Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of the Supports and Supported Cobalt Catalysts
2.2. Characterization Techniques
2.3. Evaluation of the Catalytic Performance
3. Results
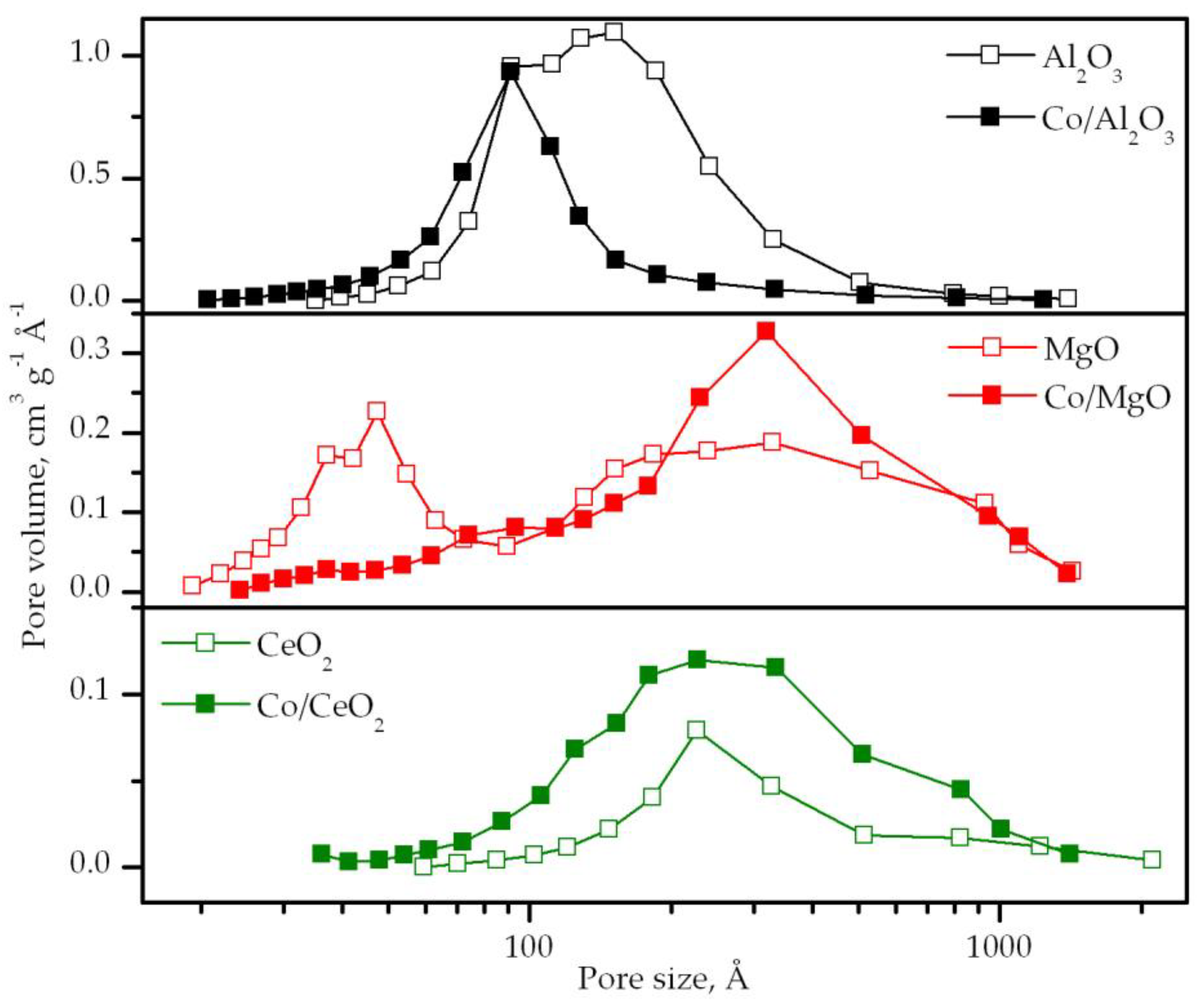
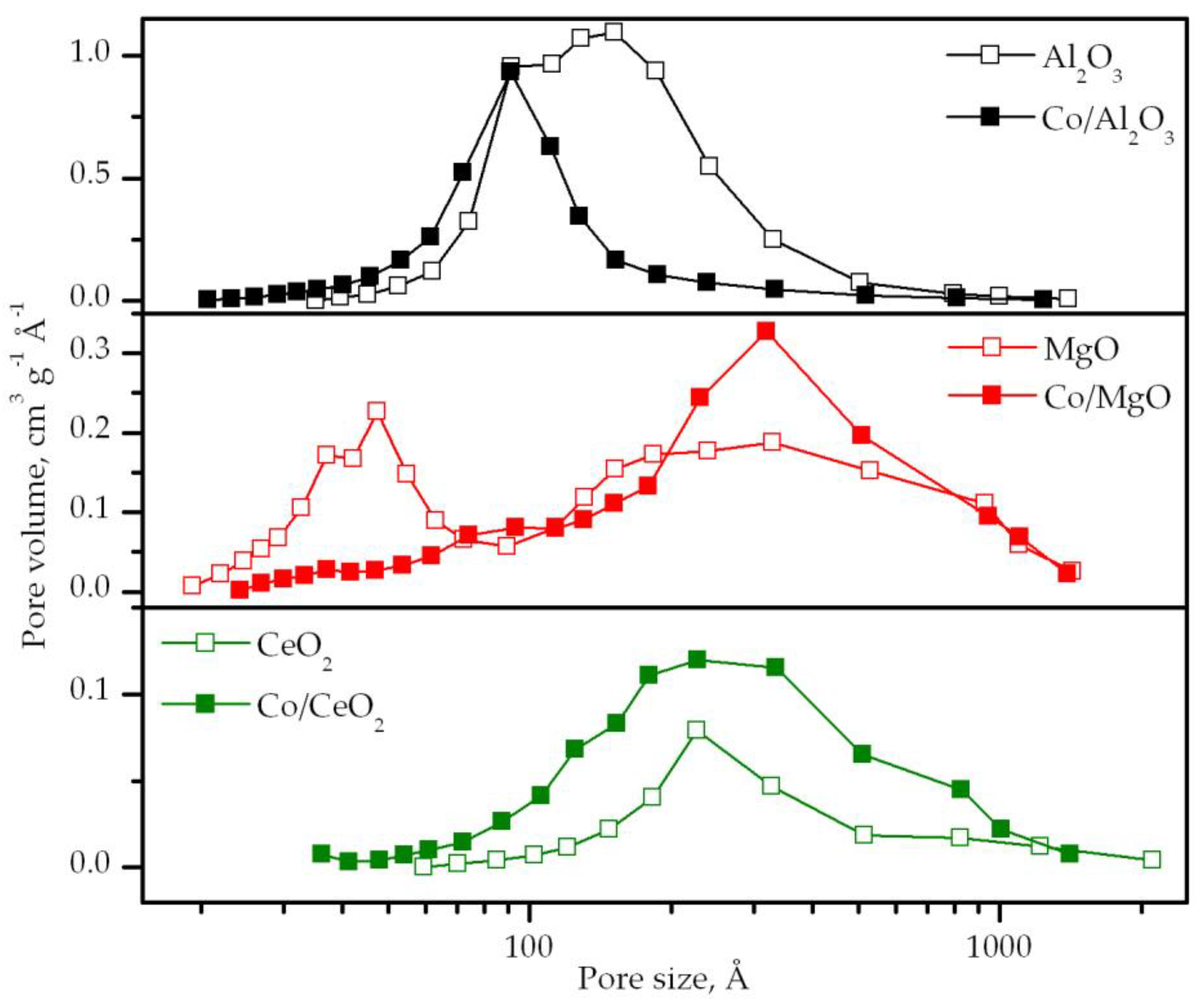
3.1. Characterization of the Supports
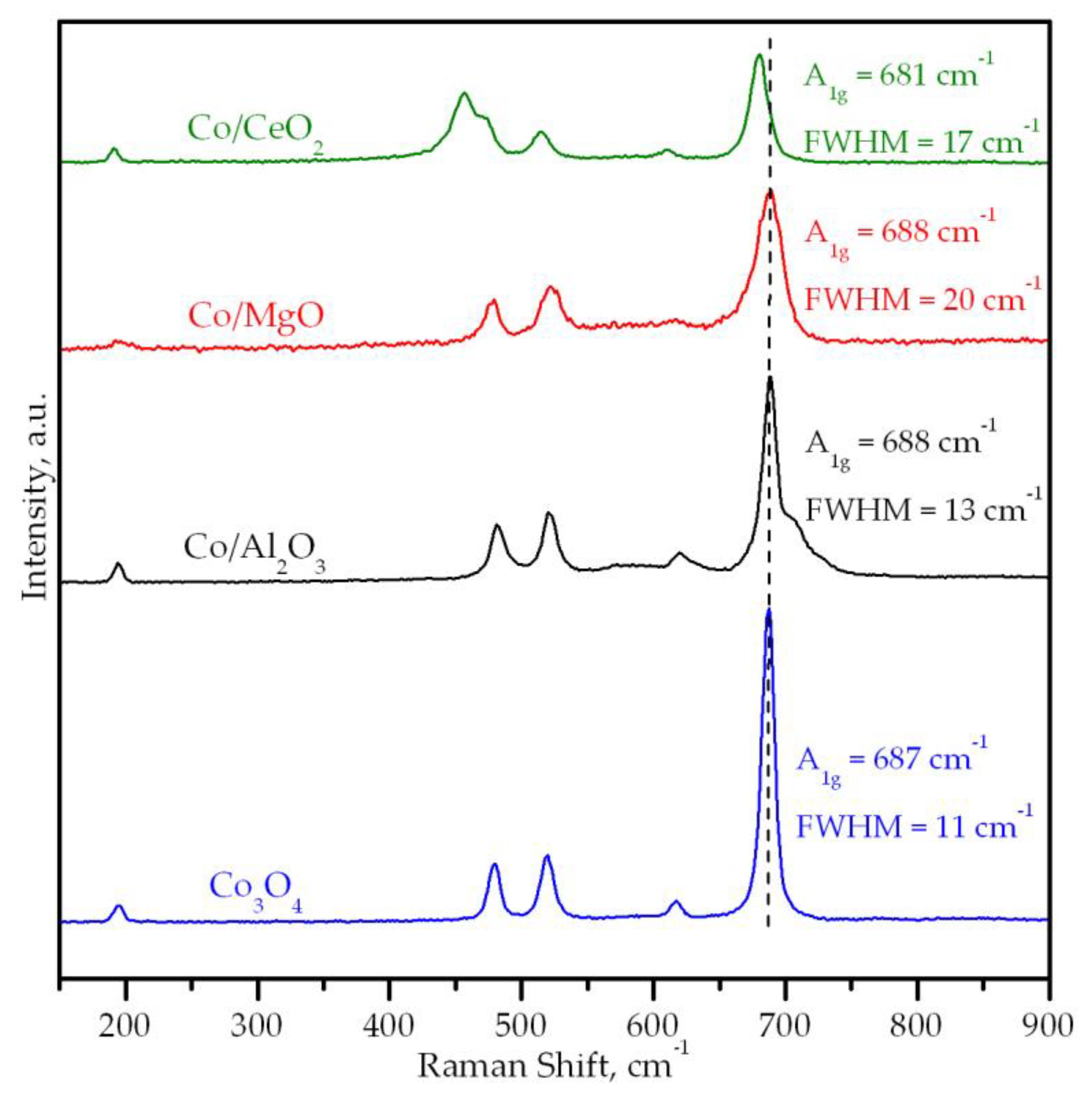
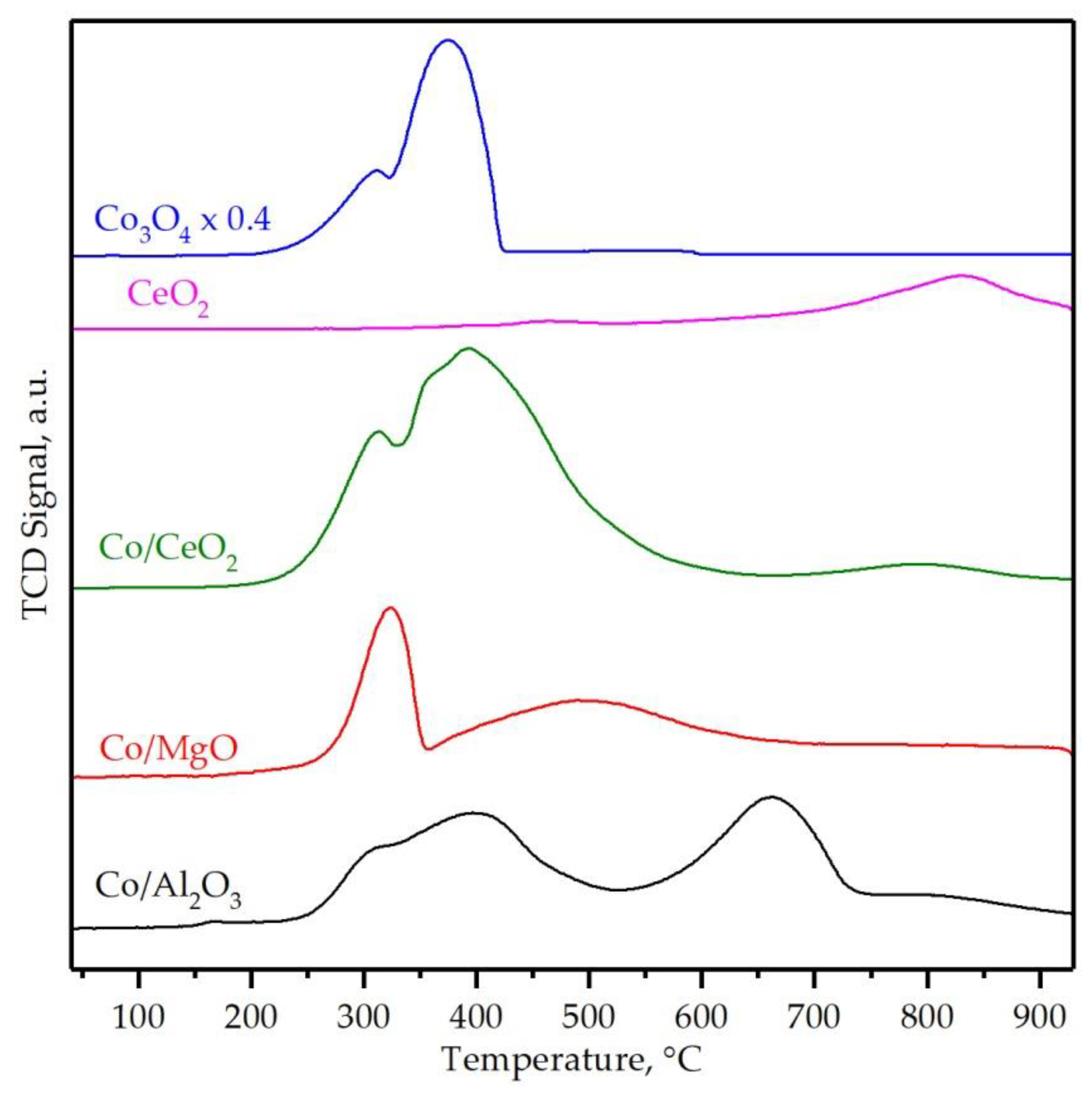
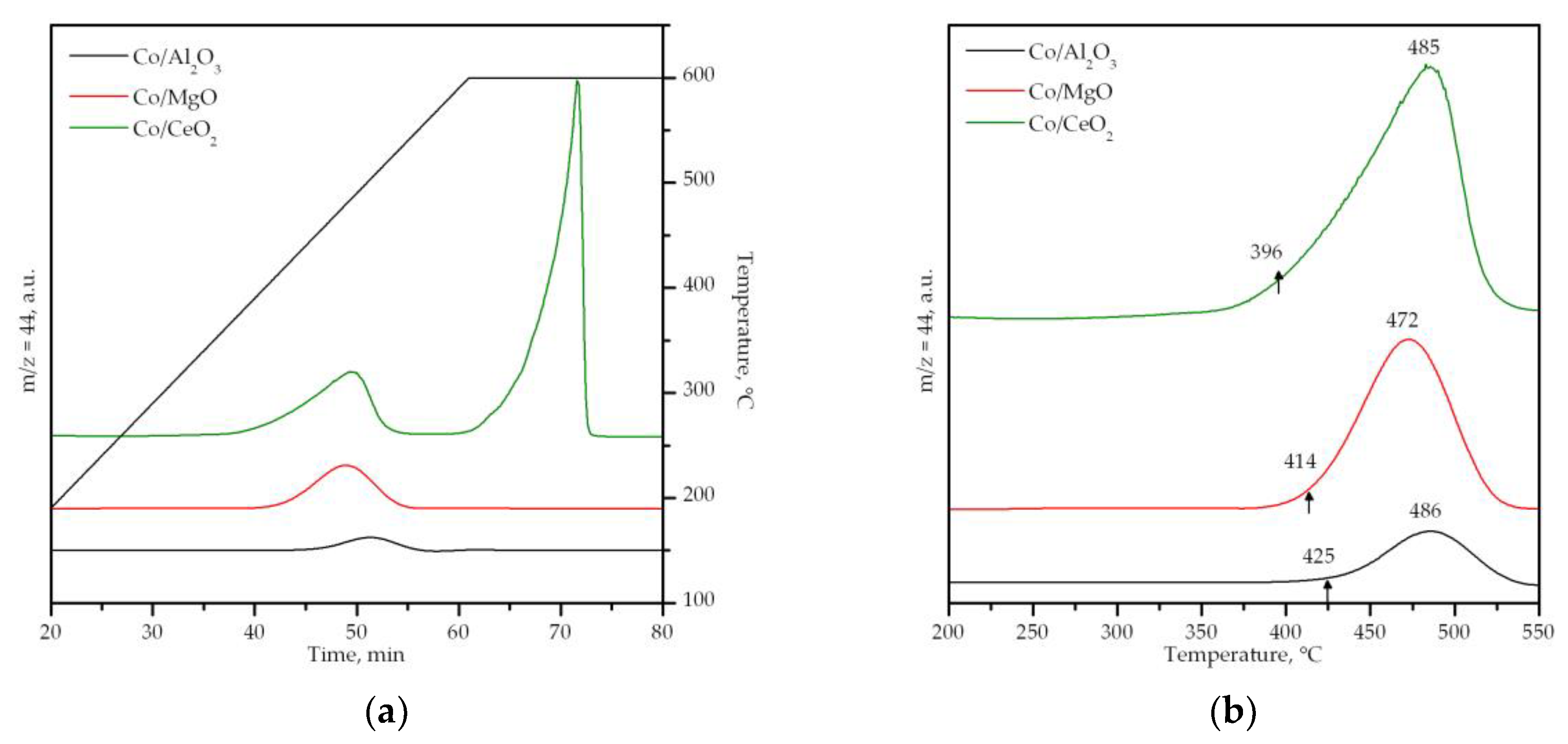
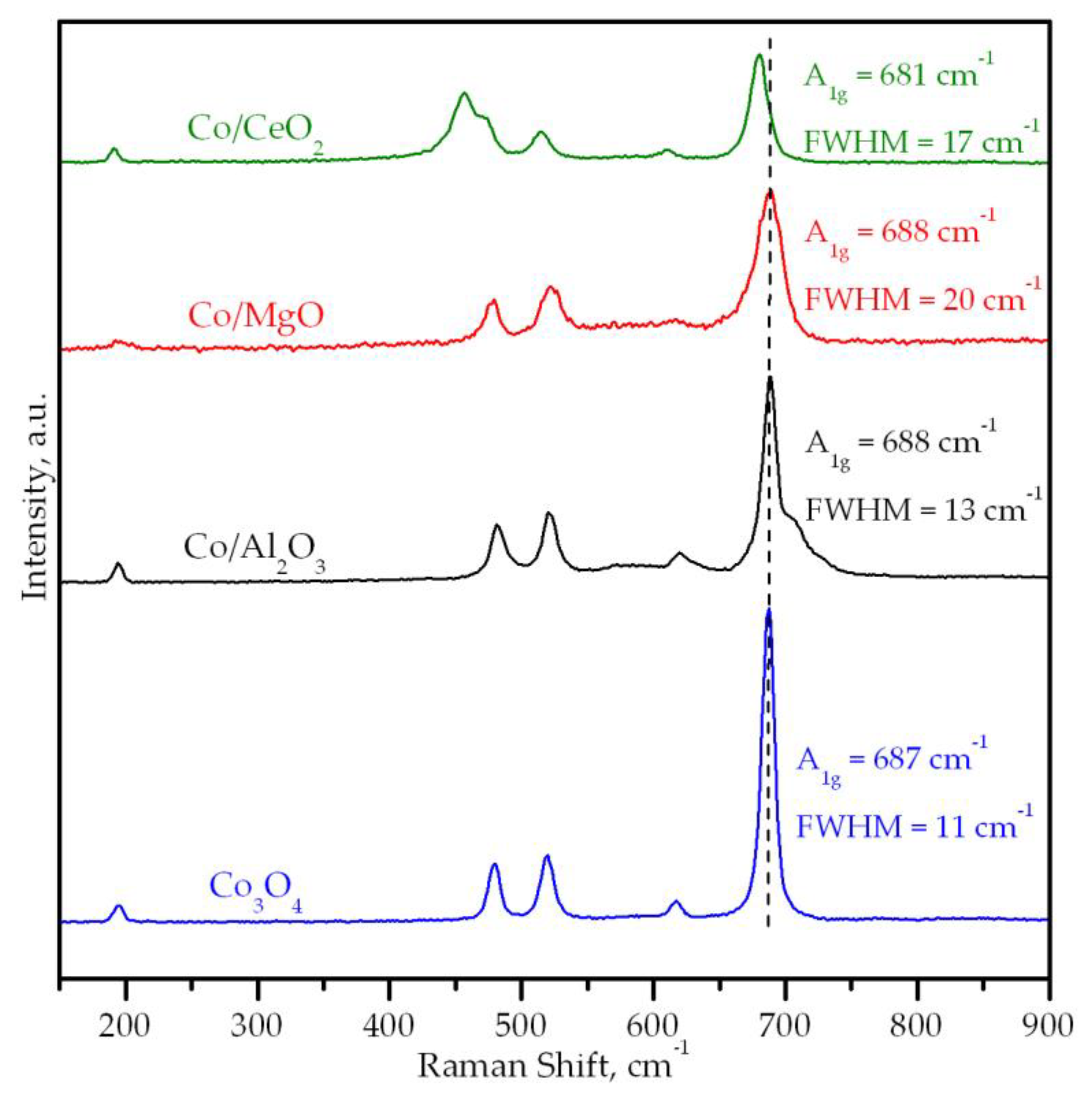
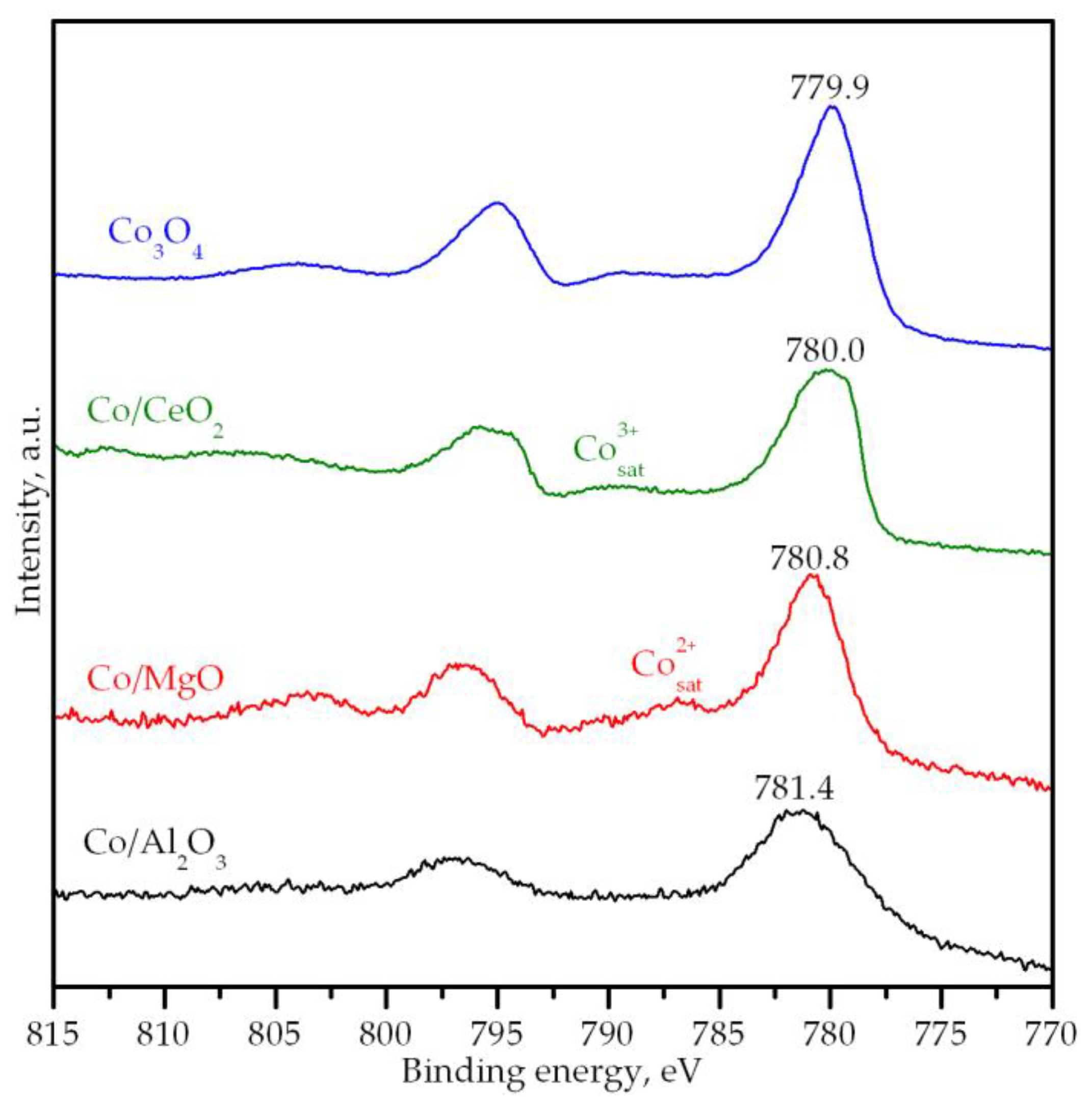
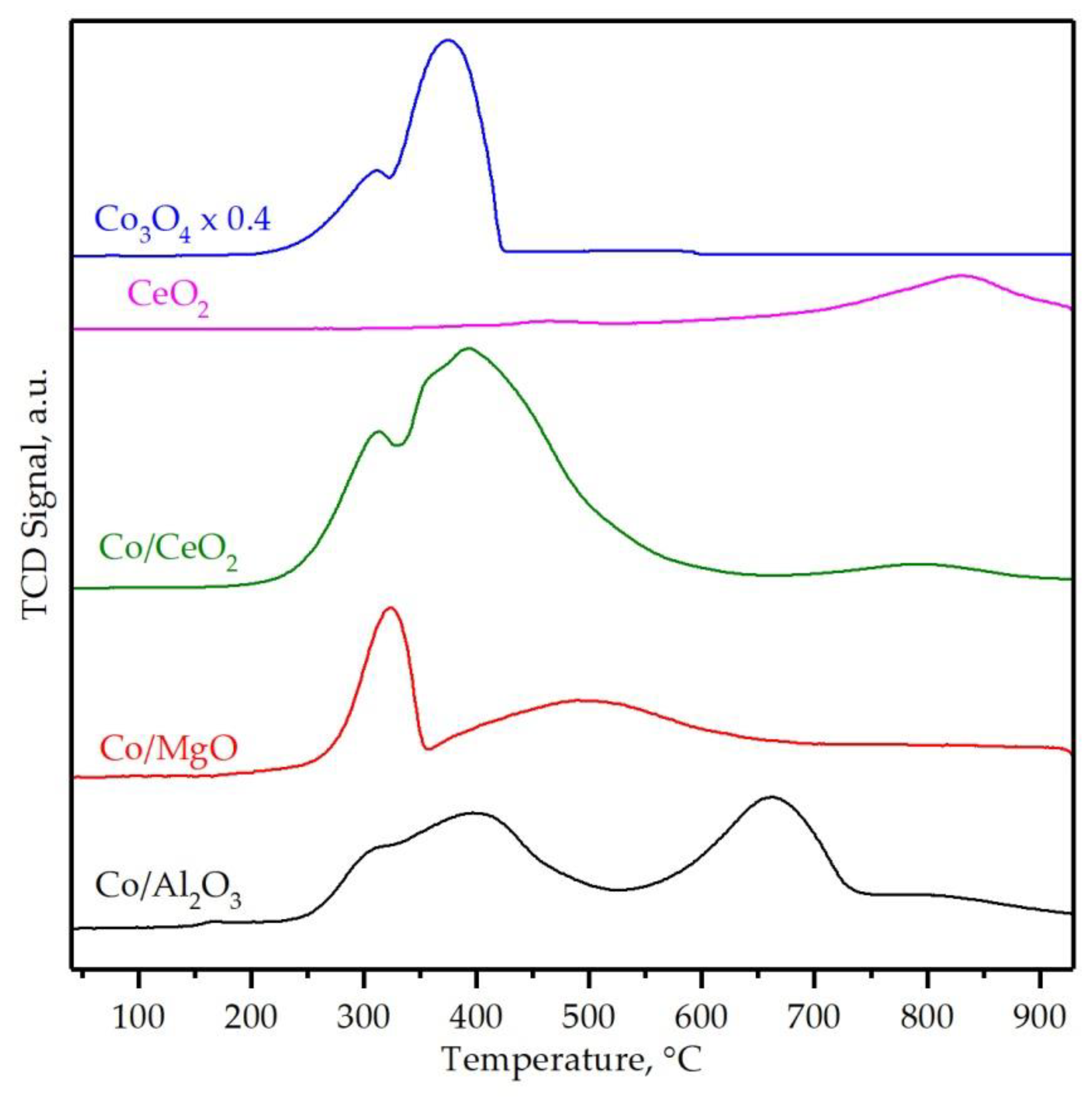
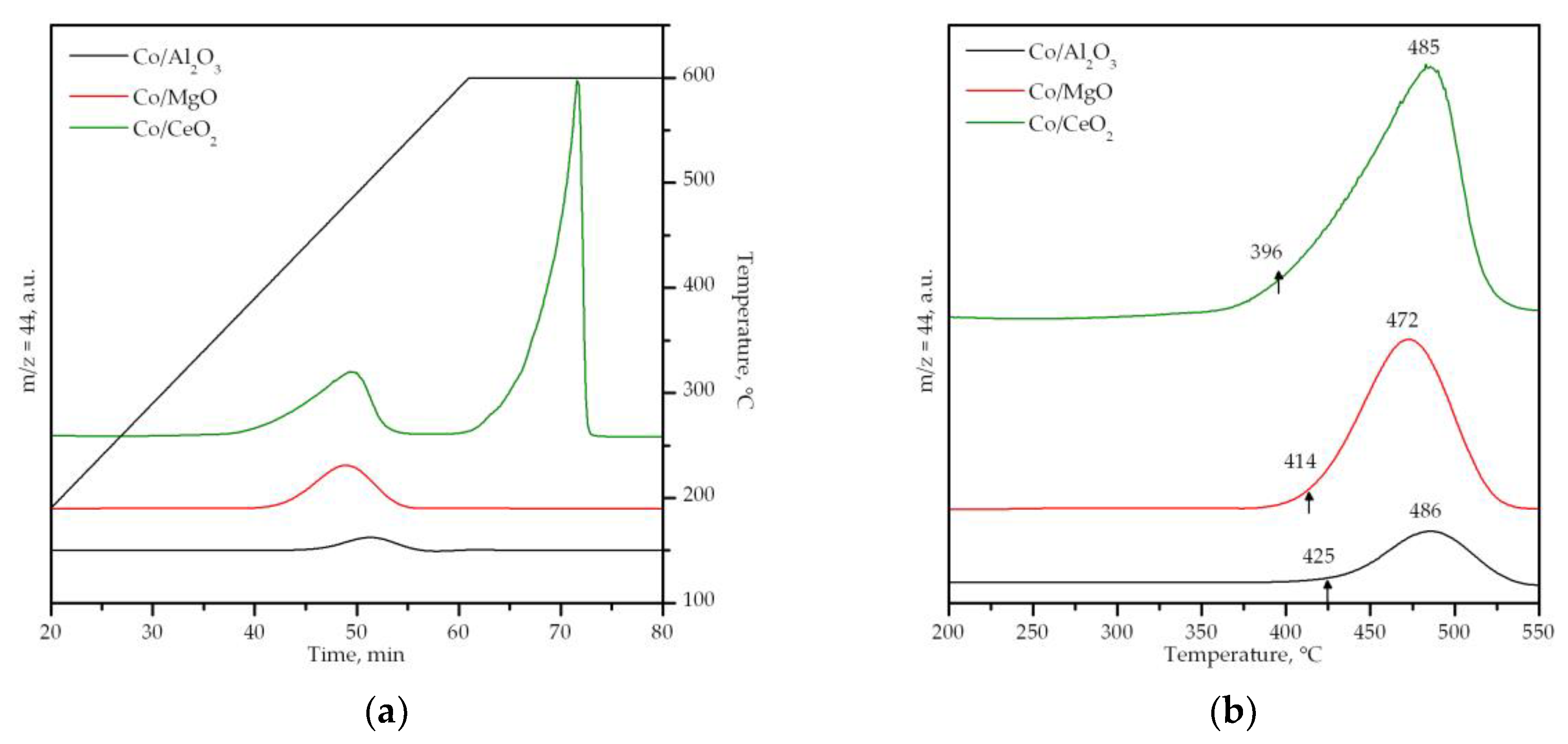
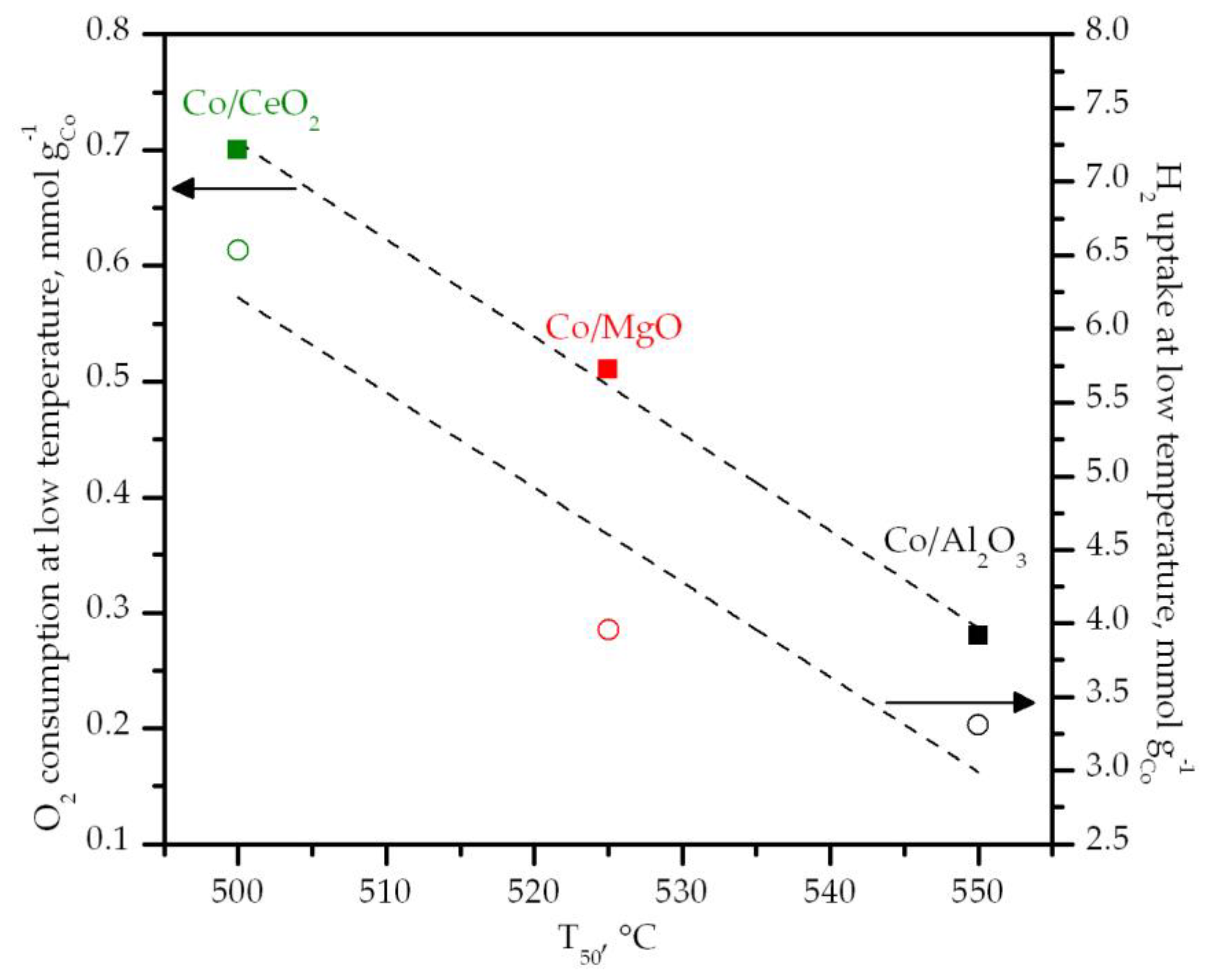
3.2. Characterization of the Supported Cobalt Catalysts
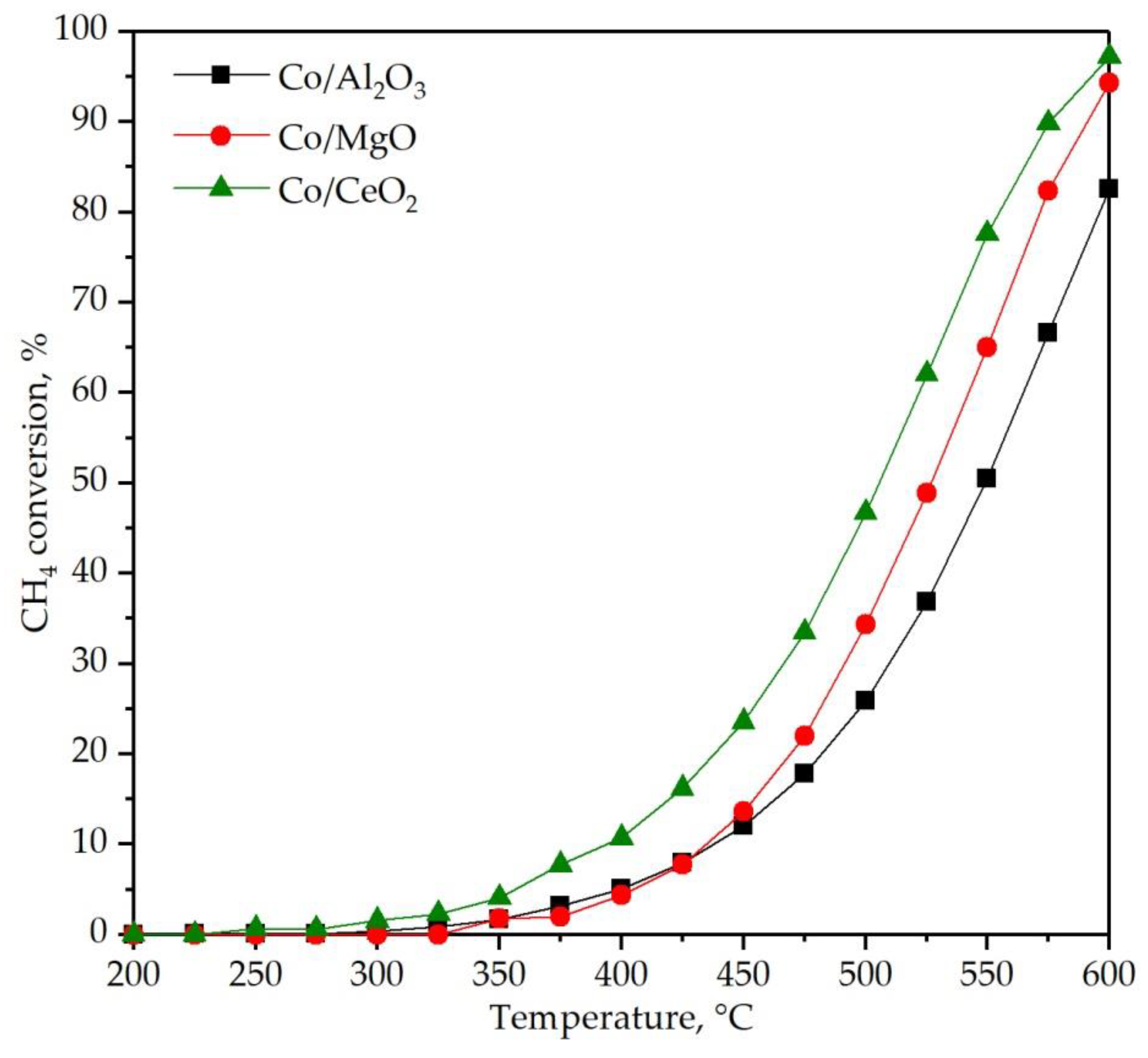
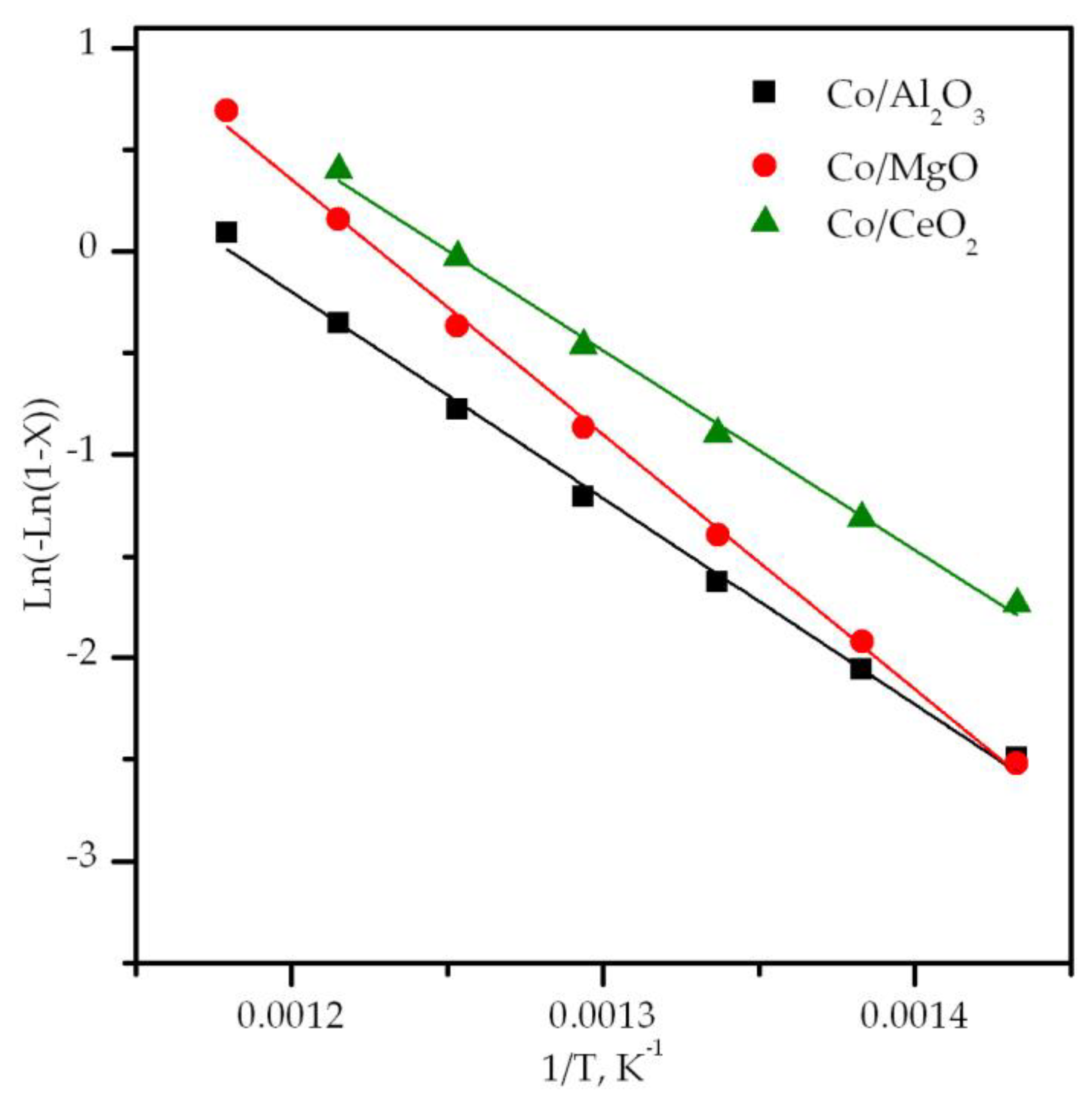
3.3. Performance of the Supported Cobalt Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yuan, Z.; Ou, X.; Peng, T.; Yan, X. Life cycle greenhouse gas emissions of multi-pathways natural gas vehicles in china considering methane leakage. Appl. Energy 2019, 253, 113472. [Google Scholar] [CrossRef]
- Vinoth Kanna, I.; Arulprakasajothi, M.; Eliyas, S. A detailed study of IC engines and a novel discussion with comprehensive view of alternative fuels used in petrol and diesel engines. Int. J. Ambient Energy 2019, 1–9. [Google Scholar] [CrossRef]
- Rink, M.; Eigenberger, G.; Nieken, U. Comparison of two different heat-integrated exhaust purification devices for monovalent CNG engines. Top. Catal. 2013, 56, 421–426. [Google Scholar] [CrossRef]
- Kim, J.; Kim, E.; Han, J.; Han, H.S. Pt/Pd bimetallic catalyst with improved activity and durability for lean-burn CNG engines. SAE Int. J. Fuels Lubr. 2013, 6, 651–656. [Google Scholar] [CrossRef]
- Cai, T.; Huang, H.; Deng, W.; Dai, Q.; Liu, W.; Wang, X. Catalytic combustion of 1,2-dichlorobenzene at low temperature over Mn-modified Co3O4 catalysts. Appl. Catal. B Environ. 2015, 166, 393–405. [Google Scholar] [CrossRef]
- Bai, B.; Arandiyan, H.; Li, J. Comparison of the performance for oxidation of formaldehyde on nano-Co3O4, 2D-Co3O4, and 3D-Co3O4 catalysts. Appl. Catal. B Environ. 2013, 142, 677–683. [Google Scholar] [CrossRef]
- Tian, Z.; Tchoua Ngamou, P.H.; Vannier, V.; Kohse-Höinghaus, K.; Bahlawane, N. Catalytic oxidation of VOCs over mixed Co-Mn oxides. Appl. Catal. B Environ. 2012, 117, 125–134. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, S.; Liu, W.; Gao, X.; Gao, D.; Wang, M.; Wang, S. Morphology-dependent performance of Co3O4 via facile and controllable synthesis for methane combustion. Appl. Catal. A Gen. 2016, 525, 94–102. [Google Scholar] [CrossRef]
- Setiawan, A.; Kennedy, E.M.; Dlugogorski, B.Z.; Adesina, A.A.; Stockenhuber, M. The stability of Co3O4, Fe2O3, Au/Co3O4 and Au/Fe2O3 catalysts in the catalytic combustion of lean methane mixtures in the presence of water. Catal. Today 2015, 258, 276–283. [Google Scholar] [CrossRef]
- Ercolino, G.; Stelmachowski, P.; Kotarba, A.; Specchia, S. Reactivity of mixed iron–cobalt spinels in the lean methane combustion. Top. Catal. 2017, 60, 1370–1379. [Google Scholar] [CrossRef]
- Chen, J.; Arandiyan, H.; Gao, X.; Li, J. Recent advances in catalysts for methane combustion. Catal. Surv. Asia 2015, 19, 140–171. [Google Scholar] [CrossRef]
- Wójcik, S.; Grzybek, G.; Gryboś, J.; Kotarba, A.; Sojka, Z. Designing, optimization and performance evaluation of the K-Zn0.4Co2.6O4|α-Al2O3|cordierite catalyst for low-temperature N2O decomposition. Catal. Commun. 2018, 110, 64–67. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, Y.; Fu, J.; Kyzas, G.Z.; Billah, S.M.R.; An, S. Synthesis, characterization, and catalytic evaluation of Co3O4/γ-Al2O3 as methane combustion catalysts: Significance of Co species and the redox cycle. Appl. Catal. B Environ. 2015, 168, 42–50. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X. MgO modifying Al2O3 to load cobalt oxide for catalytic N2O decomposition. Catal. Lett. 2019, 149, 1856–1863. [Google Scholar] [CrossRef]
- Zacharaki, I.; Kontoyannis, C.G.; Boghosian, S.; Lycourghiotis, A.; Kordulis, C. Cobalt oxide supported on alumina catalysts prepared by various methods for use in catalytic afterburner of PEM fuel cell. Catal. Today 2009, 143, 38–44. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Iwasaki, Y. Fabrication of Macroporous Co3O4–MgO composite catalysts for methylene blue degradation using ozone as an oxidant. J. Chem. Eng. Jpn. 2017, 50, 821–826. [Google Scholar] [CrossRef]
- Ulla, M.A.; Spretz, R.; Lombardo, E.; Daniell, W.; Knözinger, H. Catalytic combustion of methane on Co/MgO: Characterisation of active cobalt sites. Appl. Catal. B Environ. 2001, 29, 217–229. [Google Scholar] [CrossRef]
- Ercolino, G.; Stelmachowski, P.; Specchia, S. Catalytic performance of Pd/Co3O4 on SiC and ZrO2 open cell foams for process intensification of methane combustion in lean conditions. Ind. Eng. Chem. Res. 2017, 56, 6625–6636. [Google Scholar] [CrossRef]
- Bai, L.; Wyrwalski, F.; Safariamin, M.; Bleta, R.; Lamonier, J.; Przybylski, C.; Monflier, E.; Ponchel, A. Cyclodextrin-cobalt (II) molecule-ion pairs as precursors to active Co3O4/ZrO2 catalysts for the complete oxidation of formaldehyde: Influence of the cobalt source. J. Catal. 2016, 341, 191–204. [Google Scholar] [CrossRef]
- Pudukudy, M.; Yaakob, Z. Methane decomposition over Ni, Co and Fe based monometallic catalysts supported on sol gel derived SiO2 microflakes. Chem. Eng. J. 2015, 262, 1009–1021. [Google Scholar] [CrossRef]
- Jozwiak, W.K.; Szubiakiewicz, E.; Góralski, J.; Klonkowski, A.; Paryjczak, T. Physico-chemical and catalytic study of the Co/SiO2 catalysts. Kinet. Catal. 2004, 45, 247–255. [Google Scholar] [CrossRef]
- Grzybek, G.; Stelmachowski, P.; Gudyka, S.; Indyka, P.; Sojka, Z.; Guillén-Hurtado, N.; Rico-Pérez, V.; Bueno-López, A.; Kotarba, A. Strong dispersion effect of cobalt spinel active phase spread over ceria for catalytic N2O decomposition: The role of the interface periphery. Appl. Catal. B Environ. 2016, 180, 622–629. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Deganello, G. Catalytic performance of Co3O4/CeO2 and Co3O4/CeO2–ZrO2 composite oxides for methane combustion: Influence of catalyst pretreatment temperature and oxygen concentration in the reaction mixture. Appl. Catal. B Environ. 2007, 70, 314–322. [Google Scholar] [CrossRef]
- Klegova, A.; Inayat, A.; Indyka, P.; Gryboś, J.; Sojka, Z.; Pacultová, K.; Schwieger, W.; Volodarskaja, A.; Kuśtrowski, P.; Rokicińska, A.; et al. Cobalt mixed oxides deposited on the SiC open-cell foams for nitrous oxide decomposition. Appl. Catal. B Environ. 2019, 255, 117745. [Google Scholar] [CrossRef]
- Song, H.; Zhao, Q.; Zhou, X.; Cao, Z.; Luo, M. Selection of highly active and stable Co supported SiC catalyst for Fischer-Tropsch synthesis: Effect of the preparation method. Fuel 2018, 229, 144–150. [Google Scholar] [CrossRef]
- Zhu, Z.; Lu, G.; Zhang, Z.; Guo, Y.; Guo, Y.; Wang, Y. Highly active and stable Co3O4/ZSM-5 catalyst for propane oxidation: Effect of the preparation method. ACS Catal. 2013, 3, 1154–1164. [Google Scholar] [CrossRef]
- Laugel, G.; Arichi, J.; Bernhardt, P.; Molière, M.; Kiennemann, A.; Garin, F.; Louis, B. Preparation and characterisation of metal oxides supported on SBA-15 as methane combustion catalysts. C. R. Chim. 2009, 12, 731–739. [Google Scholar] [CrossRef]
- Jirátová, K.; Balabánová, J.; Kovanda, F.; Klegová, A.; Obalová, L.; Fajgar, R. Cobalt oxides supported over ceria–zirconia coated cordierite monoliths as catalysts for deep oxidation of ethanol and N2O decomposition. Catal. Lett. 2017, 147, 1379–1391. [Google Scholar] [CrossRef]
- Bahlawane, N. Kinetics of methane combustion over CVD-made cobalt oxide catalysts. Appl. Catal. B Environ. 2006, 67, 168–176. [Google Scholar] [CrossRef]
- Grzybek, G.; Ciura, K.; Wójcik, S.; Gryboś, J.; Indyka, P.; Inger, M.; Antoniak-Jurak, K.; Kowalik, P.; Kotarba, A.; Sojka, Z. On the selection of the best polymorph of Al2O3 carriers for supported cobalt nano-spinel catalysts for N2O abatement: An interplay between preferable surface spreading and damaging active phase-support interaction. Catal. Sci. Technol. 2017, 7, 5723–5732. [Google Scholar] [CrossRef]
- Solsona, B.; Davies, T.E.; Garcia, T.; Vázquez, I.; Dejoz, A.; Taylor, S.H. Total oxidation of propane using nanocrystalline cobalt oxide and supported cobalt oxide catalysts. Appl. Catal. B Environ. 2008, 84, 176–184. [Google Scholar] [CrossRef]
- Yung, M.M.; Holmgreen, E.M.; Ozkan, U.S. Cobalt-based catalysts supported on titania and zirconia for the oxidation of nitric oxide to nitrogen dioxide. J. Catal. 2007, 247, 356–367. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, Y.H.; Yie, J.E.; Park, E.D. NO oxidation over supported cobalt oxide catalysts. Korean J. Chem. Eng. 2010, 27, 49–54. [Google Scholar] [CrossRef]
- Wyrwalski, F.; Giraudon, J.-M.; Lamonier, J.-F. Synergistic coupling of the redox properties of supports and cobalt oxide Co3O4 for the complete oxidation of volatile organic compounds. Catal. Lett. 2010, 137, 141–149. [Google Scholar] [CrossRef]
- Zhang, W.; Tay, H.L.; Lim, S.S.; Wang, Y.; Zhong, Z.; Xu, R. Supported cobalt oxide on MgO: Highly efficient catalysts for degradation of organic dyes in dilute solutions. Appl. Catal. B Environ. 2010, 95, 93–99. [Google Scholar] [CrossRef]
- Lukashuk, L.; Yigit, N.; Rameshan, R.; Kolar, E.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; Föttinger, K.; Rupprechter, G. Operando insights into CO oxidation on cobalt oxide catalysts by NAP-XPS, FTIR, and XRD. ACS Catal. 2018, 8, 8630–8641. [Google Scholar] [CrossRef] [PubMed]
- Zasada, F.; Janas, J.; Piskorz, W.; Gorczynska, M.; Sojka, Z. Total oxidation of lean methane over cobalt spinel nanocubes controlled by the self-adjusted redox state of the catalyst: Experimental and theoretical account for interplay between the Langmuir-Hinshelwood and Mars-Van Krevelen mechanisms. ACS Catal. 2017, 7, 2853–2867. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Oxidation of residual methane from VNG vehicles over Co3O4-based catalysts: Comparison among bulk, Al2O3-supported and Ce-doped catalysts. Appl. Catal. B Environ. 2018, 237, 844–854. [Google Scholar] [CrossRef]
- Dou, J.; Tang, Y.; Nie, L.; Andolina, C.M.; Zhang, X.; House, S.; Li, Y.; Yang, J.; Tao, F. Complete oxidation of methane on Co3O4/CeO2 nanocomposite: a synergic effect. Catal. Today 2018, 311, 48–55. [Google Scholar] [CrossRef]
- Kumar, M.; Rattan, G.; Prasad, R. Optimisation of cobalt loading on γ-Al2O3 for total oxidation of methane. Indian Chem. Eng. 2017, 59, 161–176. [Google Scholar] [CrossRef]
- Jodłowski, P.J.; Kryca, J.; Iwaniszyn, M.; Jȩdrzejczyk, R.; Thomas, J.; Kołodziej, A.; Łojewska, J. Methane combustion modelling of wire gauze reactor coated with Co3O4–CeO2, Co3O4–PdO catalysts. Catal. Today 2013, 216, 276–282. [Google Scholar] [CrossRef]
- EUROKIN Spreadsheet on Requirements for Measurement of Intrinsic Kinetics in The Gas-Solid Fixed-Bed Reactor. Available online: http://eurokin.org/ (accessed on 24 July 2019).
- Aranzabal, A.; González-Marcos, J.A.; Ayastuy, J.L.; González-Velasco, J.R. Kinetics of Pd/alumina catalysed 1,2-dichloroethane gas-phase oxidation. Chem. Eng. Sci. 2006, 61, 3564–3576. [Google Scholar] [CrossRef]
- Li, D.; Ding, Y.; Wei, X.; Xiao, Y.; Jiang, L. Cobalt-aluminum mixed oxides prepared from layered double hydroxides for the total oxidation of benzene. Appl. Catal. A Gen. 2015, 507, 130–138. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, L.; Yu, W.; Liu, L.; Deng, Y.; Liu, B.; Wan, H.; Gao, F.; Sun, K.; Dong, L. Effect of cobalt precursors on the dispersion, reduction, and CO oxidation of CoOx/γ-Al2O3 catalysts calcined in N2. J. Colloid Interface Sci. 2011, 355, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, L.C.; Chen, M.; Cao, Y.; He, H.Y.; Fan, K.N. Dry citrate-precursor synthesized nanocrystalline cobalt oxide as highly active catalyst for total oxidation of propane. J. Catal. 2009, 263, 104–113. [Google Scholar] [CrossRef]
- Jiang, X.; Ma, Y.; Chen, Y.; Li, Y.; Ma, Q.; Zhang, Z.; Wang, C.; Yang, Y. Raman analysis of cobalt blue pigment in blue and white porcelain: A reassessment. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2018, 190, 61–67. [Google Scholar] [CrossRef] [PubMed]
- D’Ippolito, V.; Andreozzi, G.B.; Bersani, D.; Lottici, P.P. Raman fingerprint of chromate, aluminate and ferrite spinels. J. Raman Spectrosc. 2015, 46, 1255–1264. [Google Scholar] [CrossRef]
- Wu, M.; Fu, Y.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, Y.; Lu, G. Catalytic performance of MgO-supported Co catalyst for the liquid phase oxidation of cyclohexane with molecular oxygen. Catalysts 2017, 7, 155. [Google Scholar] [CrossRef]
- Cazzanelli, E.; Kuzmin, A.; Mariotto, G.; Mironova-Ulmane, N. Study of vibrational and magnetic excitations in NicMg1-cO solid solutions by Raman spectroscopy. J. Phys. Condens. Matter 2003, 15, 2045–2052. [Google Scholar] [CrossRef]
- Zou, G.; Xu, Y.; Wang, S.; Chen, M.; Shangguan, W. The synergistic effect in Co-Ce oxides for catalytic oxidation of diesel soot. Catal. Sci. Technol. 2015, 5, 1084–1092. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Yang, J.; Liu, H.; Martens, W.N.; Frost, R.L. Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs. J. Phys. Chem. C 2010, 114, 111–119. [Google Scholar] [CrossRef]
- Duan, X.; Pan, M.; Yu, F.; Yuan, D. Synthesis, structure and optical properties of CoAl2O4 spinel nanocrystals. J. Alloy. Compd. 2011, 509, 1079–1083. [Google Scholar] [CrossRef]
- Deng, J.; Zhang, L.; Dai, H.; Xia, Y.; Jiang, H.; Zhang, H.; He, H. Ultrasound-assisted nanocasting fabrication of ordered mesoporous MnO2 and Co3O4 with high surface areas and polycrystalline walls. J. Phys. Chem. C 2010, 114, 2694–2700. [Google Scholar] [CrossRef]
- Liotta, L.F.; Wu, H.; Pantaleo, G.; Venezia, A.M. Co3O4 nanocrystals and Co3O4–MOx binary oxides for CO, CH4 and VOC oxidation at low temperatures: A review. Catal. Sci. Technol. 2013, 3, 3085–3102. [Google Scholar] [CrossRef]
- Ji, S.F.; Xiao, T.C.; Wang, H.T.; Flahaut, E.; Coleman, K.S.; Green, M.L.H. Catalytic combustion of methane over cobalt-magnesium oxide solid solution catalysts. Catal. Lett. 2001, 75, 65–71. [Google Scholar] [CrossRef]
- Florea, M.; Matei-Rutkovska, F.; Postole, G.; Urda, A.; Neatu, F.; Pârvulescu, V.I.; Gelin, P. Doped ceria prepared by precipitation route for steam reforming of methane. Catal. Today 2018, 306, 166–171. [Google Scholar] [CrossRef]
- Rotaru, C.G.; Postole, G.; Florea, M.; Matei-Rutkovska, F.; Pârvulescu, V.I.; Gelin, P. Dry reforming of methane on ceria prepared by modified precipitation route. Appl. Catal. A Gen. 2015, 494, 29–40. [Google Scholar] [CrossRef]
- Zeng, S.; Fu, X.; Zhou, T.; Wang, X.; Su, H. Influence of pore distribution on catalytic performance over inverse CeO2/Co3O4 catalysts for CH4/CO2 reforming. Fuel Process. Technol. 2013, 114, 69–74. [Google Scholar] [CrossRef]
- Budiman, A.W.; Song, S.H.; Chang, T.S.; Shin, C.H.; Choi, M.J. Dry reforming of methane over cobalt catalysts: A literature review of catalyst development. Catal. Surv. Asia 2012, 16, 183–197. [Google Scholar] [CrossRef]
- Gil-Calvo, M.; Jiménez-González, C.; De Rivas, B.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. Hydrogen production by reforming of methane over NiAl2O4/CexZr1-xO2 catalysts. Chem. Eng. Trans. 2017, 57, 901–906. [Google Scholar] [CrossRef]
- Zasada, F.; Grybos, J.; Budiyanto, E.; Janas, J.; Sojka, Z. Oxygen species stabilized on the cobalt spinel nano-octahedra at various reaction conditions and their role in catalytic CO and CH4 oxidation, N2O decomposition and oxygen isotopic exchange. J. Catal. 2019, 371, 224–235. [Google Scholar] [CrossRef]
- Fei, Z.; He, S.; Li, L.; Ji, W.; Au, C.T. Morphology-directed synthesis of Co3O4 nanotubes based on modified Kirkendall effect and its application in CH4 combustion. Chem. Commun. 2012, 48, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Stefanov, P.; Todorova, S.; Naydenov, A.; Tzaneva, B.; Kolev, H.; Atanasova, G.; Stoyanova, D.; Karakirova, Y.; Aleksieva, K. On the development of active and stable Pd-Co/γ-Al2O3 catalyst for complete oxidation of methane. Chem. Eng. J. 2015, 266, 329–338. [Google Scholar] [CrossRef]
- Xiao, T.C.; Ji, S.F.; Wang, H.T.; Coleman, K.S.; Green, M.L.H. Methane combustion over supported cobalt catalysts. J. Mol. Catal. A Chem. 2001, 175, 111–123. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Arandiyan, H.; Peng, Y.; Chang, H.; Li, J. Low temperature complete combustion of methane over cobalt chromium oxides catalysts. Catal. Today 2013, 201, 12–18. [Google Scholar] [CrossRef]
- Darda, S.; Pachatouridou, E.; Lappas, A.; Iliopoulou, E. Effect of preparation method of Co-Ce catalysts on CH4 combustion. Catalysts 2019, 9, 219. [Google Scholar] [CrossRef]
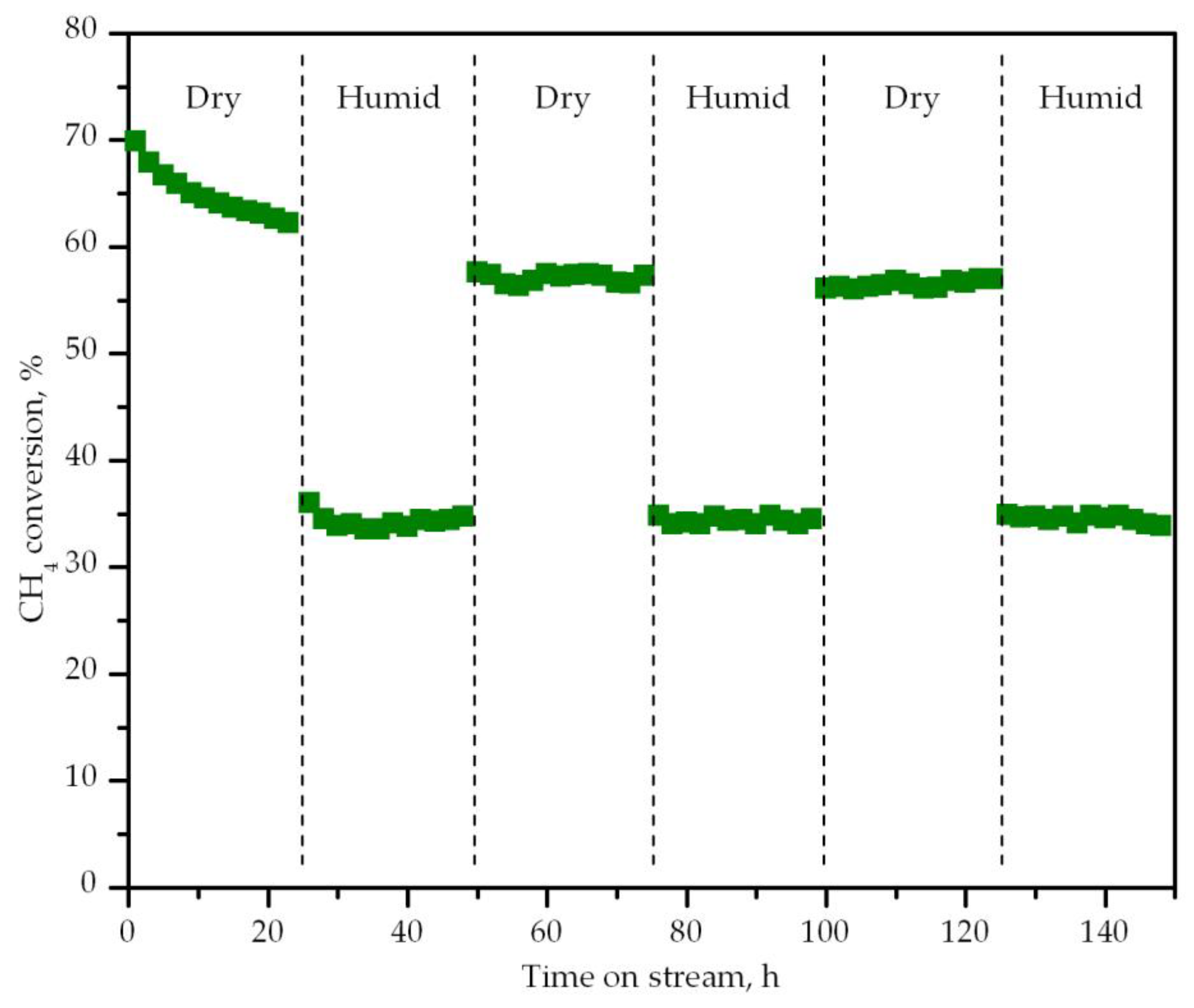
- Geng, H.; Yang, Z.; Zhang, L.; Ran, J.; Yan, Y. Methane oxidation with low O2/CH4 ratios in the present of water: Combustion or reforming. Energy Convers. Manag. 2017, 132, 339–346. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
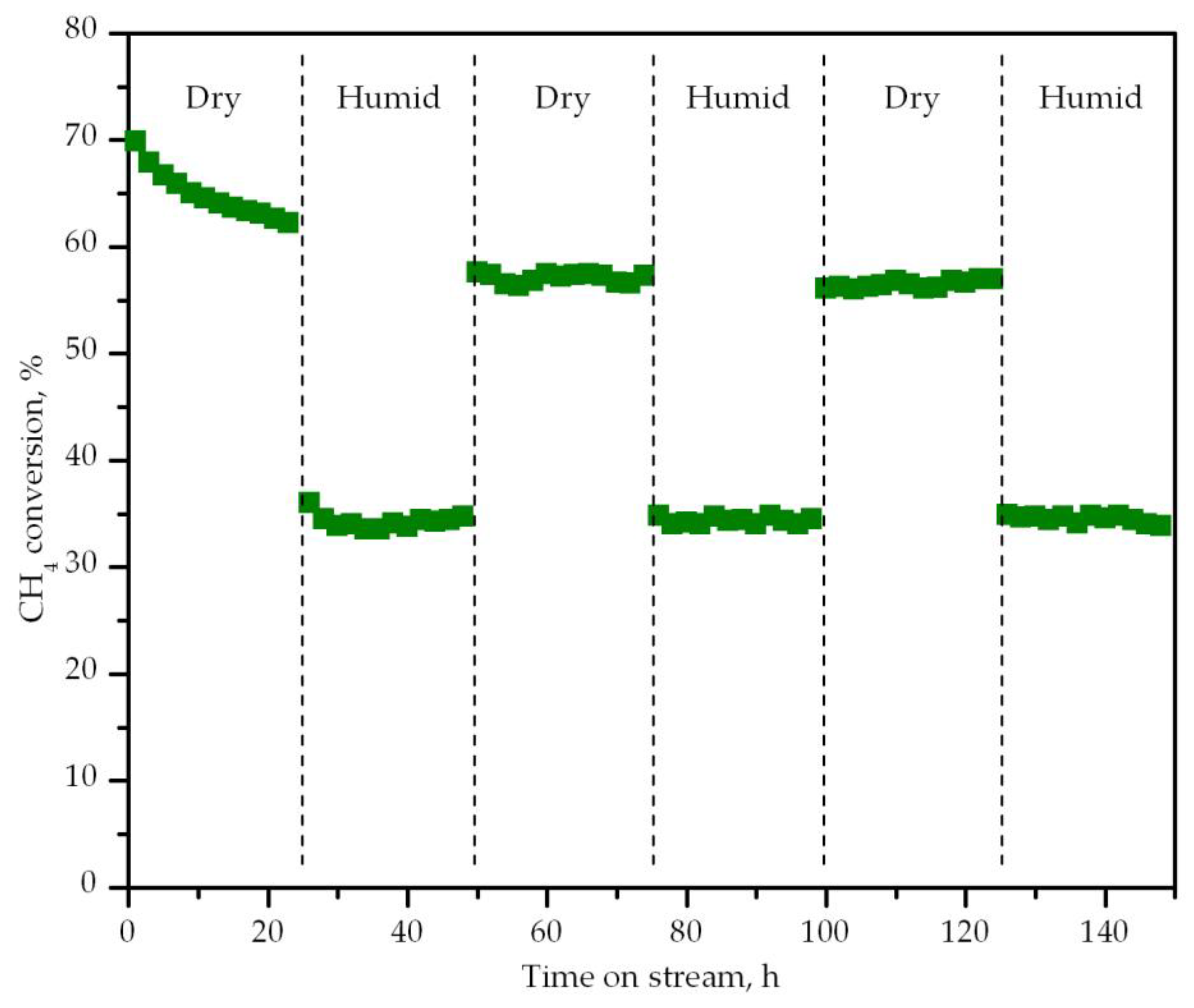{kind=link}
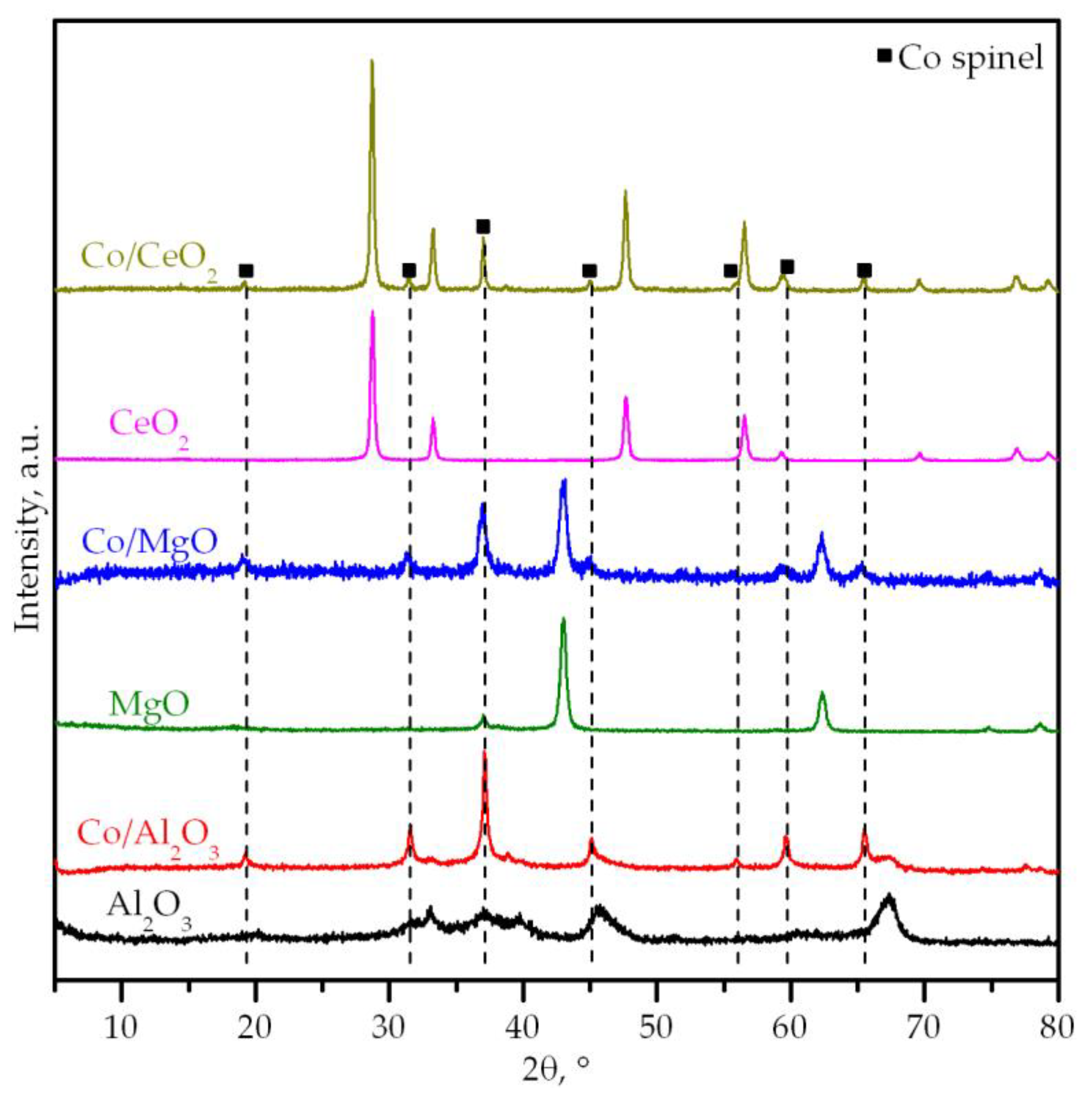
| Sample | Surface Area, m2 g−1 | Pore Volume, cm3 g−1 | Mean Pore Diameter, Å | Cobalt Content, wt.% | Support Crystallite Size, nm | Co3O4 Crystallite Size, nm | Co/M Molar Ratio1 |
|---|---|---|---|---|---|---|---|
| Al2O3 | 136 | 0.55 | 123 | - | 5 | - | - |
| MgO | 80 | 0.19 | 106 | - | 21 | - | - |
| CeO2 | 8 | 0.03 | 230 | - | 32 | - | - |
| Co/Al2O3 | 108 | 0.29 | 89 | 27.9 | 6 | 292 | 0.43 (0.39) |
| Co/MgO | 47 | 0.16 | 204 | 31.9 | 16 | 17 | 0.12 (0.38) |
| Co/CeO2 | 18 | 0.07 | 225 | 28.9 | 33 | 44 | 3.15 (1.39) |
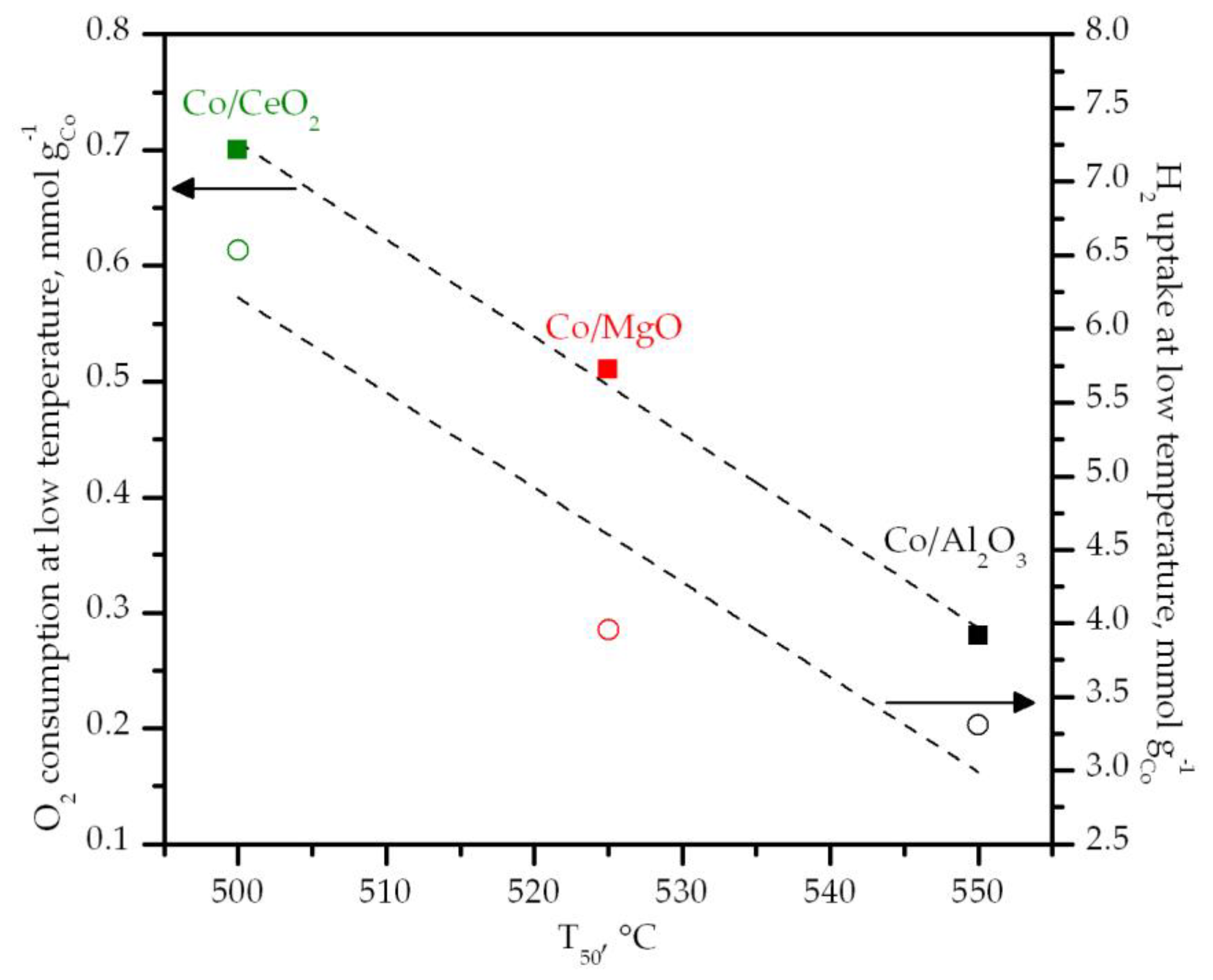
| Sample | Total H2 Uptake1 mmol gCo−1 | H2 Uptake at Low Temperature1 mmol gCo−1 | Degree of Co Reduction2 % | O2 Consumption at Low Temperature (CH4-TPR) mmol gCo−1 |
|---|---|---|---|---|
| Co/Al2O3 | 18.7 (5.6) | 9.8 (2.7) | 88 | 0.28 |
| Co/MgO | 14.5 (4.6) | 9.9 (3.2) | 64 | 0.51 |
| Co/CeO2 | 26.2 (7.6) | 23.4 (6.8) | 100 | 0.70 |
| CeO2 | - (1.5) | - | - | - |
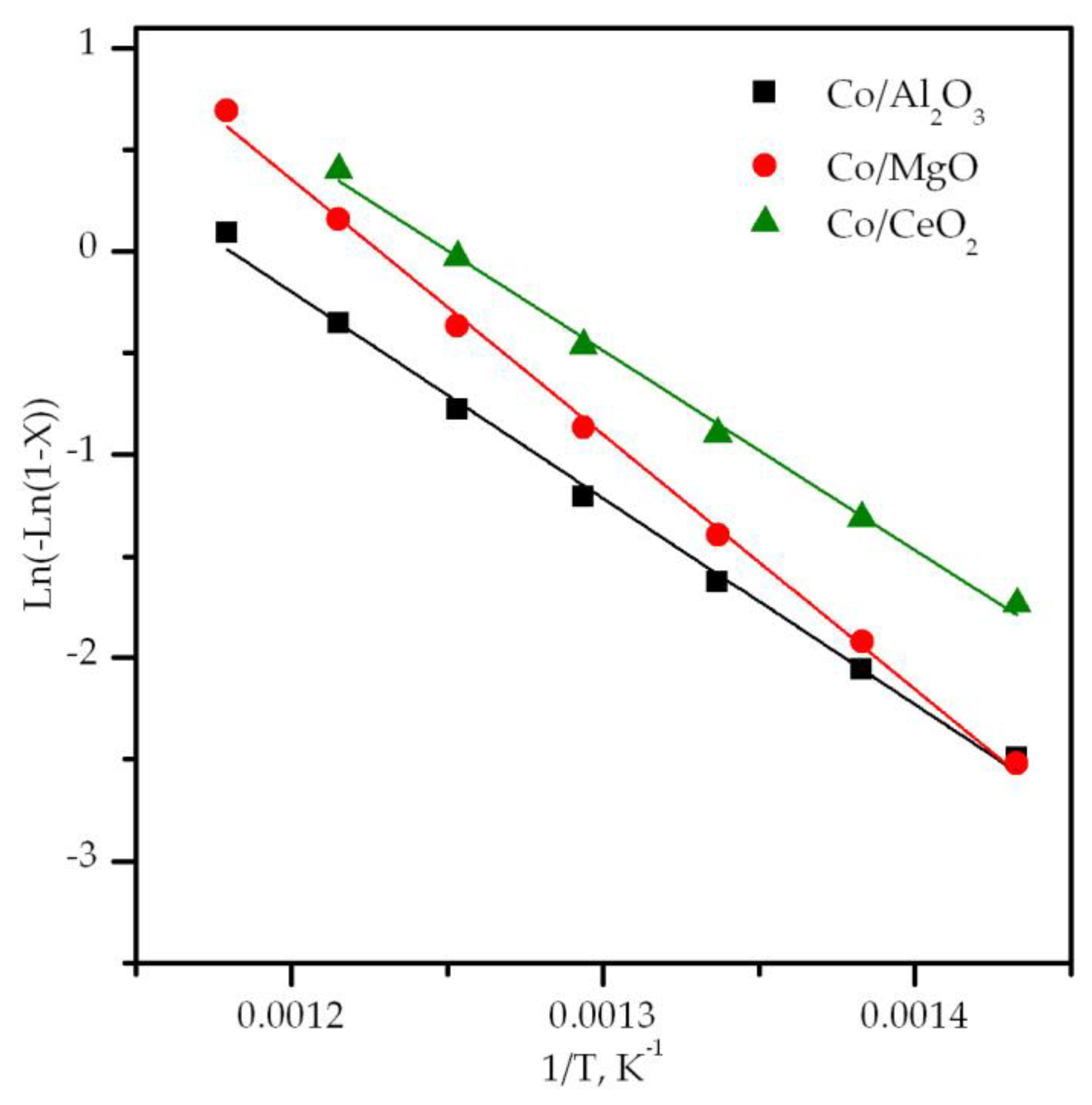
| Sample | T50 °C | Specific Rate at 425 °C mmol CH4 gCo−1 h−1 | Ea kJ mol−1 | ΔH‡ kJ mol−1 | ΔS‡ J mol−1 K−1 |
|---|---|---|---|---|---|
| Co/Al2O3 | 550 | 1.8 | 90 | 84 | −166 |
| Co/MgO | 525 | 1.4 | 102 | 98 | −139 |
| Co/CeO2 | 500 | 3.1 | 82 | 78 | −161 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choya, A.; de Rivas, B.; Gutiérrez-Ortiz, J.I.; González-Velasco, J.R.; López-Fonseca, R. Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures. Materials 2019, 12, 3174. https://doi.org/10.3390/ma12193174
Choya A, de Rivas B, Gutiérrez-Ortiz JI, González-Velasco JR, López-Fonseca R. Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures. Materials. 2019; 12(19):3174. https://doi.org/10.3390/ma12193174
Chicago/Turabian StyleChoya, Andoni, Beatriz de Rivas, Jose Ignacio Gutiérrez-Ortiz, Juan Ramón González-Velasco, and Rubén López-Fonseca. 2019. "Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures" Materials 12, no. 19: 3174. https://doi.org/10.3390/ma12193174
APA StyleChoya, A., de Rivas, B., Gutiérrez-Ortiz, J. I., González-Velasco, J. R., & López-Fonseca, R. (2019). Synthesis, Characterization and Kinetic Behavior of Supported Cobalt Catalysts for Oxidative after-Treatment of Methane Lean Mixtures. Materials, 12(19), 3174. https://doi.org/10.3390/ma12193174