Potential of Green Ceramics Waste for Alkali Activated Foams



Abstract
1. Introduction
- AAM’s main ingredient can be inexpensive waste material in powder form containing enough amorphous Al and Si, which form the aluminosilicate network (ASN), when waste material is activated with alkali (usually alkali hydroxide and/or alkali silicate), as one-part (alkali and precursor are mixed in dry state, water is added later [13]), or two-part system (precursor is mixed with liquid alkali).
- Waste material can be inert or containing small amount of environmentally problematic, hazardous, and even radioactive elements, which get completely immobilized in ASN.
- With AAM synthesis, i.e., upcycling of waste material contributes to waste-free circular economy.
- Has potential to lower building industry’s CO2 emission when compared to materials from Portland cement, since (almost) no carbon footprint is attributed to waste precursors.
- Have lower energy needs for synthesis when compared to products from Portland cement.
- AAM can have high temperature resistance.
- AAM can be chemically stable and resistant.
- Precursor fly ash, or metakaolin with fly ash, or blast furnace slag, or blast furnace slag with calcined reservoir sludge, or metakaolin were mainly used,
- Activators Na-water glass and NaOH were used by majority, while some just used NaOH, or NaAlO2 and NaOH, or K-water glass and KOH,
- Foaming agents Al, H2O2 were used, while there is also some research done with using Na-perborate, Micro Air 210, oleic acid, Na-lauryl sulfate, silica fume were used for foaming agents; while there was research performed also with mechanical foaming,
- Stabilizing agent Na-dodecyl-sulphate, Sika® Lightcrete 02 were used.
2. Materials and Methods
2.1. Characterization
2.2. Preparation of Alkali Activated Samples
2.3. Preparation of Alkali Activated Foams
3. Results and Discussion
3.1. Analysis of Precursor
3.2. Calculation of Mixtures Based on Data from 2.2 and 3.1
3.3. Analysis of Bulk AAM
3.3.1. Dependence on Curing Temperature and Amount of Substance Ratio First Group: Al:Si on Limit Value of Compressive Strength
- Calcite, magnesioferrite, and ankerite completely dissolve in alkali media.
- Quartz partially dissolves in alkali media, especially on higher temperature (90 °C), remaining crystallites grow, a bit more on milder temperature conditions, and more if the amount of added/whole H2O was smaller, i.e., mixture had higher viscosity, i.e., for mass of added/whole H2O and viscosity of liquid, size of crystallites it can be written as ζ0 > ζ5 > ζ2, while for final m% of quartz it is vice versa ζ0 < ζ5 < ζ2.
- Corundum crystallites additionally form, but are smaller compared to the corundum in the precursor. The amount of corundum is bigger when amount of added/whole H2O is higher and viscosity is lower, i.e., m% of corundum grows as ζ0 < ζ5 < ζ2, which is vice versa to quartz.
- New minerals are formed at certain conditions with alkali activation: Microcline, orthoclase, zirconia, kaolinite, zeolite, albite, and sodalite.
- ⚬
- Albite (heat treated) grows only when curing was performed on 90 °C. This is the only mineral containing Na that grows also when mixture ζ2 is used, while when mixtures ζ0 and ζ5 are used, also other minerals containing Na grow, i.e., these two mixtures have excessive amount of Na.
- ⚬
- Microcline grows only when mixture ζ0 is used, at 70 °C with a bit of Na, at 90 °C five-times bigger amount and five-times smaller crystallites just with K from the first group.
- ⚬
- Orthoclase is the only mineral that formed at all temperatures and with ζ0, ζ2, and ζ5.
- ⚬
- Kaolinite, the softest mineral according to Mohs scale that is present in XRD results of tested AAM, is not present only when mixture ζ2 is used. Small amount of kaolinite (less than 2%) in the form of bigger crystallites (too big for correct estimation of crystallites’ size with X’Pert Highscore software) is present in AAM prepared with mixture ζ5 at 90 °C with compressive strength above 30 MPa, while at 70 °C there is more than 10% in the form of 15 nm big crystallites and compressive strength around 2.5 MPa. Samples prepared with ζ0 have a bit less than 10% of kaolinite with crystallites’ size 20 nm and compressive strength around 10 MPa. Explanation for big difference in compressive strengths’ value, if we make the assumption of uniform distribution of minerals in the prism, is that for bigger amount of smaller crystallites of kaolinite there are more weakest spots through the whole prism, while for smaller amount of bigger sized crystallites there are just few weakest spots in the prism.
- When mixture ζ0 is used, the temperature has no obvious impact on the amount of amorphous phase probably due to the lowest amount of H2O present in the precursor, i.e., due to the highest viscosity that hinders speed of reaction (and evaporation of H2O) and gives time for formation and growth of minerals. However, temperature has an impact on which minerals form;
- When mixture ζ2 is used at lowest temperature conditions (room temperature for 4 h followed by curing on 70 °C for 20 h), the ions have more time to arrange into positions with lowest energy (slower evaporation of H2O) state compared to preparation of AAM just at 70 °C, i.e., amount of amorphous phase is higher at curing at just 70 °C. When curing was performed at 90 °C albite (heat treated) formed (in all XRD analyzed experiments) with which the amount of amorphous phase lowered and was comparable to AAM prepared at lowest temperature conditions. However, excluding albite (heat treated) temperature did not have significant impact on mineral formation and growth.
- When mixture ζ5 is used at 70 °C ions have more time for formation and growth of minerals compared to synthesis at 90 °C where reaction ends faster (due to forced drying with higher temperature) and leaves material with more amorphous phase. Temperature does show impact on mineral formation, i.e., at lower temperature there was much more kaolinite, at higher temperature there was also albite (heat treated).
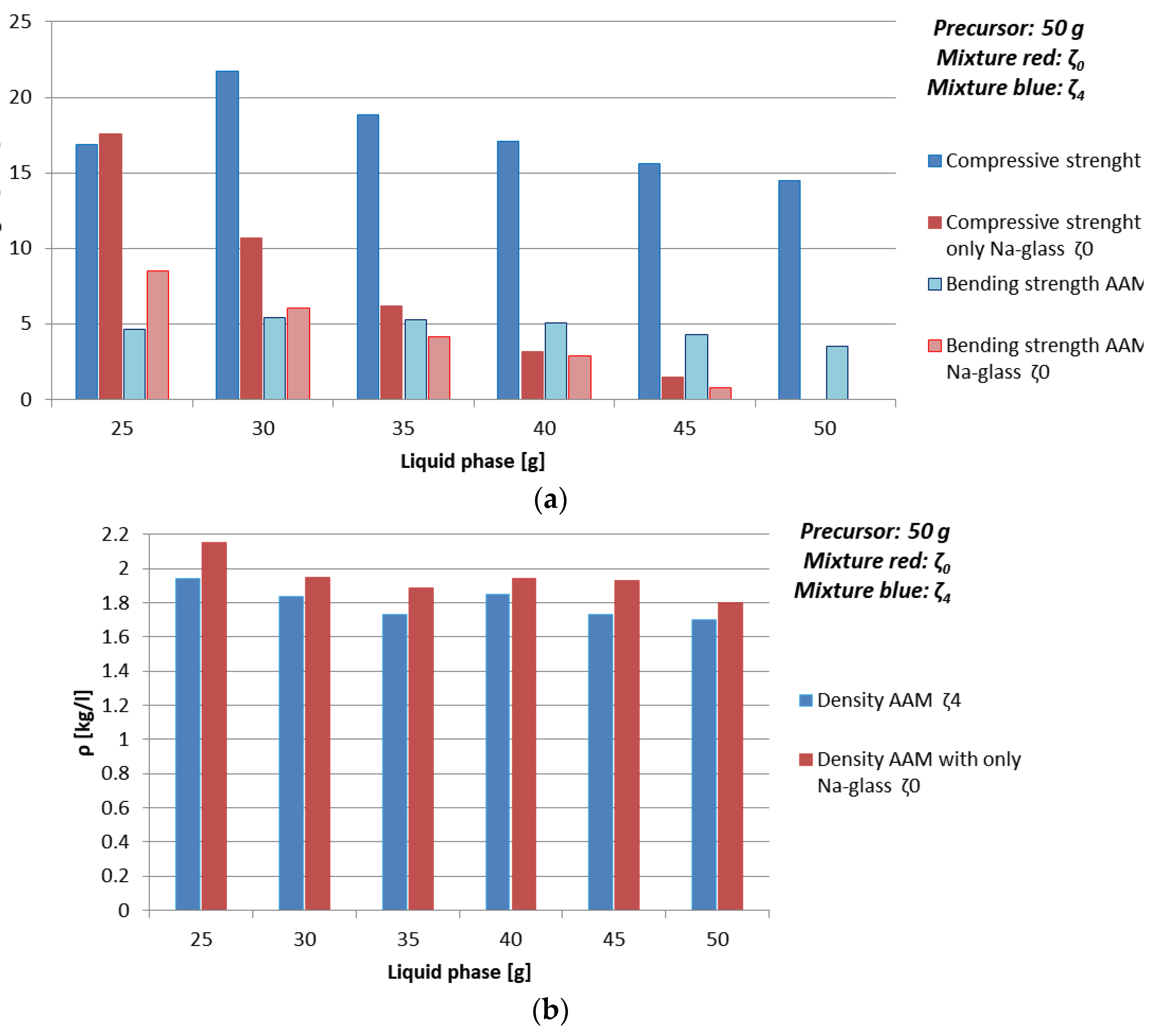
3.3.2. Dependence of Mechanical Properties on Amount of Liquid Phase and Amount of Substance Ratio First Group: Al:Si
3.3.3. Time Dependence of Mechanical Properties on Curing Temperature and Amount of Substance Ratio First Group: Al:Si
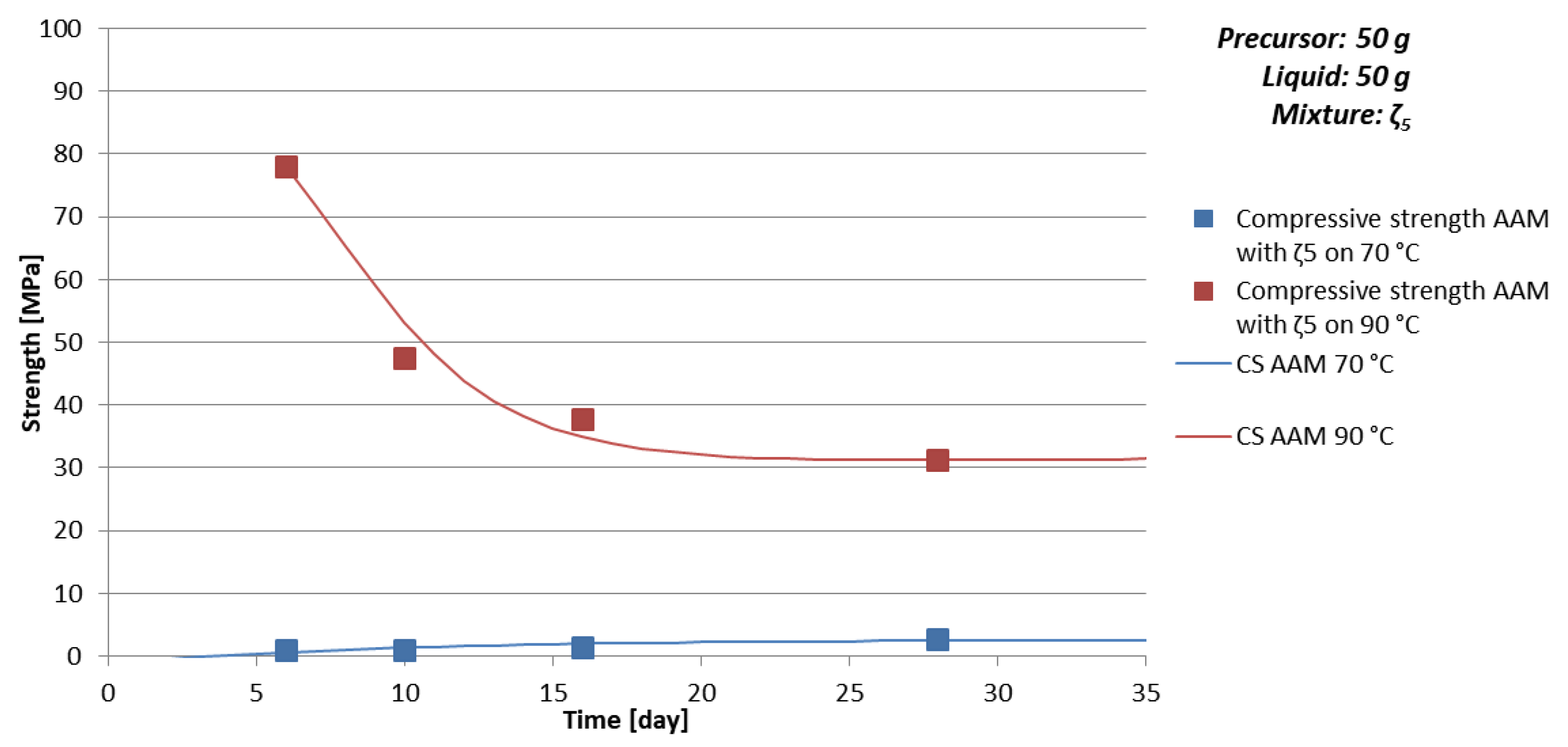
- Temperature of curing: Lower temperature results in fewer deformations in AAM, which are a consequence of physical effects—evaporation of liquid (drying of AAM); at certain temperature minerals of higher/lower hardness form-degrade, which contribute to the compressive strength value of AAM (see AAM prepared with 50 g of mixture ζ5 cured on 70 and 90 °C in Table 6 where 10% of kaolinite, mineral with lowest Mohs hardness present in the sample, form at 70 °C and just 1.5% at 90 °C),
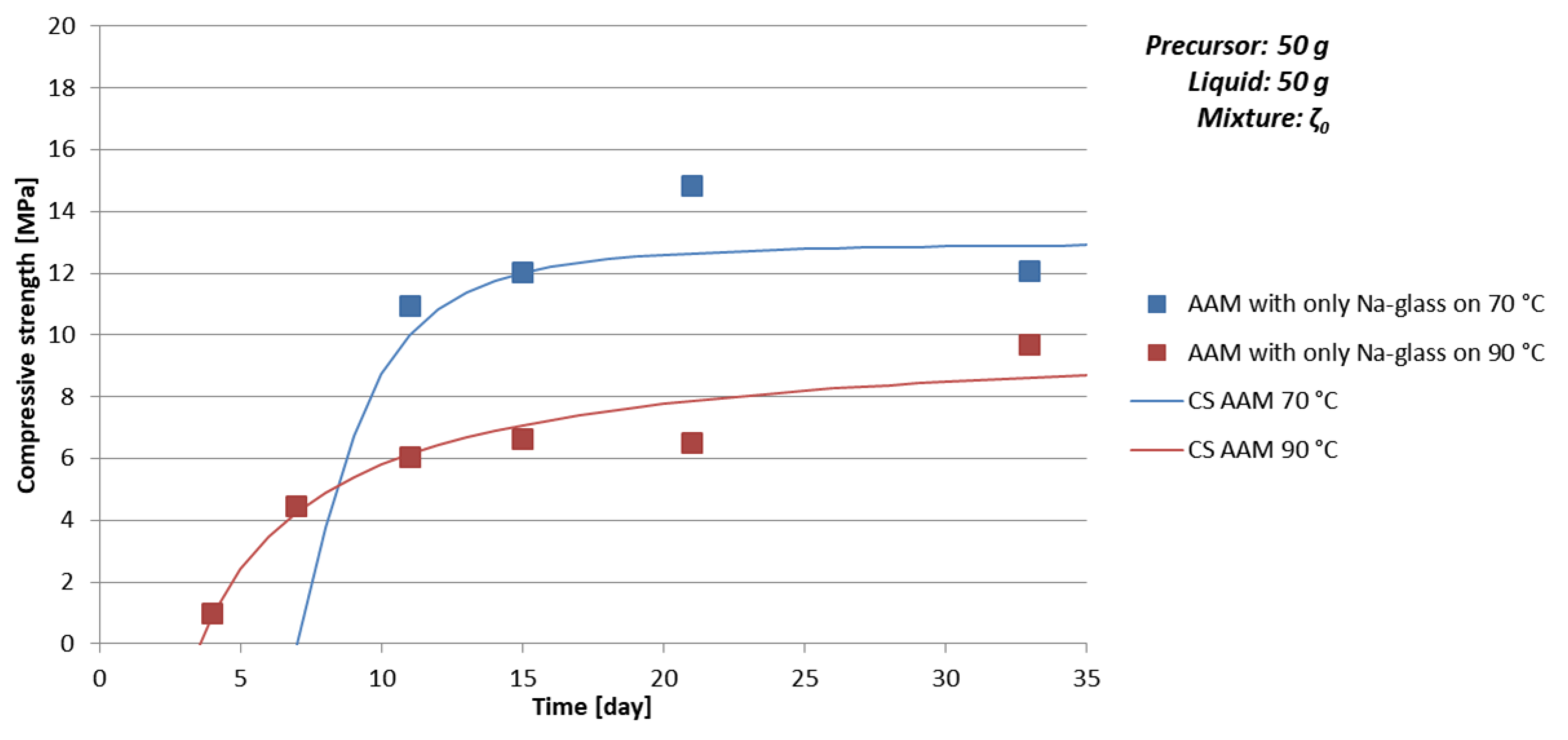
- time of curing: Too short time of curing on selected temperature can result in not finite AAM, i.e., the chemical and physical reactions are still taking place (see Figure 3a, Table 9 and Figure 4 where samples prepared with 50 g of mixture ζ0 on 70 °C do not reach measurable compressive strength in few days),
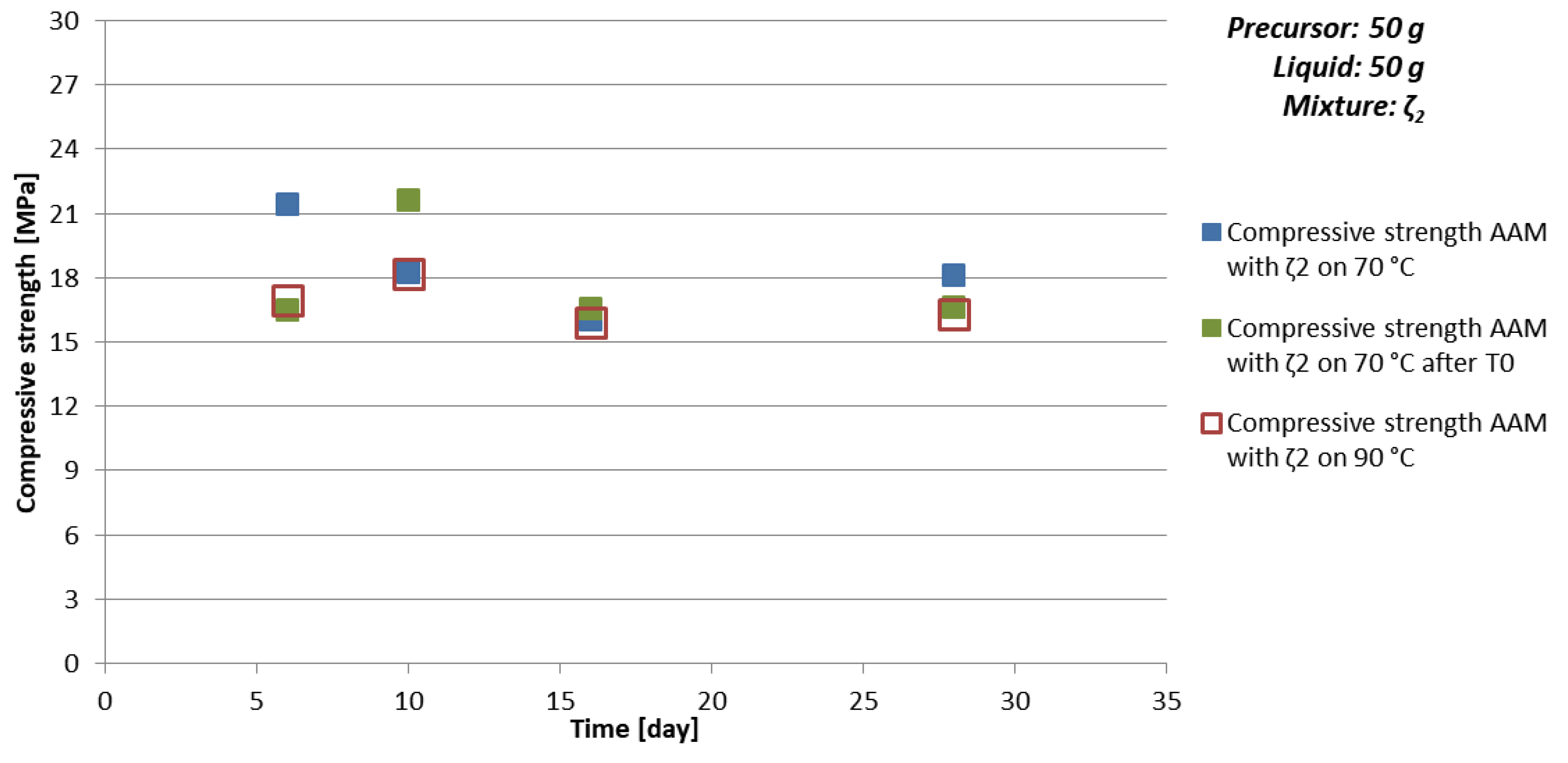
- chemistry of the selected precursor and needed additives: If the amount of additives is correct, compressive strength reaches final value soon probably due to the chemical reactions taking place mostly in the beginning of the AA (see Table 10 and Figure 5 where samples prepared with 50 g of mixture ζ2 reach limit value soon and are comparable for all curing temperatures), as well efflorescence and formation of certain minerals is avoided (see Table 6 where samples prepared with 50 g of mixture ζ2 on 70 °C did not form any mineral containing Na, while in samples with mixtures ζ0 and ζ5 they did; only samples prepared with 50 g of mixture ζ2 did not form kaolinite, i.e., mineral with lowest Mohs hardness are present in the tested samples),
- amount and viscosity of liquid phase: Too much water can lead to cracked AAM (see Table 10 where majority of samples prepared with 50 g of mixture ζ2 do not have result of the bending strength due to prisms being cracked), too high viscosity to very time-consuming reaction (see Figure 3a, Table 9 and Figure 4 where samples prepared with 50 g of mixture ζ0 on 70 °C do not reach measurable compressive strength in few days).
3.3.4. Dependence of Mechanical Properties on Amount of Substance Ratio First Group: Al:Si
3.4. Characteristics of Foamed AAM
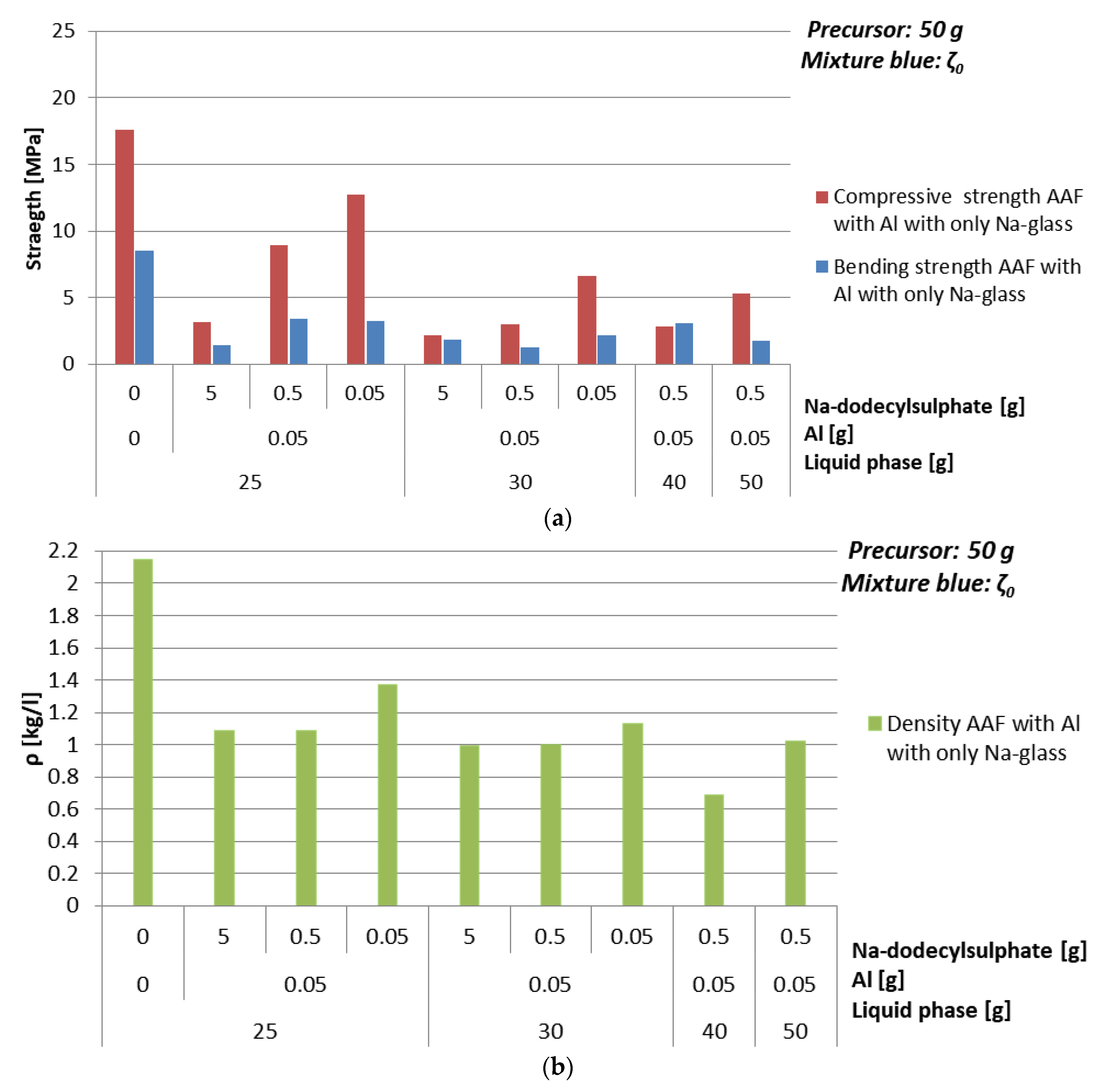
3.4.1. Foaming Agent Al
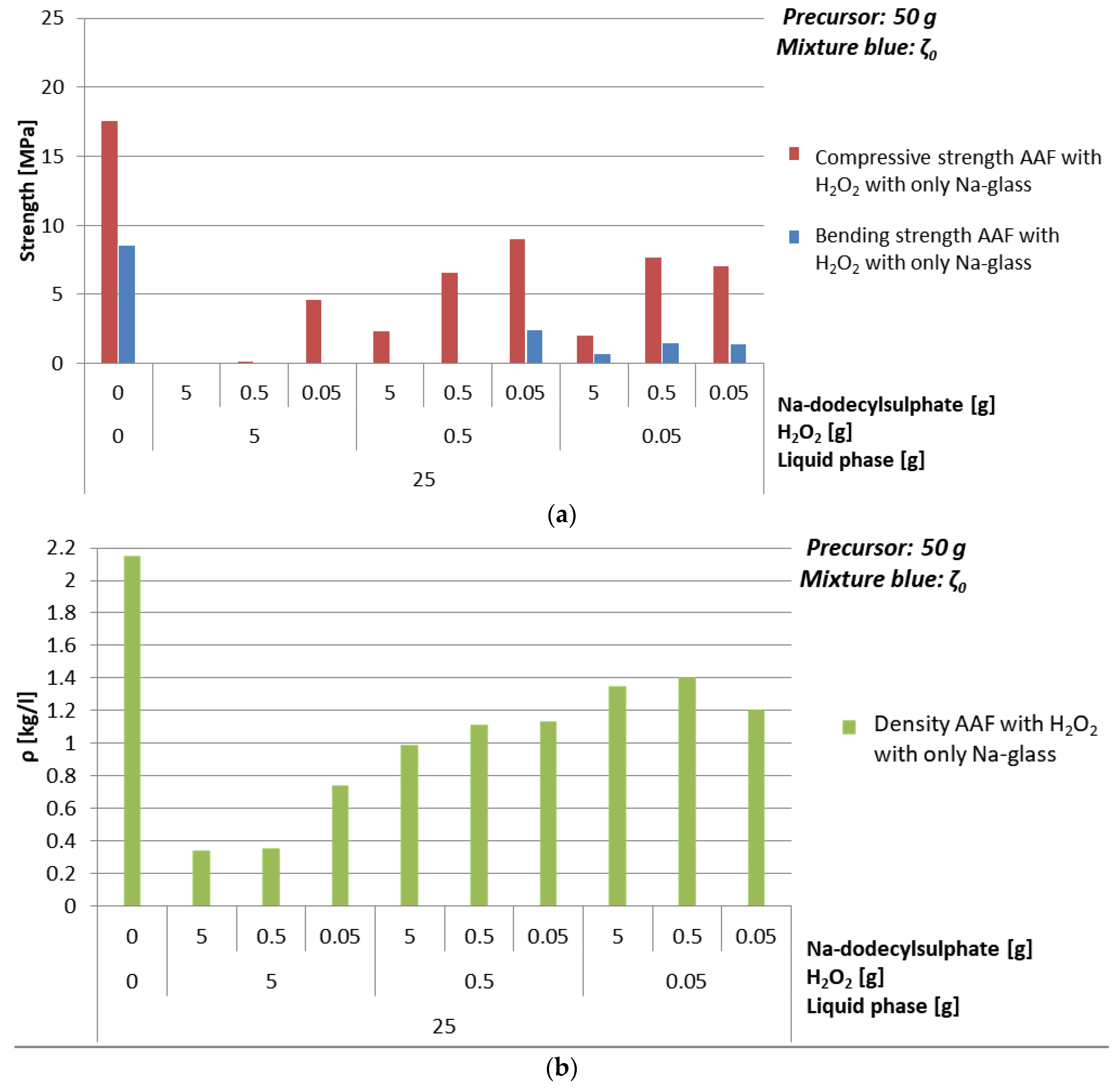
3.4.2. Foaming Agent H2O2
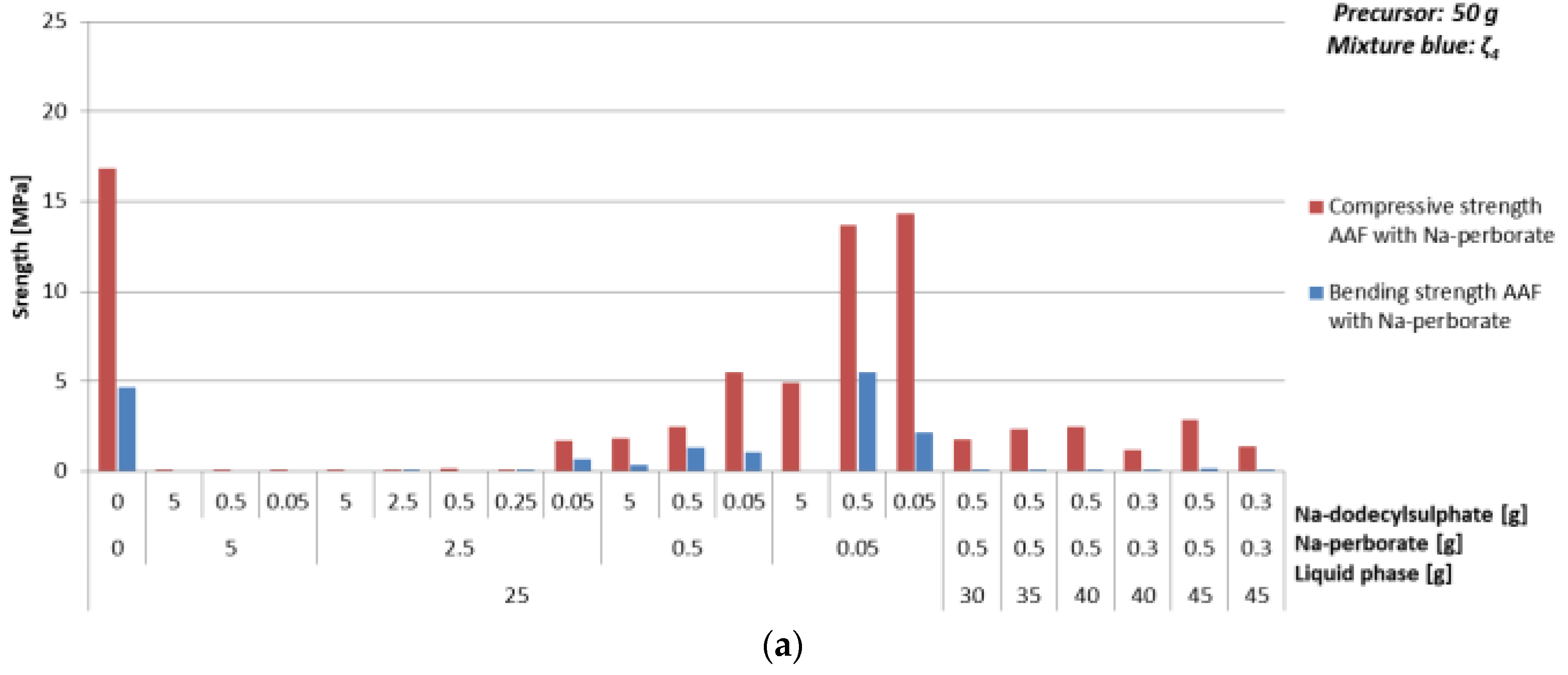
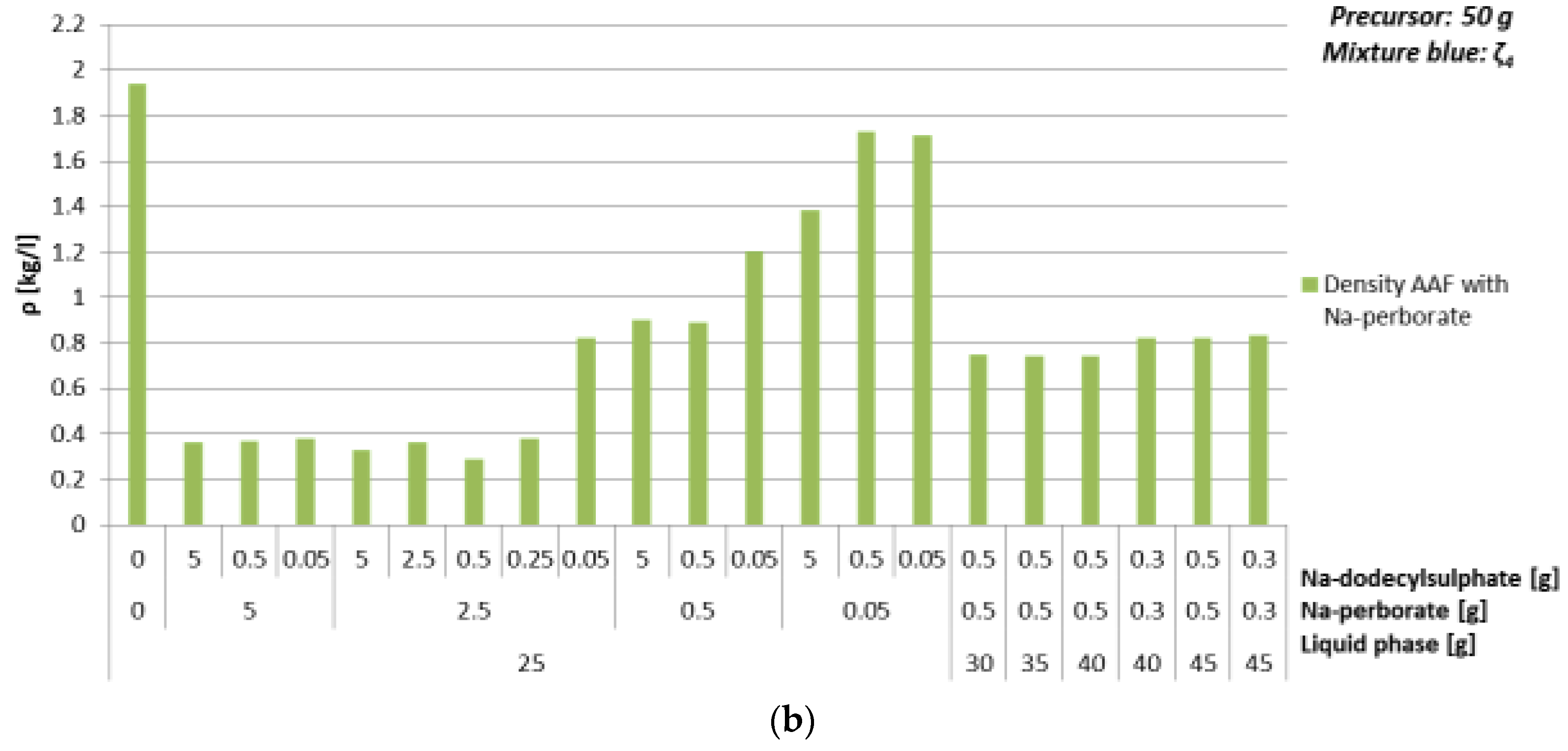
3.4.3. Foaming Agent Na-Perborate
4. Conclusions
- With Al: mixture ζ040, 0.1% of Al, 1% of Na-dodecylsulphate, ending with density 0.7 kg/l and compressive strength 2.8 MPa,
- With H2O2: mixture ζ025, 1% of H2O2, 0.1% of Na-dodecylsulphate, ending with density 0.7 kg/l and compressive strength 4.6 MPa,
- With Na-perborate:
- ⚬
- Mixture ζ025, 5% of Na-perborate, 0.1% of Na-dodecylsulphate, ending with density 0.8 kg/l and compressive strength 1.7 MPa,
- ⚬
- mixture ζ040, 0.6% of Na-perborate, 0.6% of Na-dodecylsulphate, ending with density 0.7 kg/l and compressive strength 5.1 MPa.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Provis, J.L. Geopolymers and other alkali activated materials: Why, how, and what? Mater. Struct. 2014, 47, 11–25. [Google Scholar] [CrossRef]
- Shi, C.; Jiménez, A.F.; Palomo, A. New cements for the 21st century: The pursuit of an alternative to Portland cement. Cem. Concr. Res. 2011, 41, 750–763. [Google Scholar] [CrossRef]
- Provis, J.L.; Bernal, S.A. Geopolymers and Related Alkali-Activated Materials. Annu. Rev. Mater. Res. 2014, 44, 299–327. [Google Scholar] [CrossRef]
- Palomo, A.; Grutzeck, M.W.; Blanco, M.T. Alkali-activated fly ashes A cement for the future. Cem. Concr. Res. 1999, 29, 1323–1329. [Google Scholar] [CrossRef]
- Roy, D.M. Alkali-activated cements Opportunities and challenges. Cem. Concr. Res. 1999, 29, 249–254. [Google Scholar] [CrossRef]
- Provis, J.L. Alkali-activated materials. Cem. Concr. Res. 2018, 114, 40–48. [Google Scholar] [CrossRef]
- Fernandez-Jimenez, A.; García-Lodeiro, I.; Palomo, A. Durability of alkali-activated fly ash cementitious materials. J. Mater. Sci. 2007, 42, 3055–3065. [Google Scholar] [CrossRef]
- Bernal, S.A.; Provis, J.L. Durability of Alkali-Activated Materials: Progress and Perspectives. J. Am. Ceram. Soc. 2014, 97, 997–1008. [Google Scholar] [CrossRef]
- Van Deventer, J.S.J.; Provis, J.L.; Duxson, P.; Brice, D.G. Chemical Research and Climate Change as Drivers in the Commercial Adoption of Alkali Activated Materials. Waste Biomass Valorization 2010, 1, 145–155. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Palomo, A. Composition and microstructure of alkali activated fly ash binder: Effect of the activator. Cem. Concr. Res. 2005, 35, 1984–1992. [Google Scholar] [CrossRef]
- Handbook of Alkali-Activated Cements, Mortars and Concretes; Elsevier B.V.: Amsterdam, The Netherlands, 2015.
- Palomo, A.; Palacios, M. Alkali-activated cementitious materials: Alternative matrices for the immobilisation of hazardous wastes Part II. Stabilisation of chromium and lead. Cem. Concr. Res. 2003, 33, 289–295. [Google Scholar] [CrossRef]
- Luukkonen, T.; Abdollahnejad, Z.; Yliniemi, J.; Kinnunen, P.; Illikainen, M. One-part alkali-activated materials: A review. Cem. Concr. Res. 2018, 103, 21–34. [Google Scholar] [CrossRef]
- Škvára, F. Alkali Activated Material—Geopolymer. In Proceedings of the International Conference Alkali Activated Materials–Research, Production and Utilization, Prague, Czech Republic, 21–22 June 2007. [Google Scholar]
- Ducman, V.; Korat, L. Characterization of geopolymer fly-ash based foams obtained with the addition of Al powder or H2O2 as foaming agents. Mater. Charact. 2016, 113, 207–213. [Google Scholar] [CrossRef]
- Hajimohammadi, A.; Ngo, T.; Mendis, P. How does aluminium foaming agent impact the geopolymer formation mechanism? Cem. Concr. Compos. 2017, 80, 277–286. [Google Scholar] [CrossRef]
- Korat, L.; Ducman, V. The influence of the stabilizing agent SDS on porosity development in alkali-activated fly-ash based foams. Cem. Concr. Compos. 2017, 80, 168–174. [Google Scholar] [CrossRef]
- Hlaváček, P.; Smilauer, V.; Škvára, F.; Kopecký, L.; Šulc, R. Inorganic foams made from alkali-activated fly ash: Mechanical, chemical and physical properties. J. Eur. Ceram. Soc. 2015, 35, 703–709. [Google Scholar] [CrossRef]
- Prud’Homme, E.; Michaud, P.; Joussein, E.; Peyratout, C.; Smith, A.; Arrii-Clacens, S.; Clacens, J.-M.; Rossignol, S. Silica fume as porogent agent in geo-materials at low temperature. J. Eur. Ceram. Soc. 2010, 30, 1641–1648. [Google Scholar] [CrossRef]
- Delair, S.; Prud’Homme, É.; Peyratout, C.; Smith, A.; Michaud, P.; Eloy, L.; Joussein, E.; Rossignol, S. Durability of inorganic foam in solution: The role of alkali elements in the geopolymer network. Corros. Sci. 2012, 59, 213–221. [Google Scholar] [CrossRef]
- Prud’Homme, E.; Michaud, P.; Joussein, E.; Peyratout, C.; Smith, A.; Rossignol, S. In situ inorganic foams prepared from various clays at low temperature. Appl. Clay Sci. 2011, 51, 15–22. [Google Scholar] [CrossRef]
- Budi, A.T. The combination of sodium perborate and water as intracoronal teeth bleaching agent. Dent. J. 2008, 41, 186. [Google Scholar] [CrossRef][Green Version]
- Sodium Perborate Hydrolysis. Available online: https://cellcode.us/quotes/sodium-perborate-hydrolysis.html (accessed on 3 July 2019).
- Abdollahnejad, Z.; Pacheco-Torgal, F.; Felix, T.; Tahri, W.; Aguiar, J.B. Mix design, properties and cost analysis of fly ash-based geopolymer foam. Constr. Build. Mater. 2015, 80, 18–30. [Google Scholar] [CrossRef]
- Sanjayan, J.G.; Nazari, A.; Chen, L.; Nguyen, G.H. Physical and mechanical properties of lightweight aerated geopolymer. Constr. Build. Mater. 2015, 79, 236–244. [Google Scholar] [CrossRef]
- Hajimohammadi, A.; Ngo, T.; Mendis, P.; Sanjayan, J. Regulating the chemical foaming reaction to control the porosity of geopolymer foams. Mater. Des. 2017, 120, 255–265. [Google Scholar] [CrossRef]
- Masi, G.; Rickard, W.D.; Vickers, L.; Bignozzi, M.C.; Van Riessen, A. A comparison between different foaming methods for the synthesis of light weight geopolymers. Ceram. Int. 2014, 40, 13891–13902. [Google Scholar] [CrossRef]
- Novais, R.M.; Ascensão, G.; Buruberri, L.H.; Senff, L.; Labrincha, J. Influence of blowing agent on the fresh- and hardened-state properties of lightweight geopolymers. Mater. Des. 2016, 108, 551–559. [Google Scholar] [CrossRef]
- Esmaily, H.; Nuranian, H. Non-autoclaved high strength cellular concrete from alkali activated slag. Constr. Build. Mater. 2012, 26, 200–206. [Google Scholar] [CrossRef]
- Yang, K.-H.; Lo, C.-W.; Huang, J.-S. Production and properties of foamed reservoir sludge inorganic polymers. Cem. Concr. Compos. 2013, 38, 50–56. [Google Scholar] [CrossRef]
- Garcìa-Ten, F.J.; Vázquez, M.F.Q.; Gil Albalat, C.; Villalba, D.C.; Zaera, V.; Mestre, M.C.S. Life Ceram - Zero Waste in Ceramic Tile Manufacture. Key Eng. Mater. 2015, 663, 23–33. [Google Scholar] [CrossRef]
- Ingham, B.; Toney, M. X-ray diffraction for characterizing metallic films. In Metallic Films for Electronic, Optical and Magnetic Applications; Elsevier BV.: Amsterdam, The Netherlands, 2014; pp. 3–38. [Google Scholar]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.L.; Lukey, G.C.; Mallicoat, S.W.; Kriven, W.M.; Van Deventer, J.S. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. A Physicochem. Eng. Asp. 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Mohs Hardness Scale: Testing the Resistance to Being Scratched. Available online: https://geology.com/minerals/mohs-hardness-scale.shtml (accessed on 3 June 2019).
- Speakman, S.A. Estimating Crystallite Size Using XRD. MIT Cent. Mater. Sci. Eng. 2014, 3–8. Available online: http://prism.mit.edu/xray/oldsite/CrystalSizeAnalysis.pdf (accessed on 3 June 2019).





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mass Ratio [g] | |||||||||
| Mass Ratio NaOH: Na-water Glass:H2O = 0:50:0, labeled: mixture ζ0, viscosity = 0.1731 Pa·s | |||||||||
| Precursor (s) | NaOH (s) | Na-water Glass | Added Distilled H2O | Whole H2O | Whole Liquid Phase | ||||
| All Together | Na | All Together | Na/Na2O | Si/SiO2 | H2O | ||||
| 50 | 0 | 0 | 25 | 3.1/4.2 | 3.2/6.8 | 13.9 | 0 | 13.9 | 25 |
| 50 | 0 | 0 | 30 | 3.8/5.1 | 3.9/8.3 | 16.7 | 0 | 16.7 | 30 |
| 50 | 0 | 0 | 35 | 4.4/5.9 | 4.5/9.6 | 19.5 | 0 | 19.5 | 35 |
| 50 | 0 | 0 | 40 | 5/6.7 | 5.1/10.9 | 22.2 | 0 | 22.2 | 40 |
| 50 | 0 | 0 | 45 | 5.8/7.8 | 5.8/12.4 | 25 | 0 | 25 | 45 |
| 50 | 0 | 0 | 50 | 6.3/8.5 | 6.4/13.7 | 27.8 | 0 | 27.8 | 50 |
| Mass Ratio NaOH: Na- water Glass:H2O = 2:9:39, labeled: mixture ζ2, viscosity = 0.02126 Pa·s | |||||||||
| Precursor (s) | NaOH (s) | Na-Water Glass | Added Distilled H2O | Whole H2O | Whole Liquid Phase | ||||
| All Together | Na | All Together | Na/Na2O | Si/SiO2 | H2O | ||||
| 50 | 2 | 1.2 | 9 | 1.1/1.5 | 1.2/2.6 | 5 | 39 | 44 | 50 |
| Mass Ratio NaOH: Na-water Glass:H2O = 4:20:26, labeled: mixture ζ4, viscosity = 0.02402 Pa·s | |||||||||
| Precursor (s) | NaOH (s) | Na-water Glass | Added Distilled H2O | Whole H2O | Whole Liquid Phase | ||||
| All Together | Na | All Together | Na/Na2O | Si/SiO2 | H2O | ||||
| 50 | 2 | 1.2 | 10 | 1.3/1.8 | 1.3/2.8 | 5.6 | 13 | 18.6 | 25 |
| 50 | 2.4 | 1.4 | 12 | 1.5/2.0 | 1.5/3.2 | 6.7 | 15.6 | 22.3 | 30 |
| 50 | 2.8 | 1.6 | 14 | 1.8/2.4 | 1.8/3.8 | 7.8 | 18.2 | 26 | 35 |
| 50 | 3.2 | 1.8 | 16 | 2/2.7 | 2.1/4.5 | 8.9 | 20.8 | 29.7 | 40 |
| 50 | 3.6 | 2.1 | 18 | 2.3/3.1 | 2.3/4.9 | 10 | 23.4 | 33.4 | 45 |
| 50 | 4 | 2.3 | 20 | 2.5/3.4 | 2.6/5.6 | 11.1 | 26 | 37.1 | 50 |
| Mass ratio NaOH: Na-water Glass:H2O = 5:22.5:22.5, labeled: mixture ζ5, viscosity = 0.02696 Pa·s | |||||||||
| Precursor (s) | NaOH (s) | Na-water Glass | Added Distilled H2O | Whole H2O | Whole Liquid Phase | ||||
| All Together | Na | All Together | Na/Na2O | Si/SiO2 | H2O | ||||
| 50 | 5 | 2.9 | 22.5 | 2.8/3.8 | 2.9/6.2 | 12.5 | 22.5 | 35 | 50 |
| Mass Ratio [g] | ||
|---|---|---|
| Precursor | Foaming Agent | Na-dodecylsulphate |
| 50 | 0 | 0, 0.5 |
| 0.05 | 0.05, 0.5, 5 | |
| 0.3 | 0.03, 0.05, 0.3, 5 | |
| 0.5 | 0.05, 0.5, 5 | |
| 2.5 | 0.05, 0.25, 0.5, 2.5, 5 | |
| 5 | 0.05, 0.5, 5 | |
| Elements [m%] | Na | K | Cs | Mg | Ca | Sr | Ba | Al | Si |
|---|---|---|---|---|---|---|---|---|---|
| XRF | 0.3 | 3.7 | 0 | 0.9 | 2.4 | 0.02 | 0.4 | 12.2 | 28.8 |
| XRD | 0 | 0 | 0 | 0.7 | 2.4 | 0 | 0 | 4.02 | 14.8 |
| XRF-XRD | 0.3 | 3.7 | 0 | 0.2 | 0 | 0.02 | 0.4 | 8.2 | 14.0 |
| Mass Ratio [g] | Amount of Substance Ratio in ASN | ||||||
|---|---|---|---|---|---|---|---|
| Precursor (s) | Useful Material for ASN in Precursor (without/with O) | NaOH (s) | Na- Water Glass (l) | ASN (Maximal Value) | 1st Group | Al | Si |
| 50 | 13.3/26.1 (26.1 m%/52.3 m%) | 0 | 0 | 26.1 | 0.3 | 1 | 1.6 |
| Mixture ζ0 | |||||||
| 0 | 25 | 36.2 | 1.2 | 1 | 2.4 | ||
| 0 | 30 | 38.2 | 1.4 | 1 | 2.5 | ||
| 0 | 35 | 40.2 | 1.6 | 1 | 2.7 | ||
| 0 | 40 | 42.2 | 1.8 | 1 | 2.8 | ||
| 0 | 45 | 44.2 | 2 | 1 | 3 | ||
| 0 | 50 | 46.2 | 2.1 | 1 | 3.1 | ||
| Mixture ζ2 | |||||||
| 2 | 9 | 30.9 | 1 | 1 | 1.9 | ||
| Mixture ζ4 | |||||||
| 2 | 10 | 31.3 | 1 | 1 | 1.9 | ||
| 2.4 | 12 | 32.3 | 1.1 | 1 | 2 | ||
| 2.8 | 14 | 33.4 | 1.3 | 1 | 2.1 | ||
| 3.2 | 16 | 34.4 | 1.4 | 1 | 2.1 | ||
| 3.6 | 18 | 35.4 | 1.6 | 1 | 2.2 | ||
| 4 | 20 | 36.5 | 1.7 | 1 | 2.2 | ||
| Mixture ζ5 | |||||||
| 5 | 22.5 | 38 | 2 | 1 | 2.3 | ||
| Properties | Mixture, Temperature [°C] | ||||||
|---|---|---|---|---|---|---|---|
| ζ0 | ζ2 | ζ5 | |||||
| 70 °C | 90 °C | T0, 70 °C | 70 °C | 90 °C | 70 °C | 90 °C | |
| Compressive strength after ~ 1 month [MPa] | 12.1 | 9.7 | 16.6 | 18.1 | 16.3 | 2.6 | 31.2 |
| Compressive strength’s time dependence (t) | grows | grows | const. | const. | const. | grows | falls |
| Mass of AAM in100 g [g] | 72.2 | 56 | 65 | ||||
| Mass of H2O in 100 g [g] | 27.8 | 44 | 35 | ||||
| Maximal mass of ASN [g] | 46.2 | 30.9 | 38 | ||||
| m% of ASN [%] | 64 | 55.2 | 58.5 | ||||
| Amount of substance ratio 1st group: Al:Si | 2.1:1:3.1 | 1:1:1.9 | 2:1:2.3 | ||||
| Mineral | Mixture, Temperature [°C] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ζ0 | ζ2 | ζ5 | ||||||||
| 70 °C | 90 °C | T0, 70 °C | 70 °C | 90 °C | 70 °C | 90 °C | ||||
| Geological Name | Chemical Formula | Mohs Hardness Scale [35] | Precursor m% [%] | Mineral m% [%] | ||||||
| Corundum | Al2O3 | 9 | 7.6 | 21.4 | 21.9 | 29.9 | 28.2 | 28.8 | 23.6 | 24.6 |
| Quartz | SiO2 | 7 | 31.8 | 5 | 3.6 | 9.5 | 7.6 | 7.2 | 6.9 | 6 |
| Calcite | CaCO3 | 3 | 4.1 | |||||||
| Magnesioferrite | Fe2MgO4 | 6-6.5 | 1.7 | |||||||
| Ankerite | C2CaFe0.23Mg0.77O6 | 3.5-4 | 5.2 | |||||||
| Microcline maximum | AlK0.95Na0.05Si3O8 | 6-6.5 | 3.1 | |||||||
| Microcline intermediate | AlKSi3O8 | 6-6.5 | 15.1 | |||||||
| Orthoclase | AlKSi3O8 | 6 | 14.9 | 0.9 | 15.1 | 13.1 | 12.5 | 11.8 | 13.1 | |
| Zirconia | ZrO2 | 8-8.5 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | ||
| Kaolinite 2M | H4Al2Si2O9 | 2-2.5 | 9.5 | 8.5 | 10.5 | 1.9 | ||||
| Zeolite P1 (Na-exchanged) | H24Al6Na6Si10O44 | 4-5 | 1 | |||||||
| Zeolite SUZ-4 | Al5K5O72Si31 | 4-5 | 0.8 | 0.7 | 0.8 | |||||
| Albite low | AlNaSi3O8 | 6-6.5 | 2.7 | |||||||
| Albite (heat treated) | AlNaSi3O8 | 6-6.5 | 4.2 | 5.4 | 4.9 | |||||
| Sodalite MAPO-20 | C8H24Al4Mg2N2O24P6 | 5.5-6 | 0.1 | 0.3 | 0.4 | 0.4 | ||||
| Amorphous [m%] | / | 49.5 | 45.9 | 44.9 | 44.6 | 50 | 45.3 | 43.9 | 48.9 | |
| Scherrer Size of Crystallites [nm] | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mineral | Precursor | Mixture, Temperature [°C] | ||||||
| ζ0 | ζ2 | ζ5 | ||||||
| 70 °C | 90 °C | T0, 70 °C | 70 °C | 90 °C | 70 °C | 90 °C | ||
| Corundum | 130 | 100 | 100 | 100 | 95 | 100 | 90 | 80 |
| Quartz | 95 | 150 | 140 | 110 | 100 | 100 | 125 | 120 |
| Calcite | 65 | |||||||
| Magnesioferrite | / | |||||||
| Ankerite | 50 | |||||||
| Microcline maximum | 100 | |||||||
| Microcline intermediate | 20 | |||||||
| Orthoclase | 5 | / | 25 | 20 | 30 | 15 | 10 | |
| Zirconia | / | / | / | / | / | / | ||
| Kaolinite 2M | 20 | 20 | 15 | / | ||||
| Zeolite P1 (Na-exchanged) | / | |||||||
| Zeolite SUZ-4 | / | / | / | |||||
| Albite low | 140 | |||||||
| Albite (heat treated) | 60 | 70 | 45 | |||||
| Sodalite MAPO-20 | / | / | / | / | ||||
| Mixture | Whole Liquid Phase [g] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|
| ζ0 | 25 | 2.2 | 8.5 | 17.6 |
| 30 | 2.0 | 6.0 | 10.7 | |
| 35 | 1.9 | 4.2 | 6.2 | |
| 40 | 1.9 | 2.9 | 3.2 | |
| 45 | 1.9 | 0.8 | 1.5 | |
| 50 | 1.8 | 0 | 0 | |
| ζ4 | 25 | 1.9 | 4.7 | 16.9 |
| 30 | 1.8 | 5.5 | 21.7 | |
| 35 | 1.7 | 5.3 | 18.9 | |
| 40 | 1.9 | 5.1 | 17.1 | |
| 45 | 1.7 | 4.3 | 15.6 | |
| 50 | 1.7 | 3.5 | 14.5 |
| Mixture | Curing Temperature [°C] | Time [day] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|
| ζ050 | 70 | 11 | 1.7 | 6.5 | 10.9 |
| 15 | 1.8 | 12.0 | |||
| 21 | 1.8 | 9.0 | 14.8 | ||
| 33 | 1.9 | 8.4 | 12.1 | ||
| 90 | 4 | 1.9 | 2.5 | 1.0 | |
| 7 | 1.9 | 4.5 | |||
| 11 | 1.8 | 4.0 | 6.1 | ||
| 15 | 1.8 | 6.6 | |||
| 21 | 1.8 | 2.9 | 6.5 | ||
| 33 | 1.7 | 9.7 |
| Mixture | Curing Temperature [°C] | Time [day] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|
| ζ250 | T0, later 70 | 6 | 1.8 | 10. 5 | 16.5 |
| 10 | 1.9 | 21.6 | |||
| 16 | 2.0 | 16.5 | |||
| 28 | 1.9 | 16.6 | |||
| 70 | 6 | 1.9 | 8.4 | 21.4 | |
| 10 | 1.9 | 18.2 | |||
| 16 | 1.9 | 16.0 | |||
| 28 | 1.9 | 18.1 | |||
| 90 | 6 | 1.6 | 17.0 | ||
| 10 | 1.8 | 18.2 | |||
| 16 | 1.7 | 15.9 | |||
| 28 | 1.7 | 3.6 | 16.3 |
| Mixture | Curing Temperature [°C] | Time [day] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|
| ζ550 | 70 | 6 | 1.7 | 0.02 | 0.9 |
| 10 | 1.7 | 0.9 | |||
| 16 | 1.8 | 0.3 | 1.4 | ||
| 28 | 1.7 | 2.6 | |||
| 90 | 6 | 1.8 | 11.5 | 78.0 | |
| 10 | 1.8 | 13.8 | 47.4 | ||
| 16 | 1.7 | 12.0 | 37.7 | ||
| 28 | 1.8 | 11.1 | 31.2 |
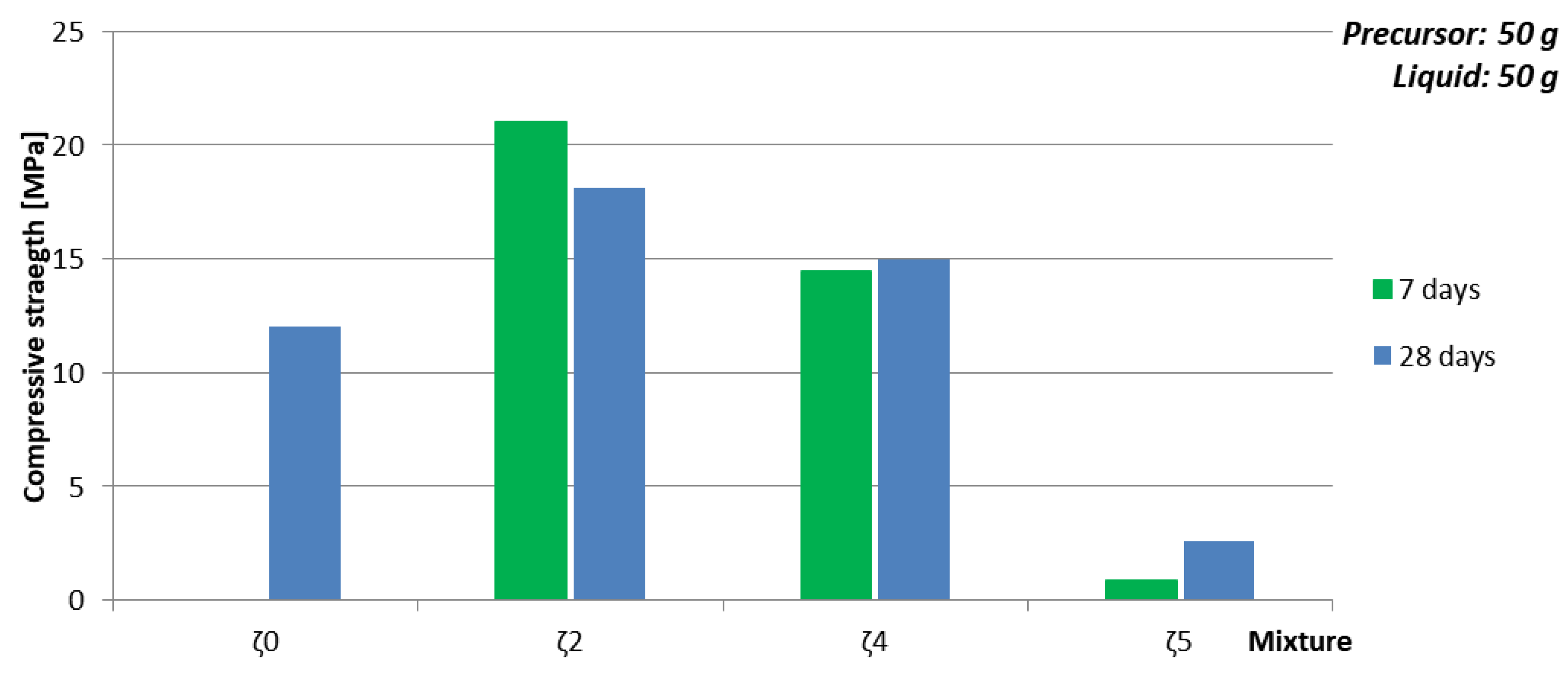
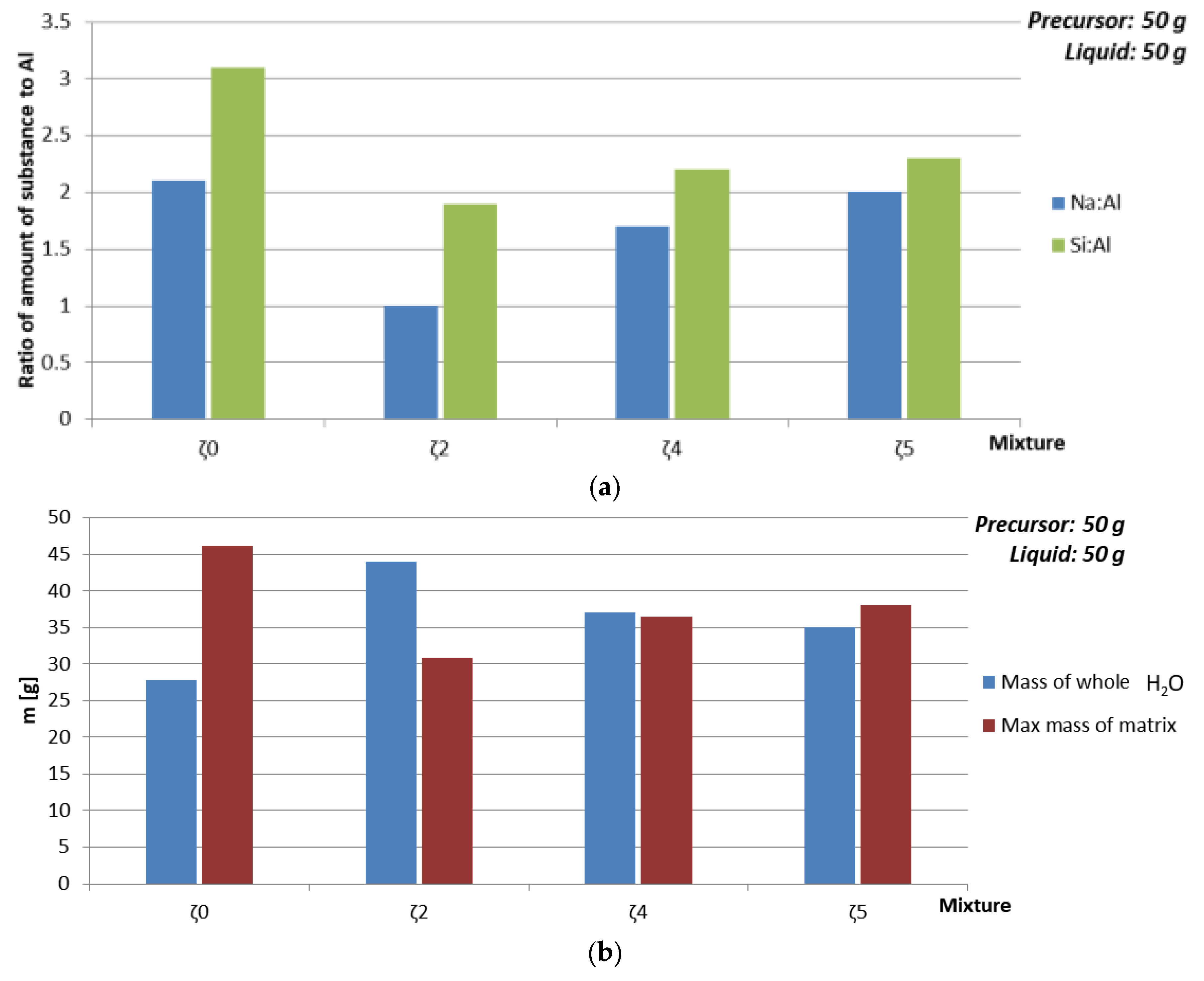
| Mixture | Viscosity [Pa·s] | Mass of Whole H2O [g] | Max Mass of ASN [g] | Amount of SubstanceNa:Al:Si | ρ 7 days [kg/l] | Bending Strength 7 days [MPa] | Compressive Strength 7 days [MPa] | Compressive Strength 28 days [MPa] |
|---|---|---|---|---|---|---|---|---|
| ζ050 | 0.1731 | 27.8 | 46.2 | 2.1:1:3.1 | 1.7 | 0 | 0 | 12.01 |
| ζ250 | 0.02126 | 44 | 30.9 | 1:1:1.9 | 1.9 | 8.35 | 21 | 18.14 |
| ζ450 | 0.02402 | 37.1 | 36.5 | 1.7:1:2.2 | 1.7 | 3.5 | 14.5 | 15 |
| ζ550 | 0.02696 | 35 | 38 | 2:1:2.3 | 1.7 | 0.015 | 0.9 | 2.55 |
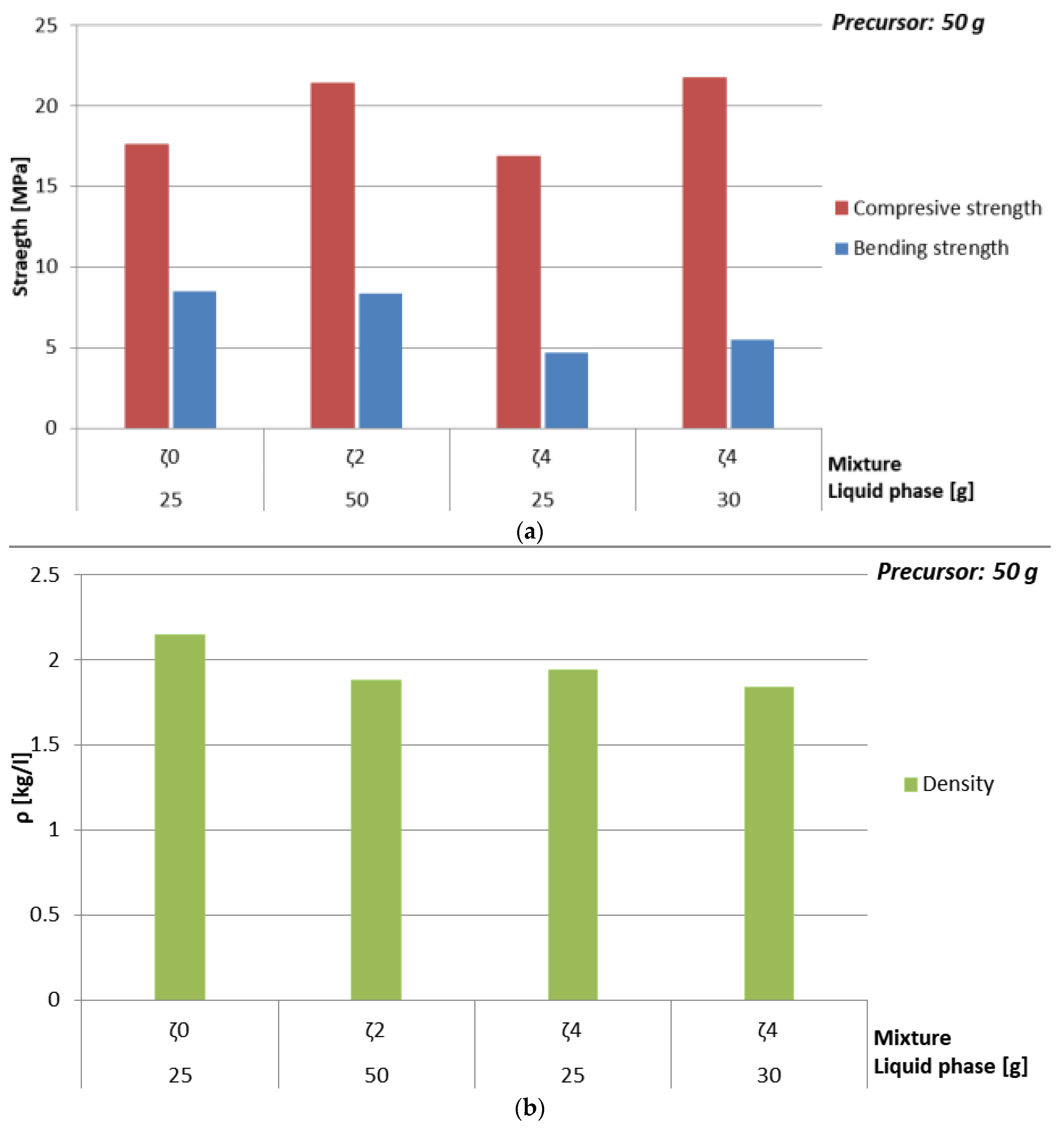
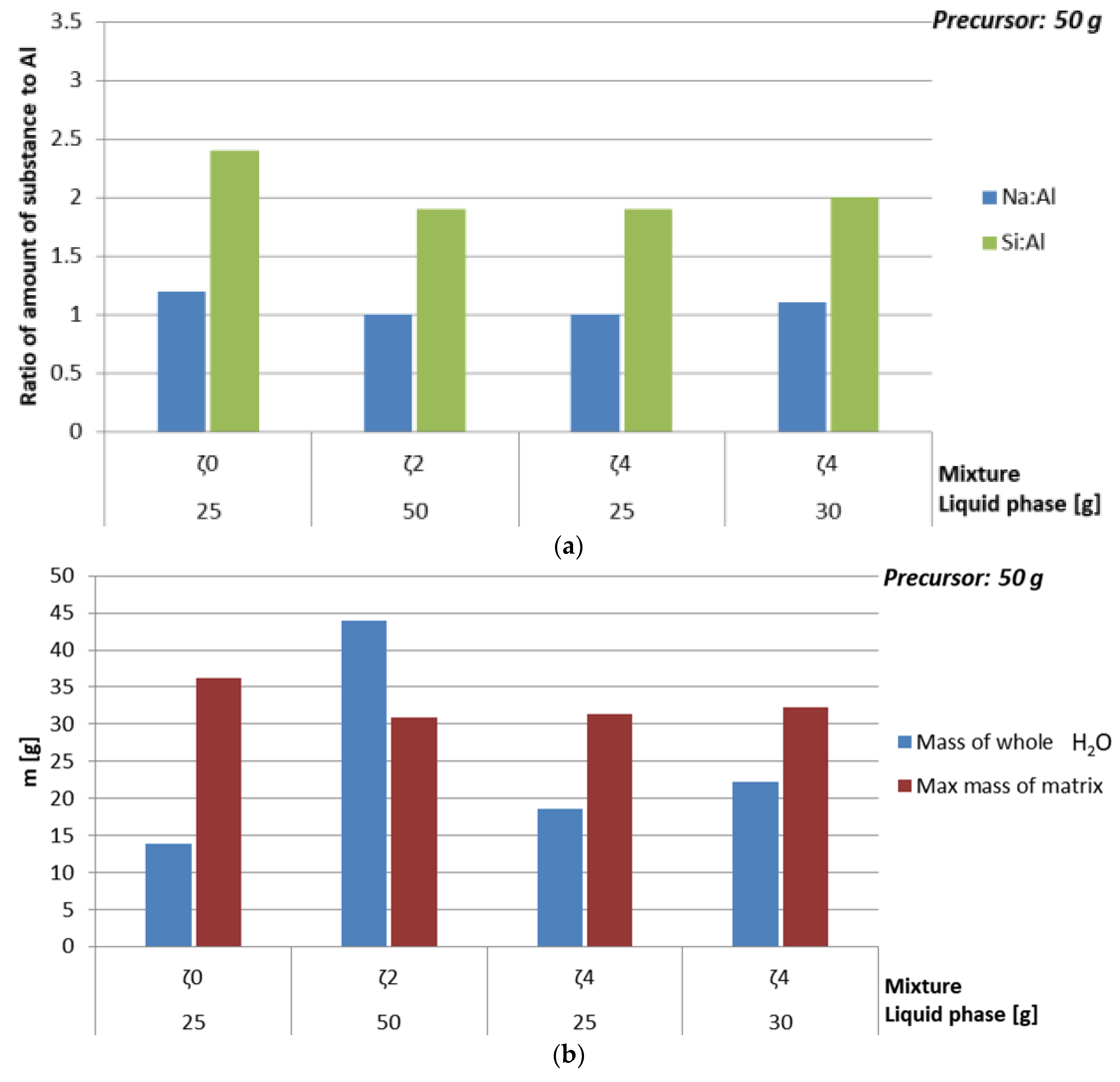
| Mixture | Mass of Whole H2O [g] | Max Mass of ASN [g] | Amount of Substance Na:Al:Si | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|---|
| ζ025 | 13.9 | 36.2 | 1.2:1:2.4 | 2.2 | 8.5 | 17.6 |
| ζ250 | 44.0 | 30.9 | 1:1:1.9 | 1.9 | 8.4 | 21.4 |
| ζ425 | 18.6 | 31.3 | 1:1:1.9 | 1.9 | 4.7 | 16.9 |
| ζ430 | 22.3 | 32.3 | 1.1:1:2 | 1.8 | 5.5 | 21.7 |
| Mixture | Al [g] | Na-dodecyl Sulphate [g] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|
| ζ025 | 0 | 0 | 2.2 | 8.5 | 17.6 |
| 0.05 | 5 | 1.1 | 1.4 | 3.2 | |
| 0.5 | 1.1 | 3.4 | 8.9 | ||
| 0.05 | 1.4 | 3.2 | 12.8 | ||
| ζ030 | 0.05 | 5 | 1.0 | 1.8 | 2.2 |
| 0.5 | 1.0 | 1.2 | 3.0 | ||
| 0.05 | 1.1 | 2.1 | 6.6 | ||
| ζ040 | 0.05 | 0.5 | 0.7 | 3.1 | 2.8 |
| ζ050 | 0.05 | 0.5 | 1.0 | 1.8 | 5.3 |
| Mixture | H2O2 [g] | Na-dodecyl Sulphate [g] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|
| ζ025 | 0 | 0 | 2.2 | 8.5 | 17.6 |
| 5 | 5 | 0.3 | 0.01 | 0.04 | |
| 0.5 | 0.4 | 0.04 | 0.1 | ||
| 0.05 | 0.7 | 0 | 4.6 | ||
| 0.5 | 5 | 1.0 | 0.04 | 2.3 | |
| 0.5 | 1.1 | 0.005 | 6.6 | ||
| 0.05 | 1.1 | 2.4 | 9.0 | ||
| 0.05 | 5 | 1.4 | 0.6 | 2.0 | |
| 0.5 | 1.4 | 1.4 | 7.6 | ||
| 0.05 | 1.2 | 1.4 | 7.0 |
| Mixture | Na-perborate [g] | Na-dodecyl Sulphate [g] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] | ||
|---|---|---|---|---|---|---|---|
| ζ425 | 0 | 0 | 1.9 | 4.7 | 16.9 | ||
| 5 | 5 | 0.4 | 0 | 0.07 | |||
| 0.5 | 0.4 | 0 | 0.06 | ||||
| 0.05 | 0.4 | 0 | 0.06 | ||||
| 2.5 | 5 | 0.3 | 0 | 0.02 | |||
| 2.5 | 0.4 | 0.07 | 0.07 | ||||
| 0.5 | 0.3 | 0 | 0.1 | ||||
| 0.25 | 0.4 | 0.07 | 0.06 | ||||
| 0.05 | 0.8 | 0.7 | 1.7 | ||||
| 0.5 | 5 | 0.9 | 0.3 | 1.8 | |||
| 0.5 | 0.9 | 1.3 | 2.5 | ||||
| 0.05 | 1.2 | 1.0 | 5.5 | ||||
| 0.05 | 5 | 1.4 | 0 | 4.9 | |||
| 0.5 | 1.7 | 5.5 | 13.7 | ||||
| 0.05 | 1.7 | 2.2 | 14.3 | ||||
| ζ430 | 0.5 | 0.5 | 0.8 | 0.02 | 1.8 | ||
| ζ435 | 0.5 | 0.5 | 0.7 | 0.02 | 2.4 | ||
| ζ440 | 0.5 | 0.5 | 0.7 | 0.05 | 2.5 | ||
| 0.3 | 0.3 | 0.8 | 0.05 | 1.2 | |||
| ζ445 | 0.5 | 0.5 | 0.8 | 0.1 | 2.9 | ||
| 0.3 | 0.3 | 0.8 | 0.02 | 1.4 | |||
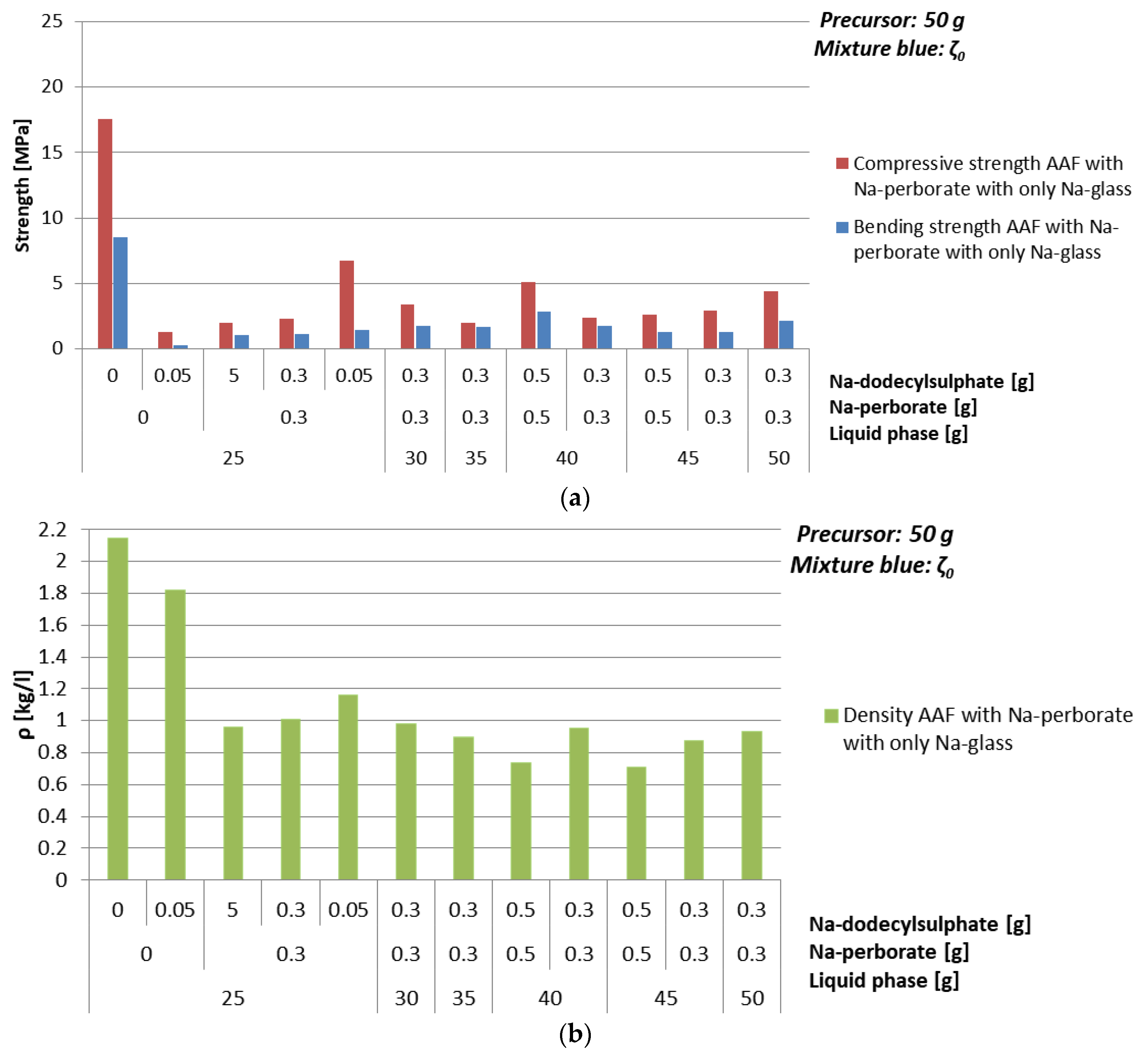
| Mixture | Na-perborate [g] | Na-dodecyl Sulphate [g] | ρ [kg/l] | Bending Strength [MPa] | Compressive Strength [MPa] |
|---|---|---|---|---|---|
| ζ025 | 0 | 0 | 2.2 | 8.5 | 17.6 |
| 0.05 | 1.8 | 0.3 | 1.3 | ||
| 0.3 | 5 | 1.0 | 1.0 | 2.0 | |
| 0.3 | 1.0 | 1.1 | 2.3 | ||
| 0.05 | 1.2 | 1.4 | 6.7 | ||
| ζ030 | 0.3 | 0.3 | 1.0 | 1.7 | 3.4 |
| ζ035 | 0.3 | 0.3 | 0.9 | 1.7 | 2.0 |
| ζ040 | 0.5 | 0.5 | 0.7 | 2.8 | 5.1 |
| 0.3 | 0.3 | 1.0 | 1.7 | 2.4 | |
| ζ045 | 0.5 | 0.5 | 0.7 | 1.3 | 2.6 |
| 0.3 | 0.3 | 0.9 | 1.3 | 2.9 | |
| ζ050 | 0.3 | 0.3 | 0.9 | 2.1 | 4.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horvat, B.; Ducman, V. Potential of Green Ceramics Waste for Alkali Activated Foams. Materials 2019, 12, 3563. https://doi.org/10.3390/ma12213563
Horvat B, Ducman V. Potential of Green Ceramics Waste for Alkali Activated Foams. Materials. 2019; 12(21):3563. https://doi.org/10.3390/ma12213563
Chicago/Turabian StyleHorvat, Barbara, and Vilma Ducman. 2019. "Potential of Green Ceramics Waste for Alkali Activated Foams" Materials 12, no. 21: 3563. https://doi.org/10.3390/ma12213563
APA StyleHorvat, B., & Ducman, V. (2019). Potential of Green Ceramics Waste for Alkali Activated Foams. Materials, 12(21), 3563. https://doi.org/10.3390/ma12213563