts for use as filler material in a thermocline storage system. Appl. Energy 2013, 109, 387–393. [Google Scholar] [CrossRef]
- Vasu, A.; Hagos, F.Y.; Noor, M.M.; Mamat, R.; Azmi, W.H.; Abdullah, A.A.; Ibrahim, T.K. Corrosion effect of phase change materials in solar thermal energy storage application. Renew. Sustain. Energy Rev. 2017, 76, 19–33. [Google Scholar] [CrossRef]
- Achchaq, F.; Palomo del Barrio, E.; Lebraud, E.; Péchev, S.; Toutain, J. Development of a new LiBr/LiOH-based alloy for thermal energy storage. J. Phys. Chem. Solids 2019, 131, 173–179. [Google Scholar] [CrossRef]
- Karakashov, B.; Toutain, J.; Achchaq, F.; Legros, P.; Fierro, V.; Celzard, A. Permeability of fibrous carbon materials. J. Mater. Sci. 2019, 54, 13537–13556. [Google Scholar] [CrossRef]
- Naito, K.; Yang, J.-M.; Xu, Y.; Kagawa, Y. Enhancing the thermal conductivity of polyacrylonitrile- and pitch-based carbon fibers by grafting carbon nanotubes on them. Carbon 2010, 6, 1849–1857. [Google Scholar] [CrossRef]
- Szabó, L.; Imanishi, S.; Tetsuo, F.; Hirose, D.; Ueda, H.; Tsukegi, T.; Ninomiya, K.; Takahashi, K. Lignin as a Functional Green Coating on Carbon Fiber Surface to Improve Interfacial Adhesion in Carbon Fiber Reinforced Polymers. Materials 2019, 12, 159. [Google Scholar] [CrossRef]
- Wietelmann, U.; Bauer, R.J. Lithium and Lithium Compounds. Ullmanns Encycl. Ind. Chem. 2000, 21, 339–363. [Google Scholar] [CrossRef]
- Linares-Solano, A.; Lozano-Castello, D.; Lillo-Rodenas, M.; Cazorla-Amoros, D. Carbon Activation by Alkaline Hydroxides. In Chemistry and Physics of Carbon; Radovic, L., Ed.; CRC Press: New York, NY, USA, 2007; pp. 1–62. [Google Scholar]
- Achchaq, F.; Risueño, E.; Mahroug, I.; Legros, P.; Lebraud, E.; Karakashov, B.; Palomo del Barrio, E.; Celzard, A.; Fierro, V.; Toutain, J. Development of a Carbon Felt/Salt-Based Hybrid Material for Thermal Energy Storage Applications. J. Energy Power Eng. 2018, 12. [Google Scholar] [CrossRef]
- Pope, C.G. X-Ray Diffraction and the Bragg Equation. J. Chem. Educ. 1997, 74, 129. [Google Scholar] [CrossRef]
- Celzard, A.; Pasc, A.; Schaefer, S.; Mandel, K.; Ballweg, T.; Li, S.; Medjahdi, G.; Nicolas, V.; Fierro, V. Floating hollow carbon spheres for improved solar evaporation. Carbon 2019, 146, 232–247. [Google Scholar] [CrossRef]
- Pimenta, M.A.; Dresselhaus, G.; Dresselhaus, M.S.; Cançado, L.G.; Jorio, A.; Saito, R. Studying disorder in graphite-based systems by Raman spectroscopy. Phys. Chem. Chem. Phys. 2007, 9, 1276–1290. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Manciu, F.S.; Oh, Y.; Barath, A.; Rusheen, A.E.; Kouzani, A.Z.; Hodges, D.; Guerrero, J.; Tomshine, J.; Lee, K.H.; Bennet, K.E. Analysis of Carbon-Based Microelectrodes for Neurochemical Sensing. Materials 2019, 12, 3186. [Google Scholar] [CrossRef]
- Chen, J. (Ed.) Activated Carbon Fiber and Textiles, 1st ed.; Woodhead Publishing: Cambridge, UK, 2016. [Google Scholar]
- Chiang, Y.-C.; Lee, C.-Y.; Lee, H.-C. Surface chemistry of polyacrylonitrile- and rayon-based activated carbon fibers after post-heat treatment. Mater. Chem. Phys. 2007, 101, 199–210. [Google Scholar] [CrossRef]
- Rahaman, M.S.A.; Ismail, A.F.; Mustafa, A. A review of heat treatment on polyacrylonitrile fiber. Polym. Degrad. Stab. 2007, 92, 1421–1432. [Google Scholar] [CrossRef]
- Nunna, S.; Maghe, M.; Rana, R.; Varley, R.J.; Knorr, D.B.; Sands, J.M.; Creighton, C.; Henderson, L.C.; Naebe, M. Time Dependent Structure and Property Evolution in Fibres during Continuous Carbon Fibre Manufacturing. Materials 2019, 12, 1069. [Google Scholar] [CrossRef]
- Tang, M.M.; Bacon, R. Carbonization of cellulose fibers—I. Low temperature pyrolysis. Carbon 1964, 2, 211–220. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, B.-J. Carbon Fibers and Their Composites. In Carbon Fibers; Springer Series in Materials Science; Springer: Dordrecht, The Netherlands, 2015; pp. 275–317. ISBN 978-94-017-9477-0. [Google Scholar]
- Al Aiti, M.; Jehnichen, D.; Fischer, D.; Brünig, H.; Heinrich, G. On the morphology and structure formation of carbon fibers from polymer precursor systems. Prog. Mater. Sci. 2018, 98, 477–551. [Google Scholar] [CrossRef]
- Li, C.; Xian, G. Experimental and Modeling Study of the Evolution of Mechanical Properties of PAN-Based Carbon Fibers at Elevated Temperatures. Materials 2019, 12, 724. [Google Scholar] [CrossRef] [PubMed]
- Fink, H.-P.; Hofmann, D.; Philipp, B. Some aspects of lateral chain order in cellulosics from X-ray scattering. Cellulose 1995, 2, 51–70. [Google Scholar] [CrossRef]
- Wada, M.; Okano, T.; Sugiyama, J. Allomorphs of native crystalline cellulose I evaluated by two equatoriald-spacings. J. Wood Sci. 2001, 47, 124–128. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Nishiyama, Y.; Wada, M.; Kuga, S. Graphitization of highly crystalline cellulose. Carbon 2001, 39, 1051–1056. [Google Scholar] [CrossRef]
- Dumanli, A.G.; Windle, A.H. Carbon fibres from cellulosic precursors: A review. J. Mater. Sci. 2012, 47, 4236–4250. [Google Scholar] [CrossRef]
- Byrne, N.; Chen, J.; Fox, B. Enhancing the carbon yield of cellulose based carbon fibres with ionic liquid impregnates. J. Mater. Chem. A 2014, 2, 15758–15762. [Google Scholar] [CrossRef]
- Carrott, P.J.M.; Nabais, J.M.V.; Ribeiro Carrott, M.M.L.; Pajares, J.A. Preparation of activated carbon fibres from acrylic textile fibres. Carbon 2001, 39, 1543–1555. [Google Scholar] [CrossRef]
- Maciá-Agulló, J.A.; Moore, B.C.; Cazorla-Amorós, D.; Linares-Solano, A. Influence of carbon fibres crystallinities on their chemical activation by KOH and NaOH. Microporous Mesoporous Mater. 2007, 101, 397–405. [Google Scholar] [CrossRef]
- Dobiášová, L.; Starý, V.; Glogar, P.; Valvoda, V. Analysis of carbon fibers and carbon composites by asymmetric X-ray diffraction technique. Carbon 1999, 37, 421–425. [Google Scholar] [CrossRef]
- Lu, L.; Sahajwalla, V.; Kong, C.; Harris, D. Quantitative X-ray diffraction analysis and its application to various coals. Carbon 2001, 39, 1821–1833. [Google Scholar] [CrossRef]
- Sonibare, O.O.; Haeger, T.; Foley, S.F. Structural characterization of Nigerian coals by X-ray diffraction, Raman and FTIR spectroscopy. Energy 2010, 35, 5347–5353. [Google Scholar] [CrossRef]
- Kong, K.; Deng, L.; Kinloch, I.A.; Young, R.J.; Eichhorn, S.J. Production of carbon fibres from a pyrolysed and graphitised liquid crystalline cellulose fibre precursor. J. Mater. Sci. 2012, 47, 5402–5410. [Google Scholar] [CrossRef]
- Cuesta, A.; Dhamelincourt, P.; Laureyns, J.; Martínez-Alonso, A.; Tascón, J.M.D. Raman microprobe studies on carbon materials. Carbon 1994, 32, 1523–1532. [Google Scholar] [CrossRef]
- Zickler, G.A.; Smarsly, B.; Gierlinger, N.; Peterlik, H.; Paris, O. A reconsideration of the relationship between the crystallite size La of carbons determined by X-ray diffraction and Raman spectroscopy. Carbon 2006, 44, 3239–3246. [Google Scholar] [CrossRef]
- Zabihi, O.; Shafei, S.; Fakhrhoseini, S.M.; Ahmadi, M.; Ajdari Nazarloo, H.; Stanger, R.; Anh Tran, Q.; Lucas, J.; Wall, T.; Naebe, M. Low-Cost Carbon Fibre Derived from Sustainable Coal Tar Pitch and Polyacrylonitrile: Fabrication and Characterisation. Materials 2019, 12, 1281. [Google Scholar] [CrossRef]
- Dresselhaus, M.S.; Dresselhaus, G.; Sugihara, K.; Spain, I.L.; Goldberg, H.A. (Eds.) Graphite Fibers and Filaments; Springer Series in Materials Science; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Zhong, S.; Padeste, C.; Kazacos, M.; Skyllas-Kazacos, M. Comparison of the physical, chemical and electrochemical properties of rayon-and polyacrylonitrile-based graphite felt electrodes. J. Power Sources 1993, 45, 29–41. [Google Scholar] [CrossRef]
- Ko, T.-H.; Liao, Y.-K.; Liu, C.-H. Effects of graphitization of PAN-based carbon fiber cloth on its use as gas diffusion layers in proton exchange membrane fuel cells. New Carbon Mater. 2007, 22, 97–101. [Google Scholar] [CrossRef]
- Dai, Z.; Shi, F.; Zhang, B.; Li, M.; Zhang, Z. Effect of sizing on carbon fiber surface properties and fibers/epoxy interfacial adhesion. Appl. Surf. Sci. 2011, 257, 6980–6985. [Google Scholar] [CrossRef]
- He, Z.; Shi, L.; Shen, J.; He, Z.; Liu, S. Effects of nitrogen doping on the electrochemical performance of graphite felts for vanadium redox flow batteries. Int. J. Energy Res. 2015, 39, 709–716. [Google Scholar] [CrossRef]
- Qian, X.; Zhang, Y.G.; Wang, X.F.; Heng, Y.J.; Zhi, J.H. Effect of carbon fiber surface functionality on the moisture absorption behavior of carbon fiber/epoxy resin composites. Surf. Interface Anal. 2016, 48, 1271–1277. [Google Scholar] [CrossRef]
- Pradere, C.; Batsale, J.C.; Goyhénèche, J.M.; Pailler, R.; Dilhaire, S. Thermal properties of carbon fibers at very high temperature. Carbon 2009, 47, 737–743. [Google Scholar] [CrossRef]
- Qiu, L.; Zheng, X.H.; Zhu, J.; Su, G.P.; Tang, D.W. The effect of grain size on the lattice thermal conductivity of an individual polyacrylonitrile-based carbon fiber. Carbon 2013, 51, 265–273. [Google Scholar] [CrossRef]
- Harris, P.J.F. Rosalind Franklin’s work on coal, carbon, and graphite. Interdiscip. Sci. Rev. 2001, 26, 204–210. [Google Scholar] [CrossRef]
- Jagiello, J.; Olivier, J.P. 2D-NLDFT adsorption models for carbon slit-shaped pores with surface energetical heterogeneity and geometrical corrugation. Carbon 2013, 55, 70–80. [Google Scholar] [CrossRef]
- Girón, R.P.; Gil, R.R.; Suárez-Ruiz, I.; Fuente, E.; Ruiz, B. Adsorbents/catalysts from forest biomass fly ash. Influence of alkaline activating agent. Microporous Mesoporous Mater. 2015, 209, 45–53. [Google Scholar] [CrossRef]
- Mikova, N.M.; Chesnokov, N.V.; Kuznetsov, B.N. Study of High Porous Carbons Prepared by the Alkaline Activation of Anthracites. J. Sib. Fed. Univ. Chem. 2009, 1, 3–10. [Google Scholar]
- Mora, E.; Blanco, C.; Pajares, J.A.; Santamaría, R.; Menéndez, R. Chemical activation of carbon mesophase pitches. J. Colloid Interface Sci. 2006, 298, 341–347. [Google Scholar] [CrossRef]
- Sutcu, H.; Dural, A. Effect of Hydroxides on Carbonization of Bituminous Coal. Coal Prep. 2006, 26, 201–208. [Google Scholar] [CrossRef]
- Aweed, K.A. Preparation of Activated Carbon from Wastes Tires by Minerals Hydroxide (LiOH, NaOH and KOH). Tikrit J. Pure Sci. 2008, 13, 71–74. [Google Scholar]
- Tamarkina, Y.V.; Shendrik, T.G.; Kucherenko, V.A.; Khabarova, T.V. Conversion of Alexandriya brown coal into microporous carbons under alkali activation. Chemistry 2012, 5, 24–36. [Google Scholar]
- Chowdhury, Z.Z.; Hamid, S.B.A.; Das, R.; Hasan, M.R.; Zain, S.M.; Khalid, K.; Uddin, M.N. Preparation of Carbonaceous Adsorbents from Lignocellulosic Biomass and Their Use in Removal of Contaminants from Aqueous Solution. BioResources 2013, 8, 6523–6555. [Google Scholar] [CrossRef]
- Cabasso, I.; Yuan, Y. Nano Structured Activated Carbon for Hydrogen Storge; Project Final Technical Report (May 2, 2005-Dec. 31, 2012); Research Foundation of State University of New York, Polymer Research Institute, State University of New York-esf: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Kucherenko, V.A.; Tamarkina, Y.V.; Raenko, G.F.; Chernyshova, M.I. Thermolysis of brown coal in the presence of alkali metal hydroxides. Solid Fuel Chem. 2017, 51, 147–154. [Google Scholar] [CrossRef]
- Krstić, S.S.; Kragović, M.M.; Dodevski, V.M.; Marinković, A.D.; Kaluđerović, B.V.; Žerjav, G.; Pintar, A.; Pagnacco, M.C.; Stojmenović, M.D. Influence of temperature and different hydroxides on properties of activated carbon prepared from saccharose. Characterization, thermal degradation kinetic and dyes removal from water solutions. Sci. Sinter. 2018, 50, 255–273. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Z.; Xing, W.; Shen, H.; Bi, X.; Zhu, T.; Qiu, Z.; Zhuo, S. A New Approach to Tuning Carbon Ultramicropore Size at Sub-Angstrom Level for Maximizing Specific Capacitance and CO2 Uptake. Adv. Funct. Mater. 2016, 26, 7955–7964. [Google Scholar] [CrossRef]
- Celzard, A.; Fierro, V.; Marêché, J.F.; Furdin, G. Advanced Preparative Strategies for Activated Carbons Designed for the Adsorptive Storage of Hydrogen. Adsorpt. Sci. Technol. 2007, 25, 129–142. [Google Scholar] [CrossRef]
- Tamarkina, Y.V.; Maslova, L.A.; Khabarova, T.V.; Kucherenko, V.A. Adsorption properties of carbon materials produced by thermolysis of brown coal in the presence of alkali metal hydroxides. Russ. J. Appl. Chem. 2008, 81, 1167–1170. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kim, J.H.; Kim, J.S.; Lee, D.B.; Lee, J.C.; Chung, Y.J.; Lim, Y.S. Fabrication of Activated Carbon Fibers from Stabilized PAN-Based Fibers by KOH. Mater. Sci. Forum 2004, 449, 217–220. [Google Scholar] [CrossRef]
- Vilian, A.T.E.; Song, J.Y.; Lee, Y.S.; Hwang, S.-K.; Kim, H.J.; Jun, Y.-S.; Huh, Y.S.; Han, Y.-K. Salt-templated three-dimensional porous carbon for electrochemical determination of gallic acid. Biosens. Bioelectron. 2018, 117, 597–604. [Google Scholar] [CrossRef]
- Silvestre-Albero, A.; Silvestre-Albero, J.; Martinez-Escandell, M.; Molina-Sabio, M.; Kovacs, A.; Rodriguez-Reinoso, F. Novel synthesis of a micro-mesoporous nitrogen-doped nanostructured carbon from polyaniline. Microporous Mesoporous Mater. 2015, 218, 199–205. [Google Scholar] [CrossRef]
- Pampel, J.; Fellinger, T.-P. Opening of Bottleneck Pores for the Improvement of Nitrogen Doped Carbon Electrocatalysts. Adv. Energy Mater. 2016, 6. [Google Scholar] [CrossRef]
- Schiemann, M.; Bergthorson, J.; Fischer, P.; Scherer, V.; Taroata, D.; Schmid, G. A review on lithium combustion. Appl. Energy 2016, 162, 948–965. [Google Scholar] [CrossRef]
- Epstein, M.; Yogev, A.; Yao, C.; Berman, A. Carbothermal reduction of alkali hydroxides using concentrated solar energy. Energy 2001, 26, 441–455. [Google Scholar] [CrossRef]
- McKee, D.W.; Chatterji, D. The catalytic behavior of alkali metal carbonates and oxides in graphite oxidation reactions. Carbon 1975, 13, 381–390. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Cuesta, A.; Martinez-Alonso, A.; Tascon, J.M.D. Correlation between Arrhenius kinetic parameters in the reaction of different carbon materials with oxygen. Energy Fuels 1993, 7, 1141–1145. [Google Scholar] [CrossRef]
- McKee, D.W.; Chatterji, D. The catalyzed reaction of graphite with water vapor. Carbon 1978, 16, 53–57. [Google Scholar] [CrossRef]
- Kapteijn, F.; Abbel, G.; Moulijn, J.A. CO2 gasification of carbon catalysed by alkali metals: Reactivity and mechanism. Fuel 1984, 63, 1036–1042. [Google Scholar] [CrossRef]
- Sheth, A.; Agrawal, P.; Yeboah, Y.D. Catalytic Gasification of Coal using Eutectic Salt Mixtures; Federal Energy Technology Center: Morgantown, WV, USA; Pittsburgh, PA, USA, 1998. [Google Scholar] [CrossRef]
- Sevilla, M.; Mokaya, R. Energy storage applications of activated carbons: Supercapacitors and hydrogen storage. Energy Environ. Sci. 2014, 7, 1250–1280. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Final Heat Treatment | Precursor | Commercial Name | Used Sample Code |
|---|---|---|---|
| Carbonised | Rayon | Carbon (Rayon) felt CeraMaterials | CR1 |
| Graphitised | Graphite (Rayon) felt CeraMaterials | GR1 | |
| Carbonised | RSF1 Beijing Great Wall Co. | CR2 | |
| Graphitised | RSF2 Beijing Great Wall Co. | GR2 | |
| Carbonised | SIGRATHERM® KFA5 | CR3 | |
| Graphitised | SIGRATHERM® GFA10 | GR3 | |
| Graphitised | GF2 Schunk | GR4 | |
| Carbonised | PAN | PX 35 ZOLTEKTM | CP5 |
| Graphitised | GFE-1 CeraMaterials | GP6 | |
| Carbonised | BESF Beijing Great Wall Co. | CP7 | |
| Carbonised | Carbon (PAN) felt CeraMaterials | CP8 | |
| Graphitised | Graphite (PAN) felt CeraMaterials | GP8 |
| Sample Code | As-Received CFs | De-Sized CFs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| C (%) | N (%) | O (%) | H (%) | C (%) | N (%) | O (%) | H (%) | ||
| Ex-Rayon CFs | CR1 | 75.95 | 3.46 | 18.37 | 2.14 | 77.66 | 3.18 | 16.63 | 2.45 |
| CR2 | 77.55 | 3.17 | 17.15 | 2.07 | 78.45 | 2.77 | 16.40 | 2.33 | |
| CR3 | 90.94 | 0.50 | 7.64 | 0.92 | 89.16 | 0.12 | 9.27 | 1.46 | |
| GR1 | 91.27 | 0.35 | 7.46 | 0.92 | 97.83 | 0.13 | 1.92 | 0.12 | |
| GR2 | 98.70 | 0.43 | 0.78 | 0.09 | 99.01 | 0.10 | 0.81 | 0.08 | |
| GR4 | 99.53 | 0.01 | 0.39 | 0.07 | 99.66 | 0.03 | 0.25 | 0.05 | |
| GR3 | 99.66 | 0.04 | 0.24 | 0.07 | 99.72 | 0.01 | 0.22 | 0.05 | |
| Ex-PAN CFs | CP5 | 96.82 | 2.82 | 0.31 | 0.05 | 97.01 | 2.52 | 0.41 | 0.06 |
| CP8 | 95.07 | 4.09 | 0.65 | 0.19 | 95.19 | 3.91 | 0.60 | 0.23 | |
| CP7 | 97.89 | 1.59 | 0.46 | 0.06 | 97.85 | 1.71 | 0.35 | 0.09 | |
| GP6 | 99.46 | 0.03 | 0.44 | 0.06 | 99.58 | 0.02 | 0.32 | 0.08 | |
| GP8 | 99.44 | 0.04 | 0.46 | 0.06 | 99.68 | 0.04 | 0.22 | 0.06 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karakashov, B.; Fierro, V.; Mathieu, S.; Gadonneix, P.; Medjahdi, G.; Celzard, A. Structural Characterisation and Chemical Stability of Commercial Fibrous Carbons in Molten Lithium Salts. Materials 2019, 12, 4232. https://doi.org/10.3390/ma12244232
Karakashov B, Fierro V, Mathieu S, Gadonneix P, Medjahdi G, Celzard A. Structural Characterisation and Chemical Stability of Commercial Fibrous Carbons in Molten Lithium Salts. Materials. 2019; 12(24):4232. https://doi.org/10.3390/ma12244232
Chicago/Turabian StyleKarakashov, Blagoj, Vanessa Fierro, Sandrine Mathieu, Philippe Gadonneix, Ghouti Medjahdi, and Alain Celzard. 2019. "Structural Characterisation and Chemical Stability of Commercial Fibrous Carbons in Molten Lithium Salts" Materials 12, no. 24: 4232. https://doi.org/10.3390/ma12244232
APA StyleKarakashov, B., Fierro, V., Mathieu, S., Gadonneix, P., Medjahdi, G., & Celzard, A. (2019). Structural Characterisation and Chemical Stability of Commercial Fibrous Carbons in Molten Lithium Salts. Materials, 12(24), 4232. https://doi.org/10.3390/ma12244232