Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3− (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads




Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
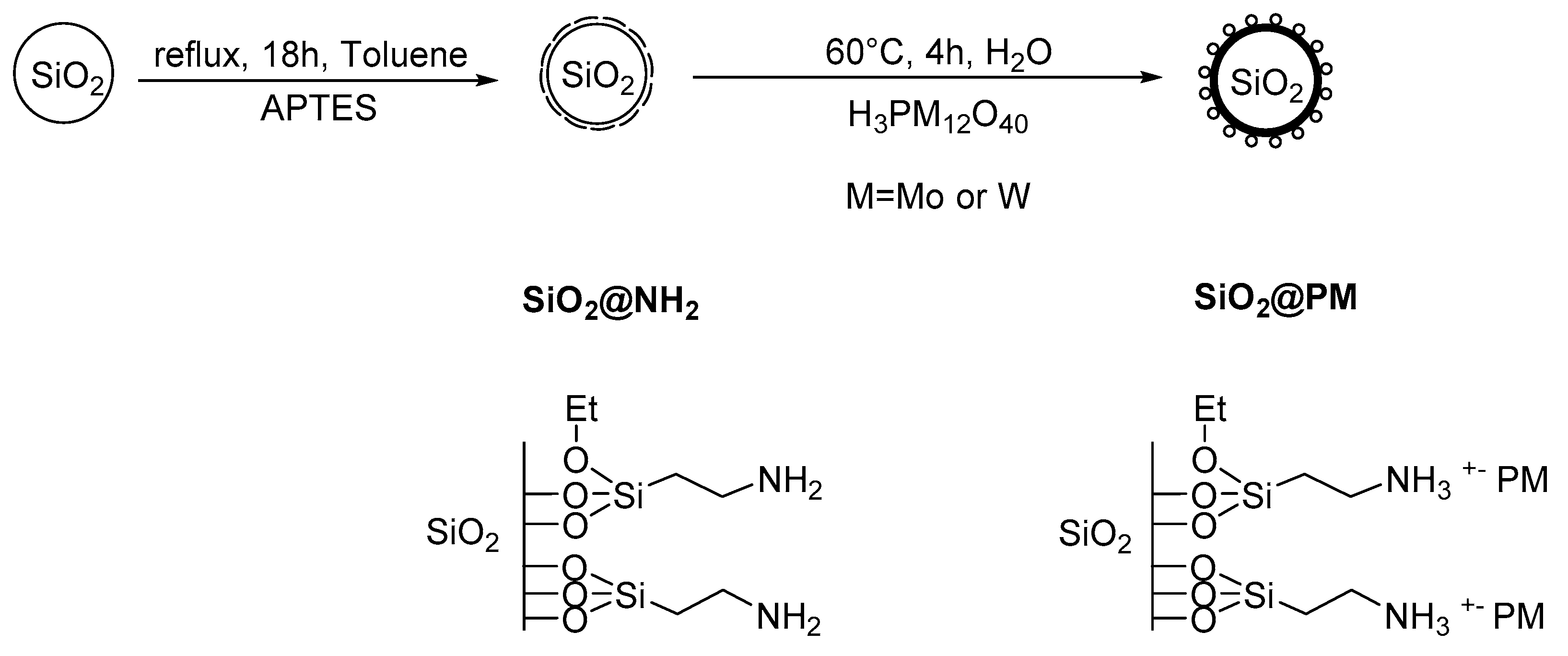
2.3. Synthesis of Nanoparticles.
2.4. Catalytic Experiments
3. Results and Discussion
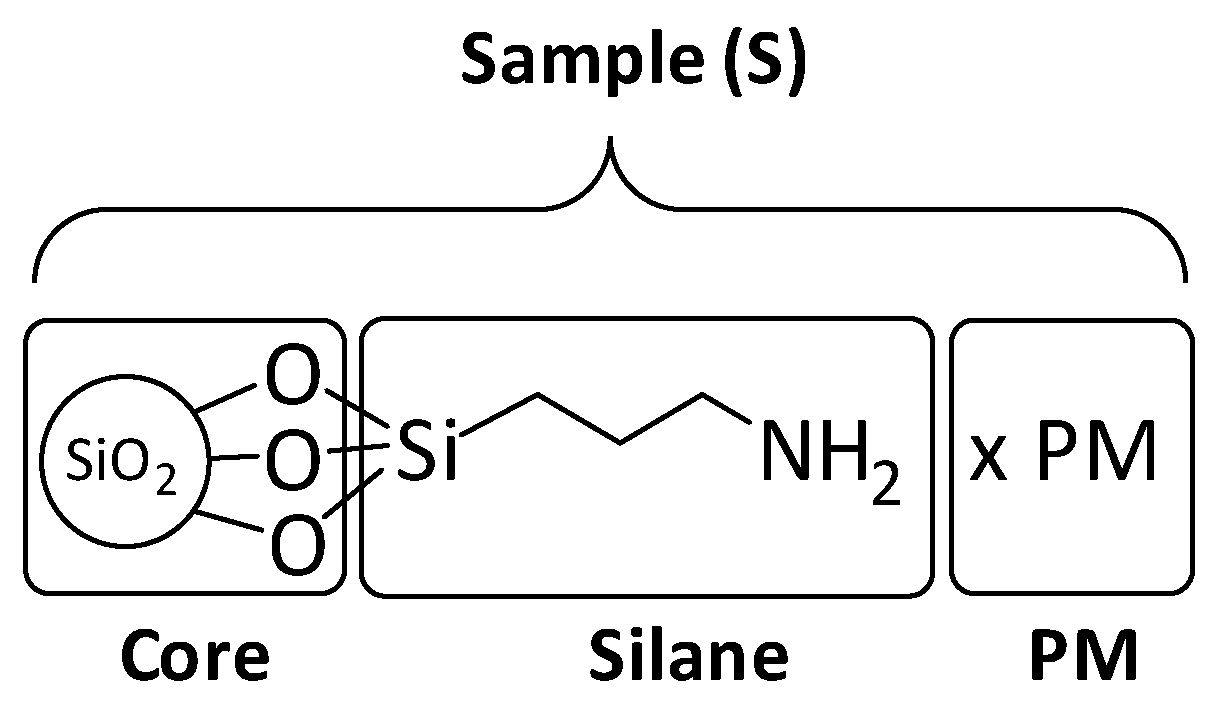
3.1. Synthesis of the Catalytic Objects.
3.2. Morphological Characterization of the Silica Beads
3.2.1. Analysis by PXRD
3.2.2. Dynamic Light Scattering (DLS) Analysis.
3.2.3. TEM Analysis
3.3. Qualitative Functional Characterization of the Silica Beads
3.3.1. Infrared Spectroscopy
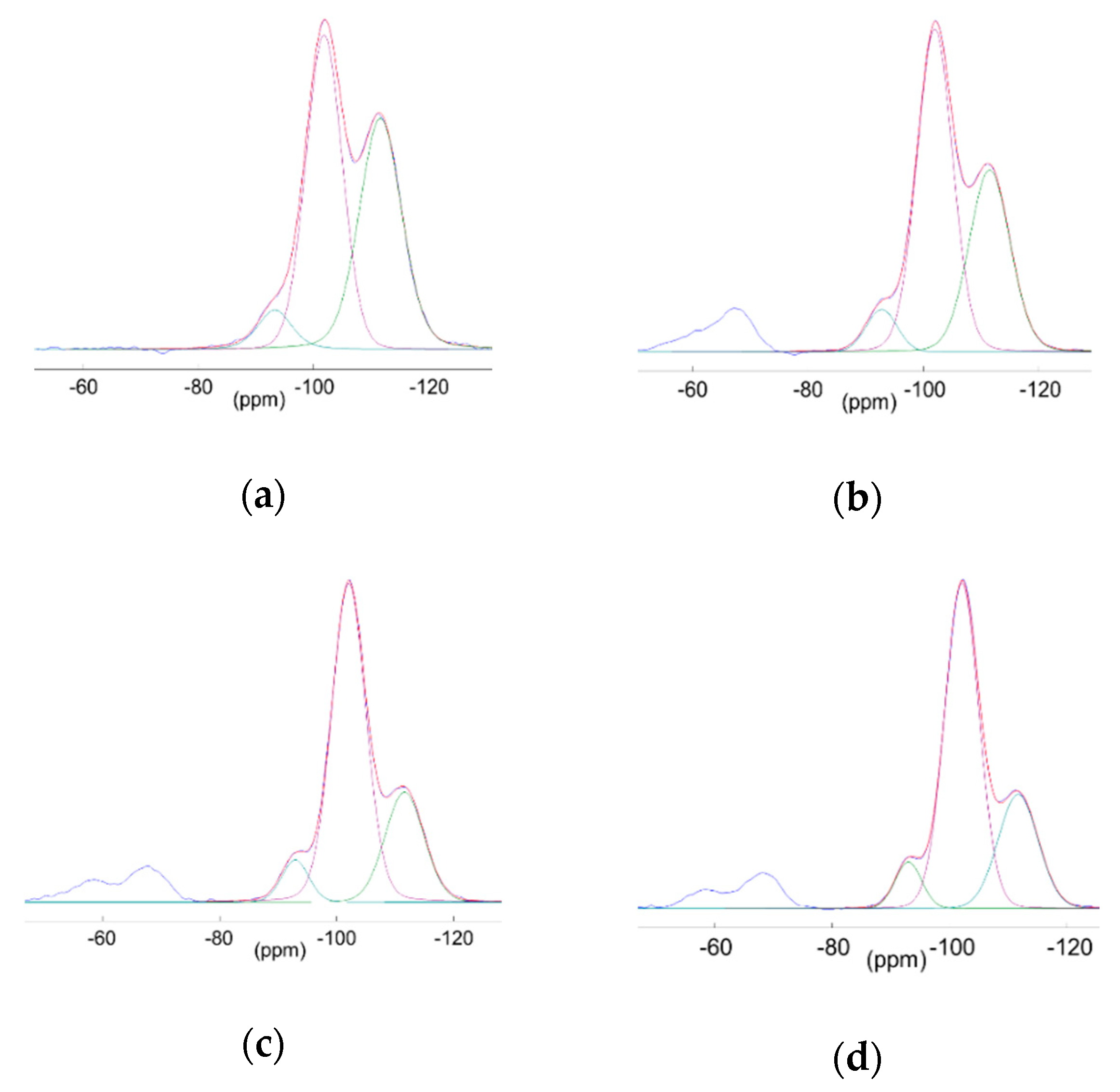
3.3.2. Multinuclear Solid State NMR
3.4. Quantitative Functional Characterization of the Silica Beads
3.4.1. Quantification by Elemental Analysis
3.4.2. Quantification of Grafted Functions and Retained POM by Liquid NMR
3.4.3. Surface Coverage of the Beads Through EA and NMR.
3.5. Catalysis
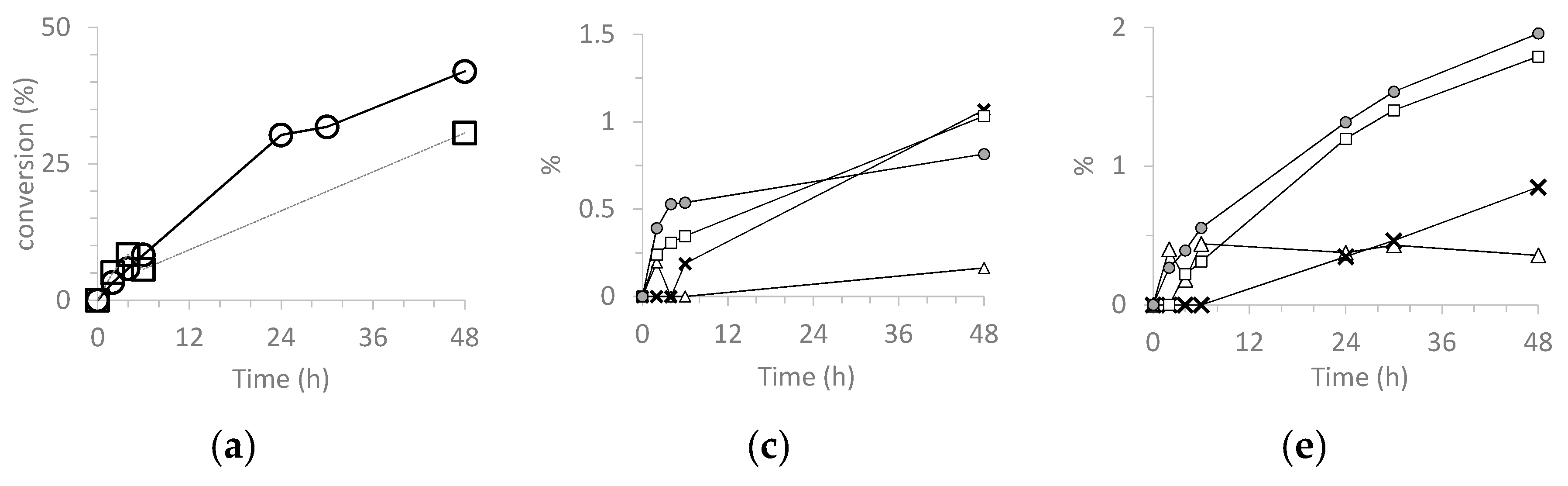
3.5.1. Cyclooctene (CO) Epoxidation.
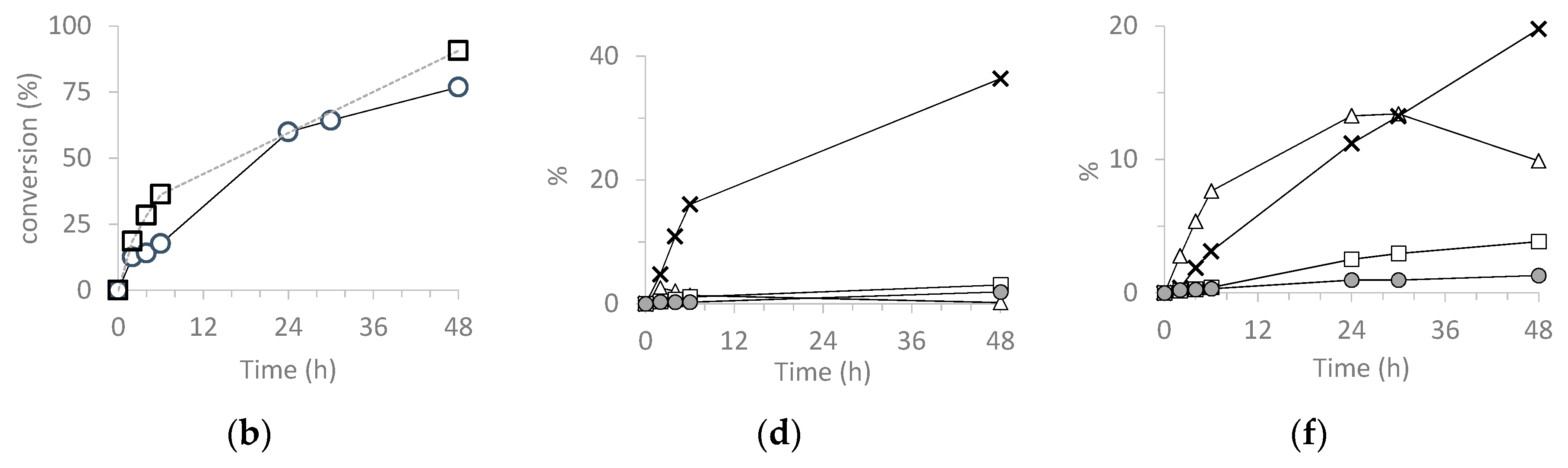
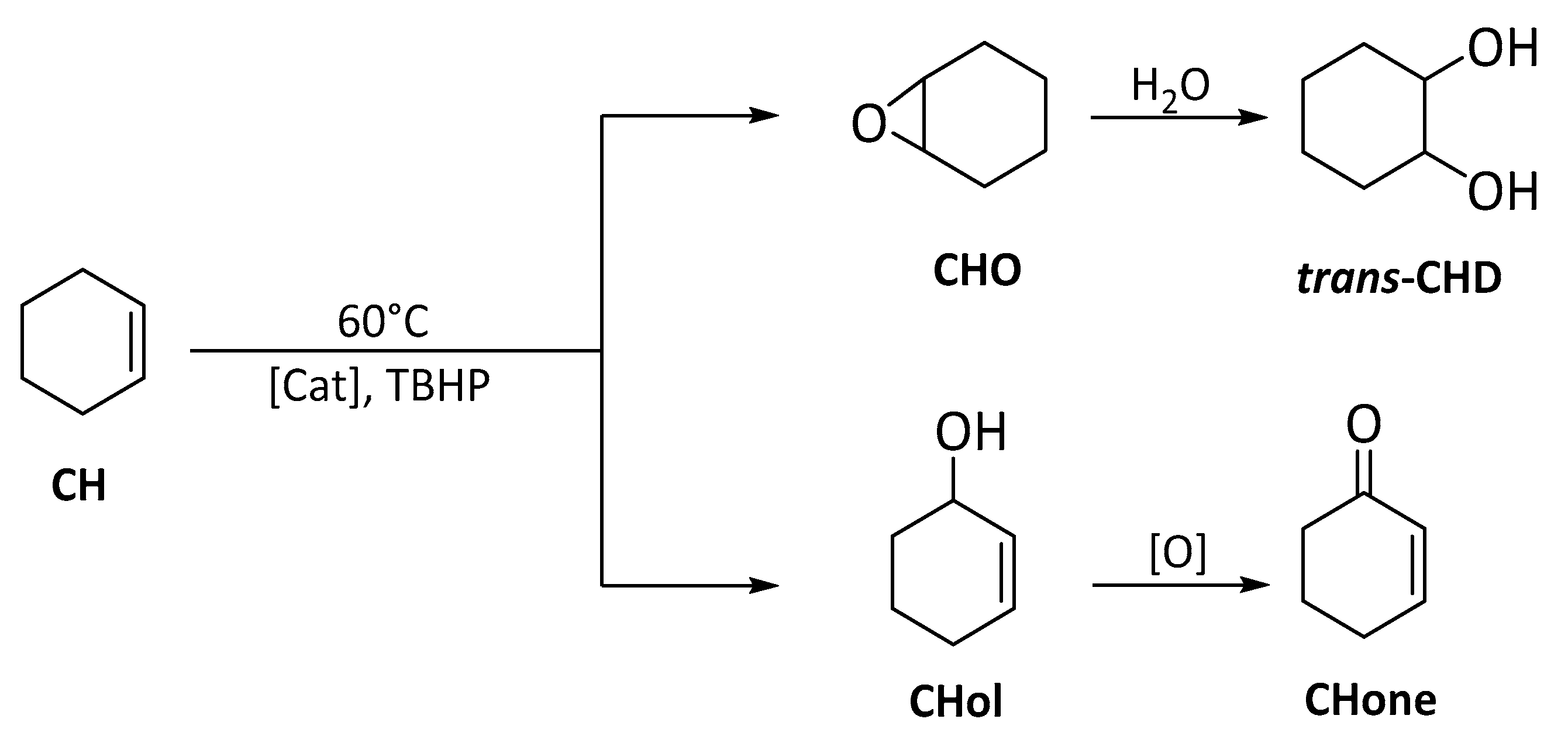
3.5.2. Cyclohexene (CH) (ep)oxidation
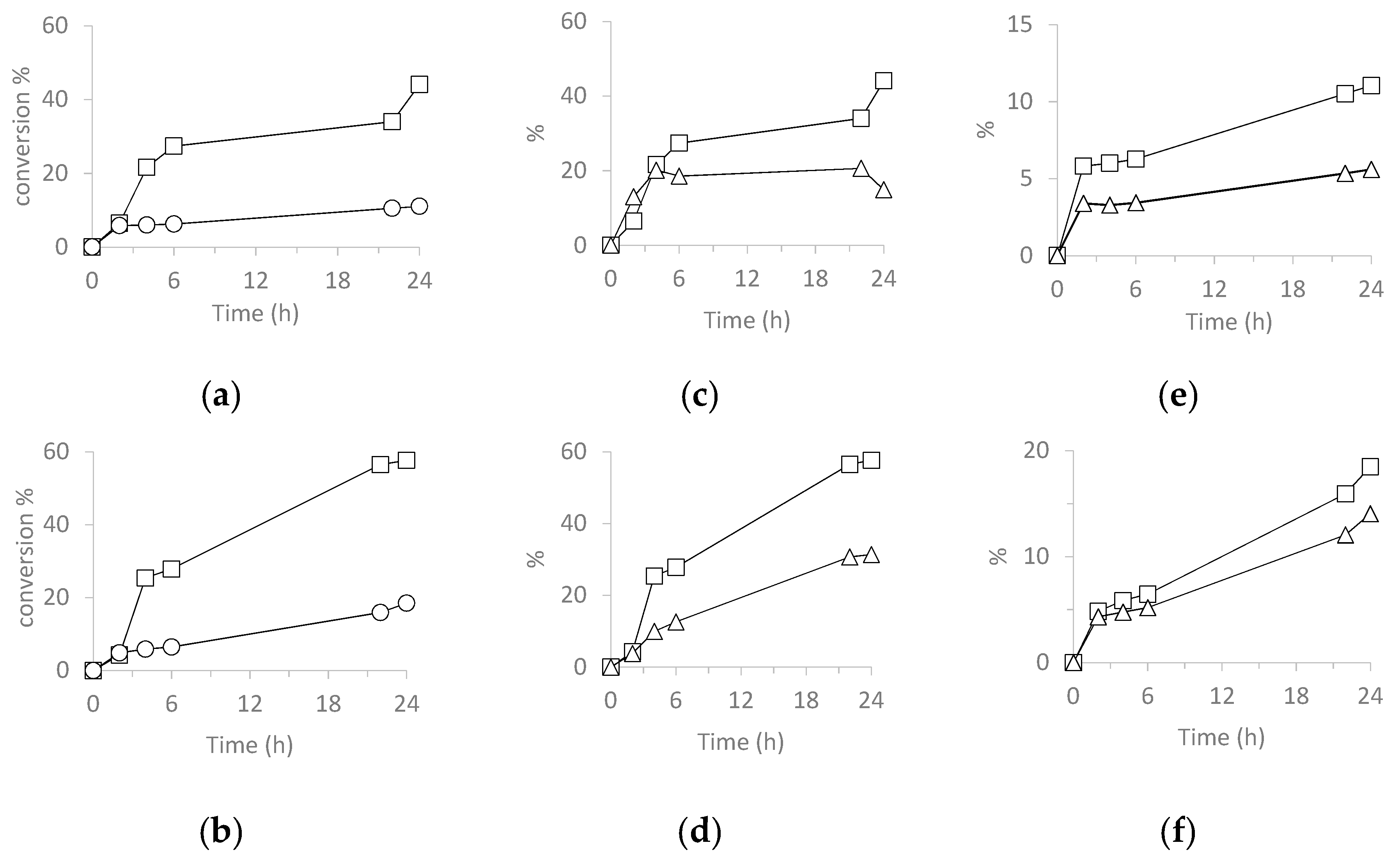
3.5.3. Catalyzed Oxidation of Limonene
3.5.4. Catalysis Oxidation of Cyclohexanol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pastor, I.M.; Yus, M. Asymmetric Ring Opening of Epoxides. Curr. Org. Chem. 2005, 9, 1–29. [Google Scholar] [CrossRef]
- Matlock, P.L.; Brown, W.L.; Clinton, N.A. Polyalkylene glycols. Chem. Ind. 1999, 77, 159–193. [Google Scholar] [CrossRef]
- Petrović, Z.S. Polyurethanes from Vegetable Oils. Polym. Rev. 2008, 48, 109–155. [Google Scholar] [CrossRef]
- McCann, S.D.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst. Acc. Chem. Res. 2015, 48, 1756–1766. [Google Scholar] [CrossRef]
- Bowman, S.; Ide, M.S.; Davis, R.J. Selective Oxidation of Alcohols and Aldehydes over Supported Metal Nanoparticles. Green Chem. 2013, 15, 17–45. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pandarus, V.; Béland, F.; Xu, Y.-J.; Pagliaro, M. Heterogeneously Catalyzed Alcohol Oxidation for the Fine Chemical Industry. Org. Process. Res. Dev. 2015, 19, 1554–1558. [Google Scholar] [CrossRef]
- Parmeggiani, C.; Matassini, C.; Cardona, F. A Step Forward towards Sustainable Aerobic Alcohol Oxidation: New and Revised Catalysts Based on Transition Metals on Solid Supports. Green Chem. 2017, 19, 2030–2050. [Google Scholar] [CrossRef]
- Sheldon, R.A. Recent Advances in Green Catalytic Oxidations of Alcohols in Aqueous Media. Catal. Today 2015, 247, 4–13. [Google Scholar] [CrossRef]
- Marais, L.; Swarts, A.J. Biomimetic Cu/Nitroxyl Catalyst Systems for Selective Alcohol Oxidation. Catalysts 2019, 9, 395. [Google Scholar] [CrossRef]
- Bai, X.; Huang, X.; Wen, L.; Song, N.; Zhang, J.; Zhang, Y.; Zhao, Y. A New Strategy for the Selective Oxidation of Alcohols Catalyzed by a Polyoxometalate-Based Hybrid Surfactant in Biphasic Systems. Chem. Commun. 2019, 55, 3598–3601. [Google Scholar] [CrossRef]
- McDonald, R.N.; Steppel, R.N.; Dorsey, J.E. m-Chloroperbenzoic acid. Org. Synth. 1970, 50, 15. [Google Scholar] [CrossRef]
- Rose, E.; Andrioletti, B.; Zrig, S.; Quelquejeu-Ethève, M. Enantioselective Epoxidation of Olefins with Chiral Metalloporphyrin Catalysts. Chem. Soc. Rev. 2005, 34, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Swern, D. Organic Peroxides; Wiley-Interscience: New York, NY, USA, 1971; Volume 2, pp. 355–533. [Google Scholar] [CrossRef]
- Plesnicar, B. Organic Chemistry, Part C; Trahanovsky, W.C., Ed.; Academic Press: Cambridge, MA, USA, 1978; pp. 211–294. [Google Scholar]
- Kobayashi, S.; Tawara, K. Method for producing epoxy compound. Patent Number JP 2007230908A.
- Sherwood, J. European Restrictions on 1,2-Dichloroethane: C−H Activation Research and Development Should Be Liberated and Not Limited. Angew. Chem. Int. Ed. 2018, 57, 14286–14290. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Anastas, P.; Warner, J. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Tundo, P.; Anastas, P.; Black, D.S.; Breen, J.; Collins, T.J.; Memoli, S.; Miyamoto, J.; Polyakoff, M.; Tumas, W. Synthetic Pathways and Processes in Green Chemistry. Introductory Overview. Pure Appl. Chem. 2000, 72, 1207–1228. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Vrdoljak, V.; Poli, R. Epoxidation Processes by Pyridoxal Dioxomolybdenum(VI) (Pre)Catalysts Without Organic Solvent. Adv. Synth. Catal. 2011, 353, 2910–2914. [Google Scholar] [CrossRef]
- Pisk, J.; Prugovečki, B.; Matković-Čalogović, D.; Poli, R.; Agustin, D.; Vrdoljak, V. Charged Dioxomolybdenum(VI) Complexes with Pyridoxal Thiosemicarbazone Ligands as Molybdenum(V) Precursors in Oxygen Atom Transfer Process and Epoxidation (Pre)Catalysts. Polyhedron 2012, 33, 441–449. [Google Scholar] [CrossRef]
- Morlot, J.; Uyttebroeck, N.; Agustin, D.; Poli, R. Solvent-Free Epoxidation of Olefins Catalyzed by “[MoO2(SAP)]”: A New Mode of Tert-Butylhydroperoxide Activation. ChemCatChem 2013, 5, 601–611. [Google Scholar] [CrossRef]
- Guérin, B.; Mesquita Fernandes, D.; Daran, J.-C.; Agustin, D.; Poli, R. Investigation of Induction Times, Activity, Selectivity, Interface and Mass Transport in Solvent-Free Epoxidation by H2O2 and TBHP: A Study with Organic Salts of the [PMo12O40]3− Anion. New J. Chem. 2013, 37, 3466–3475. [Google Scholar] [CrossRef]
- Wang, W.; Vanderbeeken, T.; Agustin, D.; Poli, R. Tridentate ONS vs. ONO Salicylideneamino(Thio)Phenolato [MoO2L] Complexes for Catalytic Solvent-Free Epoxidation with Aqueous TBHP. Catal. Commun. 2015, 63, 26–30. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Pisk, J.; Agustin, D.; Novak, P.; Vuković, J.P.; Matković-Čalogović, D. Dioxomolybdenum(VI) and Dioxotungsten(VI) Complexes Chelated with the ONO Tridentate Hydrazone Ligand: Synthesis, Structure and Catalytic Epoxidation Activity. New J. Chem. 2014, 38, 6176–6185. [Google Scholar] [CrossRef]
- Wang, W.; Daran, J.-C.; Poli, R.; Agustin, D. OH-Substituted Tridentate ONO Schiff Base Ligands and Related Molybdenum(VI) Complexes for Solvent-Free (Ep)Oxidation Catalysis with TBHP as Oxidant. J. Mol. Cat. A Chem. 2016, 416, 117–126. [Google Scholar] [CrossRef]
- Wang, W.; Agustin, D.; Poli, R. Influence of Ligand Substitution on Molybdenum Catalysts with Tridentate Schiff Base Ligands for the Organic Solvent-Free Oxidation of Limonene Using Aqueous TBHP as Oxidant. Mol. Catal. 2017, 443, 52–59. [Google Scholar] [CrossRef]
- Pisk, J.; Rubčić, M.; Kuzman, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Molybdenum(VI) Complexes of Hemilabile Aroylhydrazone Ligands as Efficient Catalysts for Greener Cyclooctene Epoxidation: An Experimental and Theoretical Approach. New J. Chem. 2019, 43, 5531–5542. [Google Scholar] [CrossRef]
- Cindrić, M.; Pavlović, G.; Katava, R.; Agustin, D. Towards a Global Greener Process: From Solvent-Less Synthesis of Molybdenum(VI) ONO Schiff Base Complexes to Catalyzed Olefin Epoxidation under Organic-Solvent-Free Conditions. New J. Chem. 2017, 41, 594–602. [Google Scholar] [CrossRef]
- Pisk, J.; Daran, J.-C.; Poli, R.; Agustin, D. Pyridoxal Based ONS and ONO Vanadium(V) Complexes: Structural Analysis and Catalytic Application in Organic Solvent Free Epoxidation. J. Mol. Cat. A Chem. 2015, 403, 52–63. [Google Scholar] [CrossRef]
- Damjanović, V.; Pisk, J.; Kuzman, D.; Agustin, D.; Vrdoljak, V.; Stilinović, V.; Cindrić, M. The Synthesis, Structure and Catalytic Properties of the [Mo7O24(μ-Mo8O26)Mo7O24]16− Anion Formed via Two Intermediate Heptamolybdates [Co(en)3]2[NaMo7O24]Cl·nH2O and (H3O)[Co(En)3]2[Mo7O24]Cl·9H2O. Dalton Trans. 2019, 48, 9974–9983. [Google Scholar] [CrossRef] [PubMed]
- Landau, R.; Sullivan, G.A.; Brown, D. Propylene Oxide by the Co-Product Processes. Chemtech 1979, 9, 602–607. [Google Scholar]
- Sutradhar, M.; Martins, L.M.; Da Silva, M.F.C.G.; Pombeiro, A.J. Vanadium complexes: Recent progress in oxidation catalysis. Coord. Chem. Rev. 2015, 301, 200–239. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Poli, R. Organic Salts and Merrifield Resin Supported [PM12O40]3− (M = Mo or W) as Catalysts for Adipic Acid Synthesis. Molecules 2019, 24, 783. [Google Scholar] [CrossRef]
- Maurya, M.R. Vanadium Complexes Based Polymer Supported Catalysts: A Brief Account of Research from Our Group. Top. Catal. 2018, 61, 1500. [Google Scholar] [CrossRef]
- Sutradhar, M.; Martins, L.M.D.R.S.; Carabineiro, S.A.C.; Da Silva, M.F.C.G.; Buijnsters, J.G.; Figueiredo, J.L.; Pombeiro, A.J.L. Oxidovanadium(V) Complexes Anchored on Carbon Materials as Catalysts for the Oxidation of 1-Phenylethanol. ChemCatChem 2016, 8, 2254–2266. [Google Scholar] [CrossRef]
- Richards, R.; Kortz, U.; Bi, L.; Zhu, K.; Suchopar, A.; Cheng, L. Supported Polyoxometallates and Process for Their Preparation. Patent Publication Number WO 2007/142727.
- Balula, S.S.; Cunha-Silva, L.; Santos, I.C.M.S.; Estrada, A.C.; Fernandes, A.C.; Cavaleiro, J.A.S.; Pires, J.; Freire, C.; Cavaleiro, A.M.V. Mono-Substituted Silicotungstates as Active Catalysts for Sustainable Oxidations: Homo- and Heterogeneous Performance. New J. Chem. 2013, 37, 2341–2350. [Google Scholar] [CrossRef]
- Panchenko, V.N.; Borbáth, I.; Timofeeva, M.N.; Gőbölös, S. Amine-Modified Silica NH2–(CH2)x–SiO2 (X=0, 2, 3) as Support for Cobalt-Substituted Polyoxometalate TBA4HPW11CoO39: Effect of the Nature of the Support on the Oxidation Activity. J. Mol. Cat. A Chem. 2010, 319, 119–125. [Google Scholar] [CrossRef]
- Li, X.; Jiang, P.; Wang, Z.; Huang, Y. Phosphotungstate-Based Ionic Silica Nanoparticles Network for Alkenes Epoxidation. Catalysts 2016, 6, 2. [Google Scholar] [CrossRef]
- Neumann, R.; Miller, H. Alkene Oxidation in Water Using Hydrophobic Silica Particles Derivatized with Polyoxometalates as Catalysts. J. Chem. Soc. Chem. Commun. 1995, 2277–2278. [Google Scholar] [CrossRef]
- Simon, A.; Cohen-Bouhacina, T.; Porté, M.C.; Aimé, J.P.; Baquey, C. Study of Two Grafting Methods for Obtaining a 3-Aminopropyltriethoxysilane Monolayer on Silica Surface. J. Colloid Interface Sci. 2002, 251, 278–283. [Google Scholar] [CrossRef]
- Cuoq, F.; Masion, A.; Labille, J.; Rose, J.; Ziarelli, F.; Prelot, B.; Bottero, J.-Y. Preparation of Amino-Functionalized Silica in Aqueous Conditions. Appl. Surf. Sci. 2013, 266, 155–160. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, T.-J.; Gao, H.; Jin, Y. High Density Silanization of Nano-Silica Particles Using γ-Aminopropyltriethoxysilane (APTES). Appl. Surf. Sci. 2015, 351, 646–654. [Google Scholar] [CrossRef]
- Okun, N.M.; Anderson, T.M.; Hill, C.L. [(FeIII(OH2)2)3(A-α-PW9O34)2]9− on Cationic Silica Nanoparticles, a New Type of Material and Efficient Heterogeneous Catalyst for Aerobic Oxidations. J. Am. Chem. Soc. 2003, 125, 3194–3195. [Google Scholar] [CrossRef] [PubMed]
- Okun, N.M.; Anderson, T.M.; Hill, C.L. Polyoxometalates on Cationic Silica: Highly Selective and Efficient O2/Air-Based Oxidation of 2-Chloroethyl Ethyl Sulfide at Ambient Temperature. J. Mol. Cat. A Chem. 2003, 197, 283–290. [Google Scholar] [CrossRef]
- Wu, W.-J.; Wang, J.; Chen, M.; Qian, D.-J.; Liu, M. Terpyridine-Functionalized NanoSiO2 Multi-Dentate Linkers: Preparation, Characterization and Luminescent Properties of Their Metal–Organic Hybrid Materials. J. Phys. Chem. C 2017, 121, 2234–2242. [Google Scholar] [CrossRef]
- Wang, J.; Hao, Q. Thin Films of Silica Particles Covered with Lanthanide Substituted Keggin Polyoxometalates and Their Optical Properties. J. Alloys Compd. 2009, 482, 235–239. [Google Scholar] [CrossRef]
- Sharma, R.K.; Sharma, S.; Gulati, S.; Pandey, A. Fabrication of a Novel Nano-Composite Carbon Paste Sensor Based on Silica-Nanospheres Functionalized with Isatin Thiosemicarbazone for Potentiometric Monitoring of Cu2+ Ions in Real Samples. Anal. Methods 2013, 5, 1414–1426. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.H.; Choi, K.; Kim, H.G.; Park, J.A.; Cho, S.H.; Hong, S.W.; Lee, J.; Lee, J.H.; Lee, S.-J.; et al. Investigation of the Mechanism of Chromium Removal in (3-Aminopropyl)Trimethoxysilane Functionalized Mesoporous Silica. Sci. Rep. 2018, 8, 12078. [Google Scholar] [CrossRef] [PubMed]
- Goscianska, J.; Olejnik, A.; Nowak, I. APTES-Functionalized Mesoporous Silica as a Vehicle for Antipyrine – Adsorption and Release Studies. Colloids Surf. A 2017, 533, 187–196. [Google Scholar] [CrossRef]
- Nogueira, L.S.; Ribeiro, S.; Granadeiro, C.M.; Pereira, E.; Feio, G.; Cunha-Silva, L.; Balula, S.S. Novel Polyoxometalate Silica Nano-Sized Spheres: Efficient Catalysts for Olefin Oxidation and the Deep Desulfurization Process. Dalton Trans. 2014, 43, 9518–9528. [Google Scholar] [CrossRef]
- Okun, N.M.; Ritorto, M.D.; Anderson, T.M.; Apkarian, R.P.; Hill, C.L. Polyoxometalates on Cationic Silica Nanoparticles. Physicochemical Properties of an Electrostatically Bound Multi-Iron Catalyst. Chem. Mater. 2004, 16, 2551–2558. [Google Scholar] [CrossRef]
- Naz, A.; Arun, S.; Narvi, S.S.; Alam, M.S.; Singh, A.; Bhartiya, P.; Dutta, P.K. Cu(II)-Carboxymethyl Chitosan-Silane Schiff Base Complex Grafted on Nano Silica: Structural Evolution, Antibacterial Performance and Dye Degradation Ability. Int. J. Biol. Macromol. 2018, 110, 215–226. [Google Scholar] [CrossRef]
- Talreja, K.; Chauhan, I.; Ghosh, A.; Majumdar, A.; Butola, B.S. Functionalization of Silica Particles to Tune the Impact Resistance of Shear Thickening Fluid Treated Aramid Fabrics. RSC Adv. 2017, 7, 49787–49794. [Google Scholar] [CrossRef]
- Van den Berg, R.; Parmentier, T.E.; Elkjær, C.F.; Gommes, C.J.; Sehested, J.; Helveg, S.; de Jongh, P.E.; de Jong, K.P. Support Functionalization To Retard Ostwald Ripening in Copper Methanol Synthesis Catalysts. ACS Catal. 2015, 5, 4439–4448. [Google Scholar] [CrossRef]
- Sándor, M.; Nistor, C.L.; Szalontai, G.; Stoica, R.; Nicolae, C.A.; Alexandrescu, E.; Fazakas, J.; Oancea, F.; Donescu, D. Aminopropyl-Silica Hybrid Particles as Supports for Humic Acids Immobilization. Materials 2016, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.; Andrews, P.C.; Fraser, B.H.; Forsyth, C.M.; Junk, P.C.; Massi, M.; Tuck, K.L. Facile Methods for the Separation of the Cis- and Trans-Diastereomers of Limonene 1,2-Oxide and Convenient Routes to Diequatorial and Diaxial 1,2-Diols. Synthesis 2007, 2007, 1523–1527. [Google Scholar] [CrossRef]
- Van der Werf, M.J.; Jongejan, H.; Franssen, M.C.R. Resolution of Limonene 1,2-Epoxide Diastereomers by Mercury(II) Ions. Tetrahedron Lett. 2001, 42, 5521–5524. [Google Scholar] [CrossRef]
- Steiner, D.; Ivison, L.; Goralski, C.T.; Appell, R.B.; Gojkovic, J.R.; Singaram, B. A Facile and Efficient Method for the Kinetic Separation of Commercially Available Cis- and Trans-Limonene Epoxide. Tetrahedron Asymmetry 2002, 13, 2359–2363. [Google Scholar] [CrossRef]
- Stöber, W.; Fink, A.; Bohn, E. Controlled Growth of Monodisperse Silica Spheres in the Micron Size Range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Bu, J.; Li, R.; Quah, C.W.; Carpenter, K.J. Propagation of PAMAM Dendrons on Silica Gel: A Study on the Reaction Kinetics. Macromolecules 2004, 37, 6687–6694. [Google Scholar] [CrossRef]
- Dolbecq, A.; Dumas, E.; Mayer, C.R.; Mialane, P. Hybrid Organic–Inorganic Polyoxometalate Compounds: From Structural Diversity to Applications. Chem. Rev. 2010, 110, 6009–6048. [Google Scholar] [CrossRef]
- Azarshin, S.; Moghadasi, J.; A Aboosadi, Z. Surface Functionalization of Silica Nanoparticles to Improve the Performance of Water Flooding in Oil Wet Reservoirs. Energy Explor. Exploit. 2017, 35, 685–697. [Google Scholar] [CrossRef]
- Gholami, T.; Salavati-Niasari, M.; Bazarganipour, M.; Noori, E. Synthesis and Characterization of Spherical Silica Nanoparticles by Modified Stöber Process Assisted by Organic Ligand. Superlattices Microstruct. 2013, 61, 33–41. [Google Scholar] [CrossRef]
- Cai, W.; Zhou, Y.; Bao, R.; Yue, B.; He, H. Catalytic Epoxidation of Cyclohexene over Mesoporous-Silica Immobilized Keggin-Type Tungstophosphoric Acid. Chin. J. Catal. 2013, 34, 193–199. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic Light Scattering: A Practical Guide and Applications in Biomedical Sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Juillerat, F.; Galletto, P.; Bowen, P.; Borkovec, M. Aggregation and Charging of Colloidal Silica Particles: Effect of Particle Size. Langmuir 2005, 21, 5761–5769. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, E.D.; Pandya, Y.; Mun, E.A.; Rogers, S.E.; Abutbul-Ionita, I.; Danino, D.; Williams, A.C.; Khutoryanskiy, V.V. Structure and Characterisation of Hydroxyethylcellulose–Silica Nanoparticles. RSC Adv. 2018, 8, 6471–6478. [Google Scholar] [CrossRef]
- Neves, C.S.; Granadeiro, C.M.; Cunha-Silva, L.; Ananias, D.; Gago, S.; Feio, G.; Carvalho, P.A.; Eaton, P.; Balula, S.S.; Pereira, E. Europium Polyoxometalates Encapsulated in Silica Nanoparticles—Characterization and Photoluminescence Studies. Eur. J. Inorg. Chem. 2013, 2013, 2877–2886. [Google Scholar] [CrossRef]
- Greasley, S.L.; Page, S.J.; Sirovica, S.; Chen, S.; Martin, R.A.; Riveiro, A.; Hanna, J.V.; Porter, A.E.; Jones, J.R. Controlling Particle Size in the Stöber Process and Incorporation of Calcium. J. Colloid Interface Sci. 2016, 469, 213–223. [Google Scholar] [CrossRef]
- Mahalingam, V.; Onclin, S.; Péter, M.; Ravoo, B.J.; Huskens, J.; Reinhoudt, D.N. Directed Self-Assembly of Functionalized Silica Nanoparticles on Molecular Printboards through Multivalent Supramolecular Interactions. Langmuir 2004, 20, 11756–11762. [Google Scholar] [CrossRef]
- Martín, S.; Takashima, Y.; Lin, C.-G.; Song, Y.-F.; Miras, H.N.; Cronin, L. Integrated Synthesis of Gold Nanoparticles Coated with Polyoxometalate Clusters. Inorg. Chem. 2019, 58, 4110–4116. [Google Scholar] [CrossRef]
- Aneja, K.S.; Bohm, S.; Khanna, A.S.; Mallika Bohm, H.L. Graphene Based Anticorrosive Coatings for Cr(VI) Replacement. Nanoscale 2015, 7, 17879–17888. [Google Scholar] [CrossRef]
- Karimpour, T.; Safaei, E.; Karimi, B.; Lee, Y.-I. Iron(III) Amine Bis(Phenolate) Complex Immobilized on Silica-Coated Magnetic Nanoparticles: A Highly Efficient Catalyst for the Oxidation of Alcohols and Sulfides. ChemCatChem 2018, 10, 1889–1899. [Google Scholar] [CrossRef]
- Tighadouini, S.; Radi, S.; Elidrissi, A.; Zaghrioui, M.; Garcia, Y. Selective Confinement of CdII in Silica Particles Functionalized with β-Keto-Enol-Bisfuran Receptor: Isotherms, Kinetic and Thermodynamic Studies. Eur. J. Inorg. Chem. 2019, 2019, 3180–3186. [Google Scholar] [CrossRef]
- Luo, X.; Yang, C. Photochromic Ordered Mesoporous Hybrid Materials Based on Covalently Grafted Polyoxometalates. Phys. Chem. Chem. Phys. 2011, 13, 7892–7902. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Landry, C.C. Immobilization of a Mo, V-Polyoxometalate on Cationically Modified Mesoporous Silica: Synthesis and Characterization Studies. Microporous Mesoporous Mater. 2007, 98, 309–316. [Google Scholar] [CrossRef]
- Misono, M.; Mizuno, N.; Katamura, K.; Kasai, A.; Konishi, Y.; Sakata, K.; Okuhara, T.; Yoneda, Y. Catalysis by Heteropoly Compounds. III. The Structure and Properties of 12-Heteropolyacids of Molybdenum and Tungsten (H3PMo12−xWxO40) and Their Salts Pertinent to Heterogeneous Catalysis. Bull. Chem. Soc. Jpn. 1982, 55, 400–406. [Google Scholar] [CrossRef]
- Trébosc, J.; Wiench, J.W.; Huh, S.; Lin, V.S.-Y.; Pruski, M. Solid-State NMR Study of MCM-41-Type Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2005, 127, 3057–3068. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.C.; Jones, C.W. Controlling the Density of Amine Sites on Silica Surfaces Using Benzyl Spacers. Langmuir 2006, 22, 2676–2681. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.O.; Granadeiro, C.M.; Almeida, P.L.; Pires, J.; Capel-Sanchez, M.C.; Campos-Martin, J.M.; Gago, S.; de Castro, B.; Balula, S.S. Oxidative Desulfurization Strategies Using Keggin-Type Polyoxometalate Catalysts: Biphasic versus Solvent-Free Systems. Catal. Today 2019, 333, 226–236. [Google Scholar] [CrossRef]
- Bentaleb, F.; Makrygenni, O.; Brouri, D.; Coelho Diogo, C.; Mehdi, A.; Proust, A.; Launay, F.; Villanneau, R. Efficiency of Polyoxometalate-Based Mesoporous Hybrids as Covalently Anchored Catalysts. Inorg. Chem. 2015, 54, 7607–7616. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Lin, K.-J.; Prasad, M.R.; Fu, S.-J.; Chang, S.-Y.; Shyu, S.-G.; Sheu, H.-S.; Chen, C.-H.; Chuang, C.-H.; Lin, M.-T. Synthesis of a Reusable Oxotungsten-Containing SBA-15 Mesoporous Catalyst for the Organic Solvent-Free Conversion of Cyclohexene to Adipic Acid. Catal. Commun. 2007, 8, 1060–1064. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, D.; Ma, N.; Hou, Z.; Hu, M.; Wang, C.; Wang, W. Covalent Immobilization of a Polyoxometalate in a Porous Polymer Matrix: A Heterogeneous Catalyst towards Sustainability. RSC Adv. 2013, 3, 21544–21551. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Molecular Design of Polyoxometalate-Based Compounds for Environmentally-Friendly Functional Group Transformations: From Molecular Catalysts to Heterogeneous Catalysts. Catal. Surv. Asia 2011, 15, 68–79. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V.; Kloetstra, K.R.; Sinnema, A.; Zandbergen, H.W.; van Bekkum, H. Study of Catalysts Comprising Heteropoly Acid H3PW12O40 Supported on MCM-41 Molecular Sieve and Amorphous Silica. J. Mol. Catal. A Chem. 1996, 114, 287–298. [Google Scholar] [CrossRef]
- Canioni, R.; Roch-Marchal, C.; Sécheresse, F.; Horcajada, P.; Serre, C.; Hardi-Dan, M.; Férey, G.; Grenèche, J.-M.; Lefebvre, F.; Chang, J.-S.; et al. Stable Polyoxometalate Insertion within the Mesoporous Metal Organic Framework MIL-100(Fe). J. Mater. Chem. 2011, 21, 1226–1233. [Google Scholar] [CrossRef]
- Crucho, C.I.C.; Baleizão, C.; Farinha, J.P.S. Functional Group Coverage and Conversion Quantification in Nanostructured Silica by 1H NMR. Anal. Chem. 2017, 89, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Nlate, S.; Kortz, U.; Bonchio, M. Polyoxometalate as Homogeneous Oxidation Catalysts. In Innovative Catalysis in Organic Synthesis: Oxidation, Hydrogenation, and C-X Bond Forming Reaction; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 3–24. [Google Scholar]
- Neumann, R. Applications of Polyoxometalates in Homogeneous Catalysis. In Polyoxometalate Molecular Science; Borrás-Almenar, J.J., Coronado, E., Müller, A., Pope, M., Eds.; NATO Science Series; Springer: Dordrecht, The Netherlands, 2003; pp. 327–349. [Google Scholar] [CrossRef]
- Rafiee, E.; Eavani, S. Polyoxometalates as Heterogeneous Catalysts for Organic Reactions. Curr. Org. Chem. 2017, 21, 752–778. [Google Scholar] [CrossRef]
- Duprez, D.; Cavani, F. Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry; World Scientific: Singapore, 2014. [Google Scholar]
- Carraro, M.; Fiorani, G.; Sartorel, A.; Bonchio, M. Polyoxometalates Catalysts for Sustainable Oxidations and Energy Applications. In Handbook of Advanced Methods and Processes in Oxidation Catalysis; Imperial College Press: London, UK, 2011; pp. 586–630. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Maksimchuk, N.V.; Maksimov, G.M. Polyoxometalate-Based Heterogeneous Catalysts for Liquid Phase Selective Oxidations: Comparison of Different Strategies. Catal. Today 2010, 157, 107–113. [Google Scholar] [CrossRef]
- Ma, X.; He, P.; Xu, B.; Lu, J.; Wan, R.; Wu, H.; Wang, Y.; Ma, P.; Niu, J.; Wang, J. Pyrazine dicarboxylate-bridged arsenotungstate: Synthesis, characterization, and catalytic activities in epoxidation of olefins and oxidation of alcohols. Dalton Trans. 2019, 48, 12956–12963. [Google Scholar] [CrossRef]
- Zhang, T.-T.; Zhang, X.; Lü, Y.; Li, G.-D.; Xiao, L.-N.; Cui, X.-B.; Xu, J.-Q. New organic–inorganic hybrid compounds based on [SiNb12V2O42]12− with high catalytic activity for styrene epoxidation. Inorg. Chem. Front. 2017, 4, 1397–1404. [Google Scholar] [CrossRef]
- Zhang, L.; Song, S.; Yang, N.; Tantai, X.; Xiao, X.; Jiang, B.; Sun, Y. Porous Hybrid Nanoflower Self-Assembled from Polyoxometalate and Polyionene for Efficient Oxidative Desulfurization. Ind. Eng. Chem. Res. 2019, 58, 3618–3629. [Google Scholar] [CrossRef]
- Masteri-Farahani, M.; Modarres, M. Wells-Dawson Heteropoly Acid Immobilized inside the Nanocages of SBA-16 with Ship-in-a-Bottle Method: A New Recoverable Catalyst for the Epoxidation of Olefins. J. Mol. Catal. A Chem. 2016, 417, 81–88. [Google Scholar] [CrossRef]
- Song, X.; Zhu, W.; Li, K.; Wang, J.; Niu, H.; Gao, H.; Gao, W.; Zhang, W.; Yu, J.; Jia, M. Epoxidation of Olefins with Oxygen/Isobutyraldehyde over Transition-Metal-Substituted Phosphomolybdic Acid on SBA-15. Catal. Today 2016, 259, 59–65. [Google Scholar] [CrossRef]
- Karimi, Z.; Mahjoub, A.R.; Harati, S.M. Polyoxometalate-Based Hybrid Mesostructured Catalysts for Green Epoxidation of Olefins. Inorg. Chim. Acta 2011, 376, 1–9. [Google Scholar] [CrossRef]
- Gao, W.; Sun, X.; Niu, H.; Song, X.; Li, K.; Gao, H.; Zhang, W.; Yu, J.; Jia, M. Phosphomolybdic Acid Functionalized Covalent Organic Frameworks: Structure Characterization and Catalytic Properties in Olefin Epoxidation. Microporous Mesoporous Mater. 2015, 213, 59–67. [Google Scholar] [CrossRef]
- Song, X.; Hu, D.; Yang, X.; Zhang, H.; Zhang, W.; Li, J.; Jia, M.; Yu, J. Polyoxomolybdic Cobalt Encapsulated within Zr-Based Metal–Organic Frameworks as Efficient Heterogeneous Catalysts for Olefins Epoxidation. ACS Sustain. Chem. Eng. 2019, 7, 3624–3631. [Google Scholar] [CrossRef]
- Mohammadi, M.; Khazaei, A.; Rezaei, A.; Huajun, Z.; Xuwei, S. Ionic-Liquid-Modified Carbon Quantum Dots as a Support for the Immobilization of Tungstate Ions (WO42–): Heterogeneous Nanocatalysts for the Oxidation of Alcohols in Water. ACS Sustain. Chem. Eng. 2019, 7, 5283–5291. [Google Scholar] [CrossRef]
- Jiménez-González, C.; Curzons, A.D.; Constable, D.J.C.; Cunningham, V.L. Expanding GSK’s Solvent Selection Guide—Application of Life Cycle Assessment to Enhance Solvent Selections. Clean Technol. Environ. Policy 2004, 7, 42–50. [Google Scholar] [CrossRef]
- Wang, J.Y.; Zhou, M.D.; Yuan, Y.G.; Fu, N.H.; Zang, S.L. Oxidation of Cyclooctene to Suberic Acid Using Perrhenate-Containing Composite Ionic Liquids as Green Catalysts. Russ. J. Gen. Chem. 2015, 85, 2378–2385. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, W.-J.; Lu, X.; Yang, H.; Chen, Z.; Liao, R.-Z.; Yin, G. Nonredox Metal Ions Promoted Olefin Epoxidation by Iron(II) Complexes with H2O2: DFT Calculations Reveal Multiple Channels for Oxygen Transfer. Inorg. Chem. 2017, 56, 15138–15149. [Google Scholar] [CrossRef]
- Liu, C.-G.; Jiang, M.-X.; Su, Z.-M. Computational Study on M1/POM Single-Atom Catalysts (M = Cu, Zn, Ag, and Au; POM = [PW12O40]3−): Metal–Support Interactions and Catalytic Cycle for Alkene Epoxidation. Inorg. Chem. 2017, 56, 10496–10504. [Google Scholar] [CrossRef]
- Kamata, K.; Kotani, M.; Yamaguchi, K.; Hikichi, S.; Mizuno, N. Olefin Epoxidation with Hydrogen Peroxide Catalyzed by Lacunary Polyoxometalate [γ-SiW10O34(H2O)2]4−. Chem. Eur. J. 2007, 13, 639–648. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, Y.; Ma, B.; Ding, Y.; Qiu, W. Oxidation of alcohols with hydrogen peroxide in water catalyzed by recyclable keggin-type tungstoborate catalyst. Catal. Commun. 2010, 11, 527–531. [Google Scholar] [CrossRef]
- Sutradhar, M.; Martins, L.M.; Da Silva, M.F.C.G.; Pombeiro, A.J. Oxidovanadium complexes with tridentate aroylhydrazone as catalyst precursors for solvent-free microwave-assisted oxidation of alcohols. Appl. Catal. A Gen. 2015, 493, 50–57. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SiO2@NH2 | SiO2@PW | SiO2@PMo |
|---|---|---|---|
| N (%) | 0.74 | 0.43 | 0.58 |
| x | 0 | 0.47 | 0.29 |
| Sample | ρ(NH2) 1 (1H NMR) | ρ(PM) 1 (31P NMR) | ρ(NH2) 1 (EA) | ρ(PM) 1 (EA) | μ(NH2) 2 |
|---|---|---|---|---|---|
| SiO2@NH2 | 0.52 | - | 0.53 | - | 6.8 |
| SiO2@PW | 0.33 | 0.15 | 0.31 | 0.14 | 6.8 |
| SiO2@PMo | 0.40 | 0.12 | 0.41 | 0.12 | 6.7 |
| Catalyst | Run | CO Conv. 2 | COE Sel. 3 | TON 4 |
|---|---|---|---|---|
| H3PW12O40 | 1 | 64 | 14 | 807 |
| SiO2@PW | 1 | 72 | 41 | 981 |
| 2 | 75 | 38 | 987 | |
| 3 | 77 | 37 | 968 | |
| H3PMo12O40 | 1 | 99 | 44 | 1712 |
| SiO2@PMo | 1 | 98 | 71 | 1693 |
| 2 | 96 | 72 | 1620 | |
| 3 | 93 | 69 | 1598 |
| Catalyst | Run | CH Conv 2 | CHO Sel 3 | CHD Sel 3 | Chol Sel 3 | Chone Sel 3 | TON 4 |
|---|---|---|---|---|---|---|---|
| H3PW12O40 | 1 | 31 | <1 | 4 | 3 | 3 | 11,307 |
| SiO2@PW | 1 | 45 | 1 | 2 | 4 | 5 | 21,458 |
| 2 | 43 | 1 | 2 | 2 | 3 | 20,649 | |
| 3 | 26 | 3 | 3 | 7 | 7 | 12,373 | |
| H3PMo12O40 | 1 | 91 | <1 | 40 | 3 | 2 | 52,728 |
| SiO2@PMo | 1 | 80 | 13 | 26 | 5 | 2 | 46,732 |
| 2 | 74 | 15 | 22 | 6 | 3 | 42,487 | |
| 3 | 60 | 26 | 20 | 4.9 | 2 | 36,345 |
| Catalyst | Run | Lim Conv 2 | cis-LO Sel 3 | trans-LO Sel 3 | ax-LD Sel 3 | eq-LD Sel 3 | Col Sel 3 | Cone Sel 3 | TON 4 |
|---|---|---|---|---|---|---|---|---|---|
| H3PW12O40 | 1 | 67 | 0 | 0 | 5 | 3 | 1 | 4 | 1287 |
| SiO2@PW | 1 | 58 | 0 | 3 | 13 | 1 | 8 | 8 | 754 |
| 2 | 59 | 0 | 3 | 13 | 1 | 10 | 8 | 768 | |
| 3 | 62 | 0 | 2 | 12 | 2 | 11 | 8 | 754 | |
| H3PMo12O40 | 1 | 99 | 0 | 0 | 18 | 10 | 1 | 2 | 1859 |
| SiO2@PMo | 1 | 91 | 0 | 0 | 36 | 11 | 4 | 3 | 1721 |
| 2 | 86 | 0 | 0 | 32 | 8 | 6 | 7 | 1626 | |
| 3 | 81 | 0 | < 1 | 20 | 5 | 6 | 6 | 1526 |
| Catalyst | Run | CYol Conversion 2 | CYone Selectivity 3 | TON 4 |
|---|---|---|---|---|
| H3PW12O40 | 1 | 44 | 34 | 525 |
| SiO2@PW | 1 | 11 | 51 | 137 |
| 2 | 8 | 97 | 101 | |
| 3 | 7 | 87 | 92 | |
| H3PMo12O40 | 1 | 58 | 54 | 728 |
| SiO2@PMo | 1 | 18 | 76 | 228 |
| 2 | 17 | 90 | 207 | |
| 3 | 20 | 75 | 249 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Gayet, F.; Guillo, P.; Agustin, D. Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3− (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads. Materials 2019, 12, 3278. https://doi.org/10.3390/ma12203278
Wang Y, Gayet F, Guillo P, Agustin D. Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3− (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads. Materials. 2019; 12(20):3278. https://doi.org/10.3390/ma12203278
Chicago/Turabian StyleWang, Yun, Florence Gayet, Pascal Guillo, and Dominique Agustin. 2019. "Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3− (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads" Materials 12, no. 20: 3278. https://doi.org/10.3390/ma12203278
APA StyleWang, Y., Gayet, F., Guillo, P., & Agustin, D. (2019). Organic Solvent-Free Olefins and Alcohols (ep)oxidation Using Recoverable Catalysts Based on [PM12O40]3− (M = Mo or W) Ionically Grafted on Amino Functionalized Silica Nanobeads. Materials, 12(20), 3278. https://doi.org/10.3390/ma12203278