Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring

 , , , , ,
, , , , ,  ,
,  ,
, 

Abstract
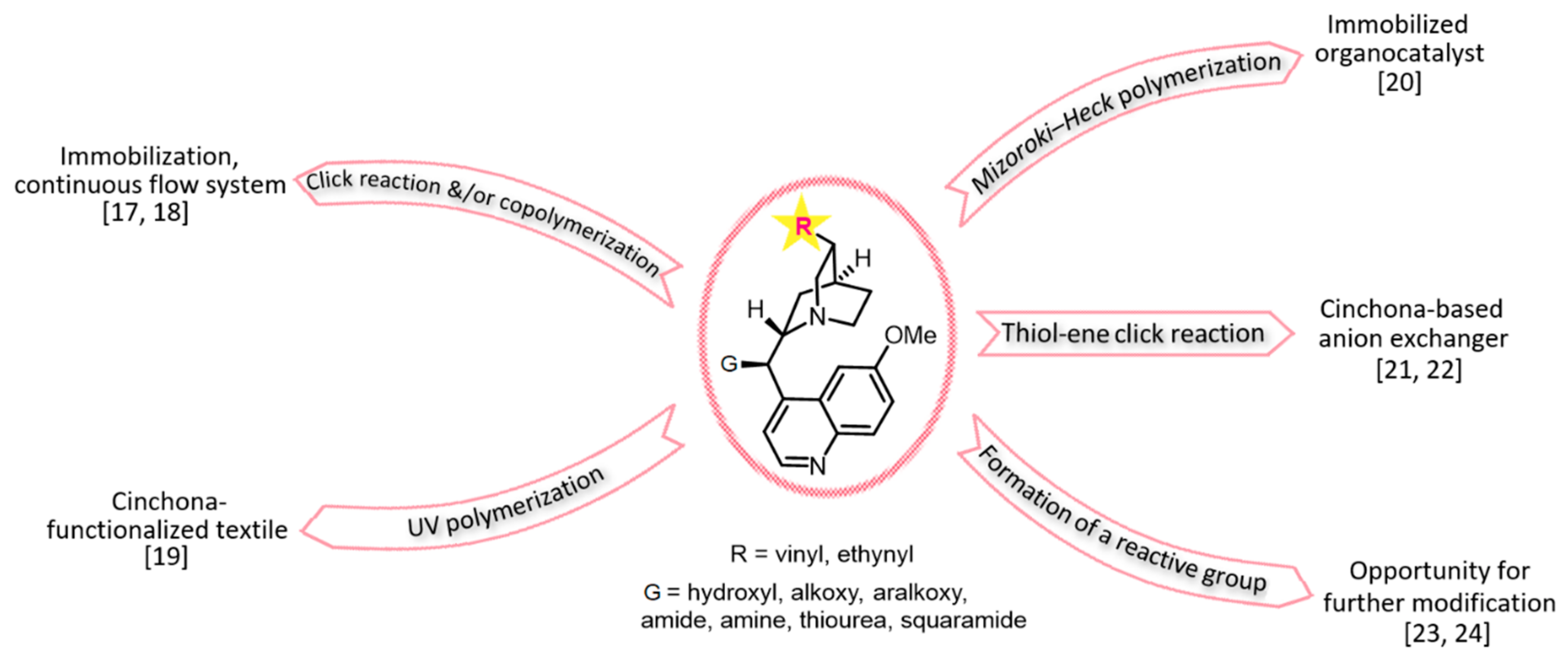
1. Introduction
2. Results and Discussion
2.1. The pKa Values of Conjugate Acid Forms of Quinuclidine
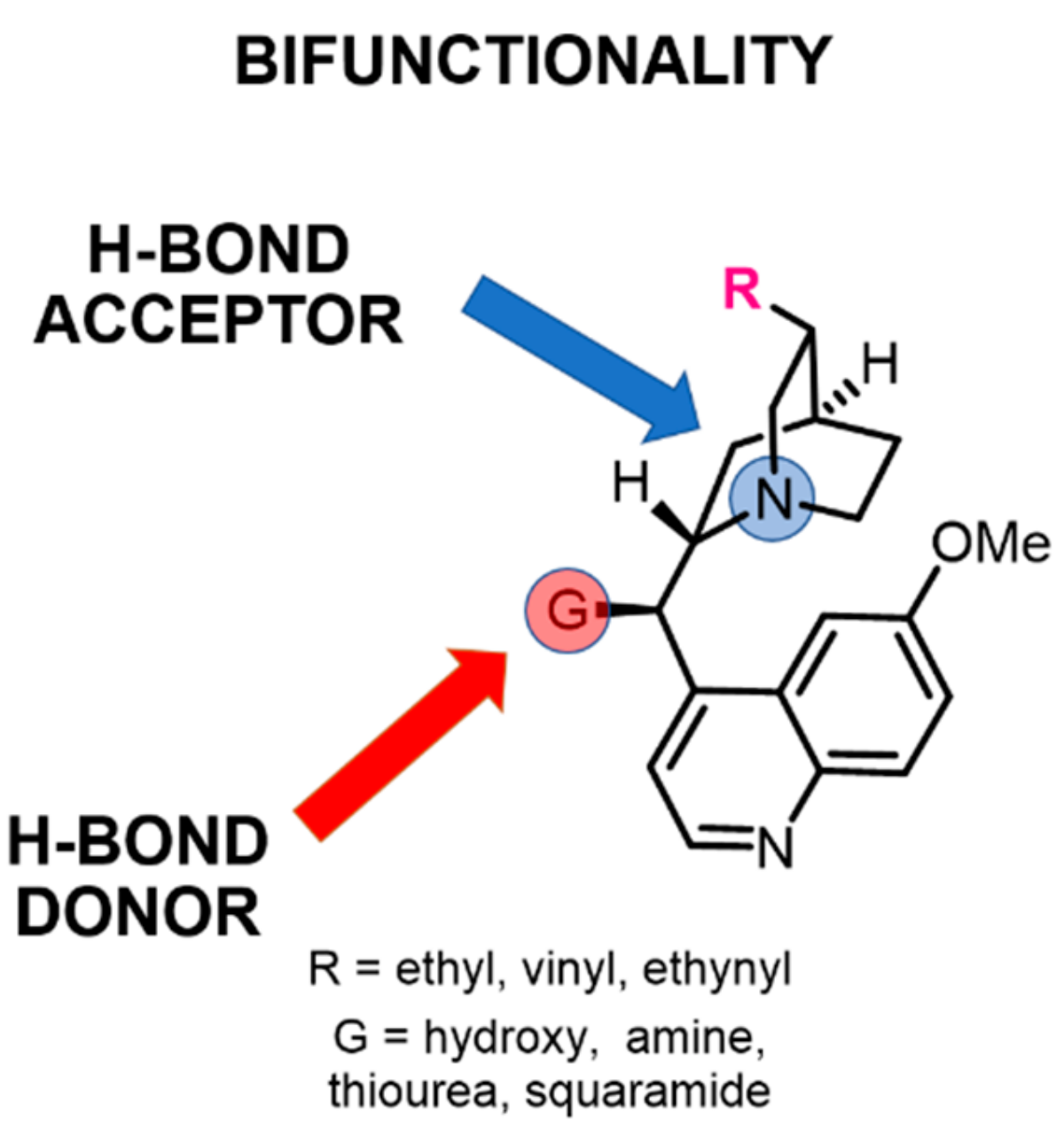
2.2. The Effect of the Substituents on Quinuclidine Basicity
2.3. The Effect of the C9 Group of Cinchona on Quinuclidine Basicity
2.4. Relationship Between pKa Values and H-Bonding Abilities
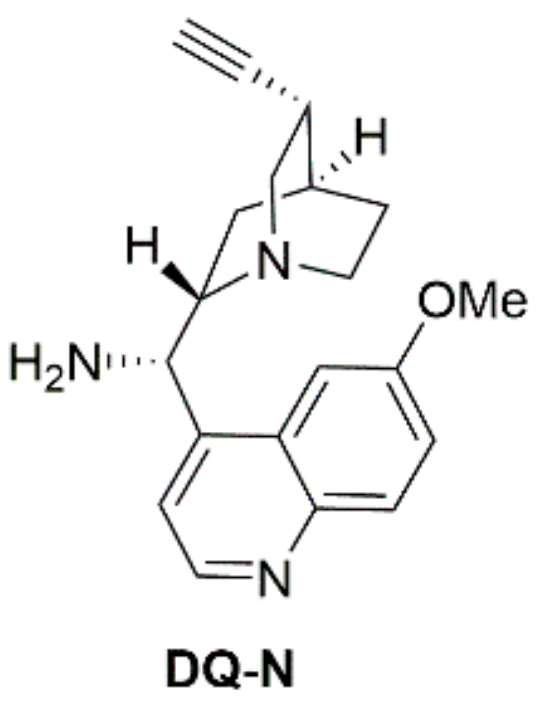
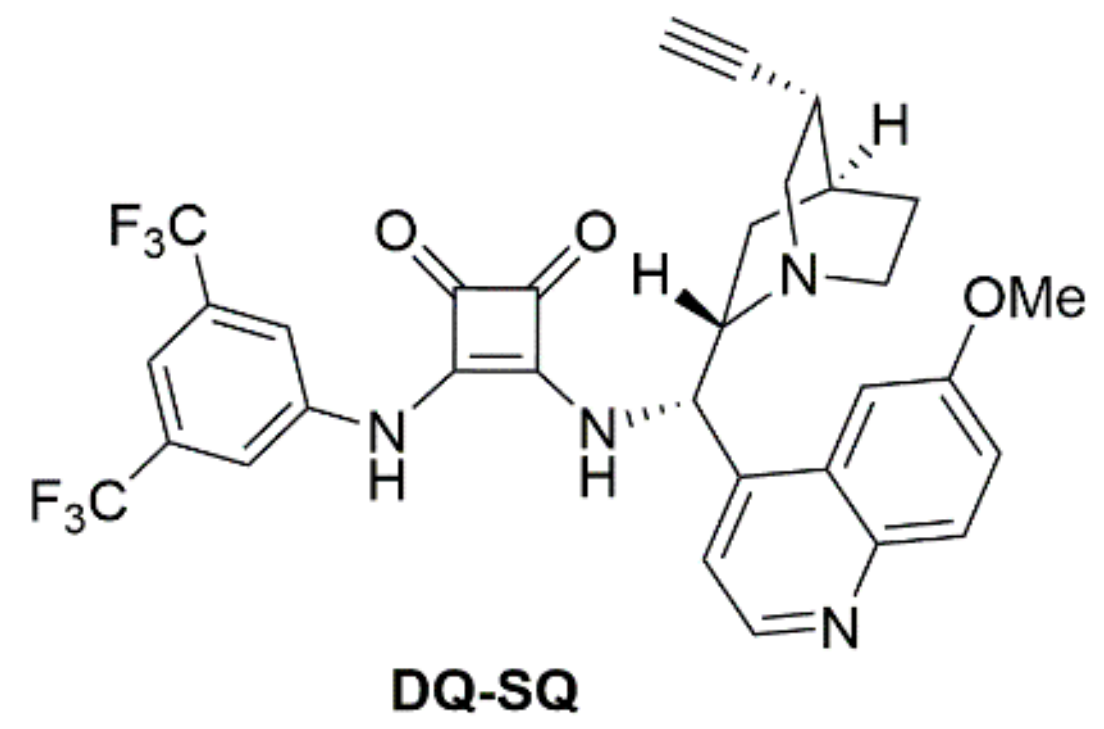
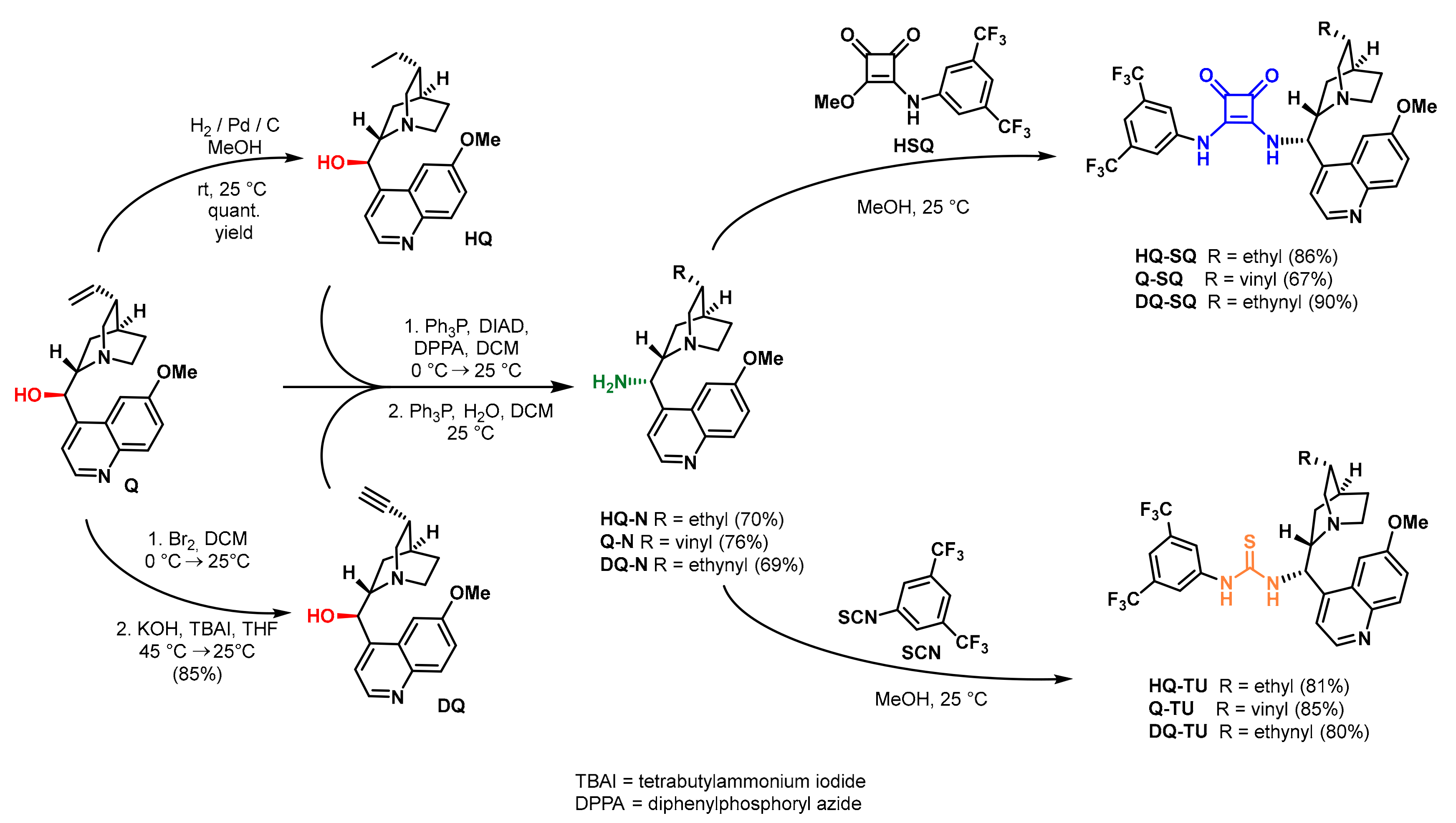
2.5. Synthesis of the Catalysts
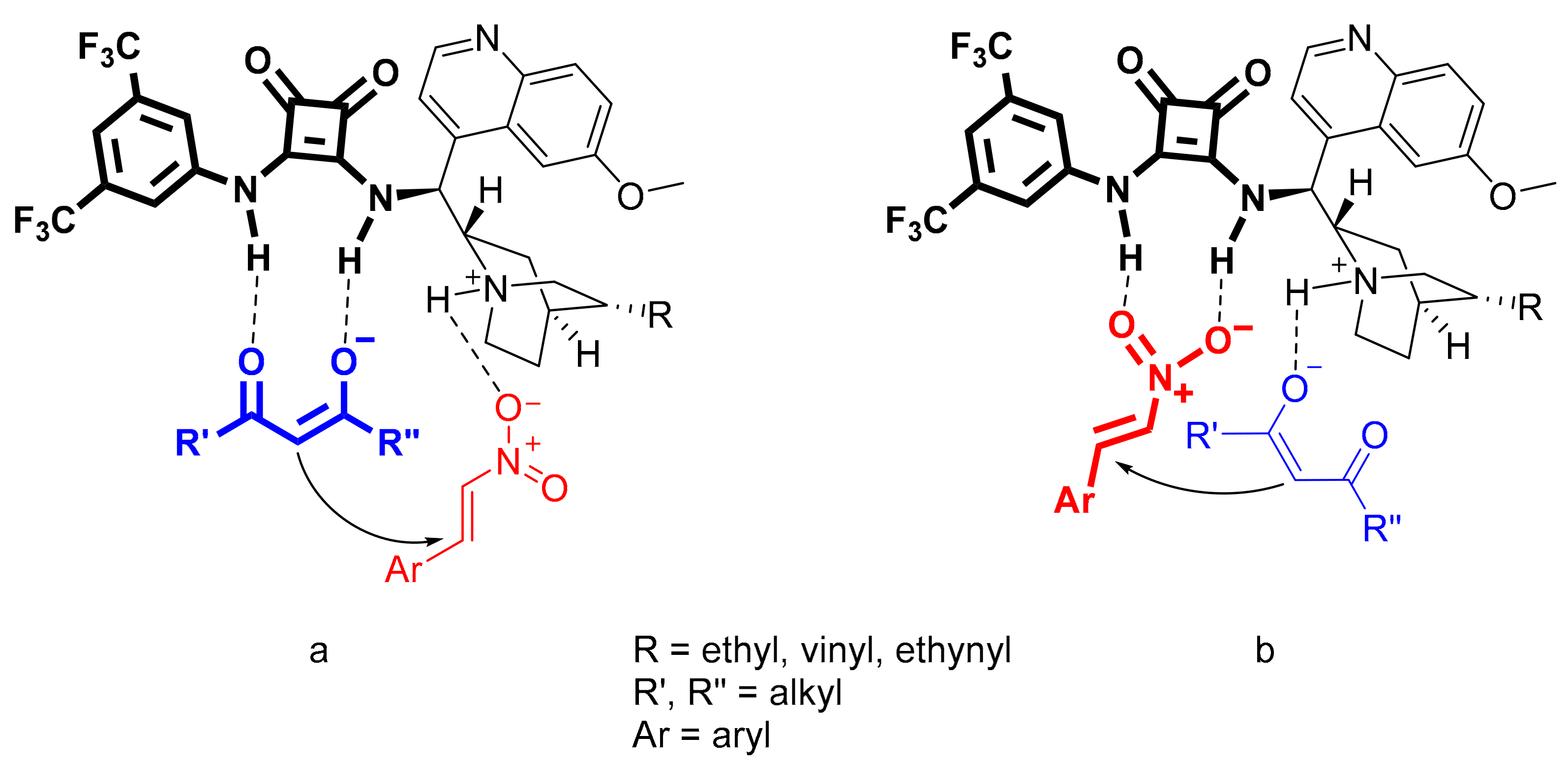
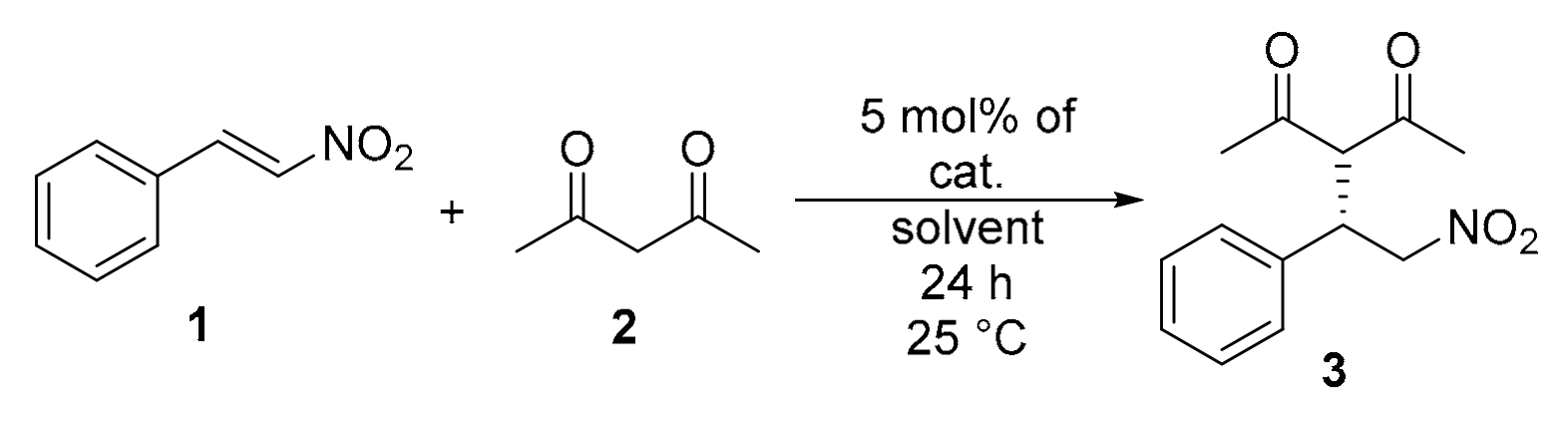
2.6. Application of the Catalysts in Asymmetric Michael Addition
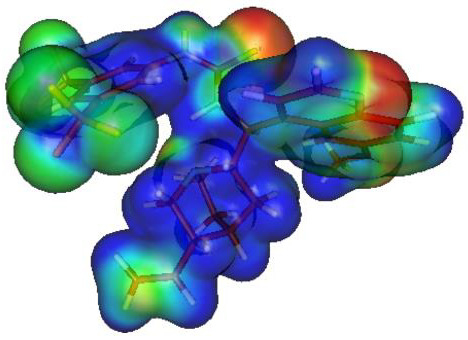
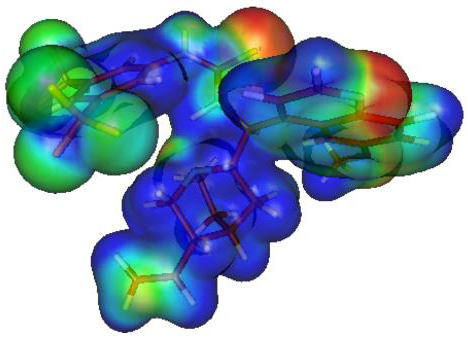
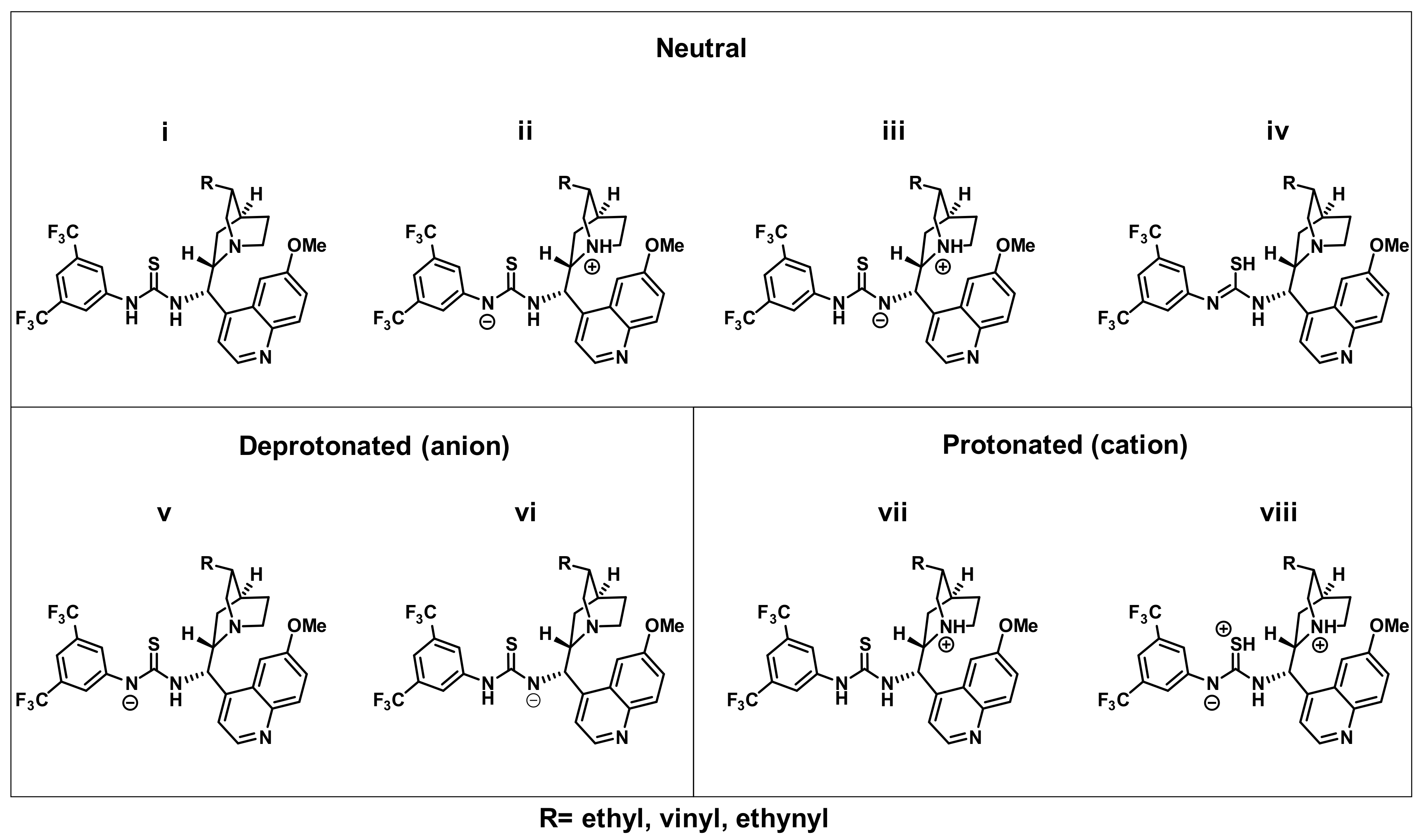
2.7. Theoretical Calculations
3. Conclusions
4. Experimental
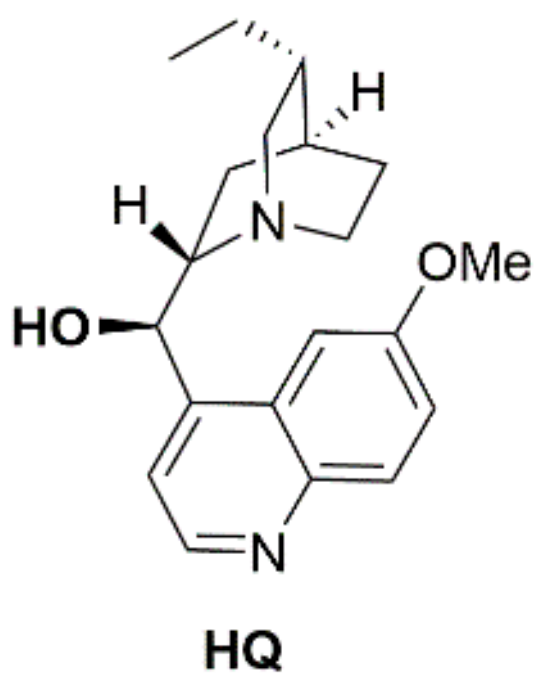
4.1. Hydroquinine
4.2. (S)-((1S,2S,4S,5S)-5-Ethynylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methanamine
4.3. 3-((3,5-Bis(trifluoromethyl)phenyl)amino)-4-(((S)-((1S,2S,4S,5R)-5-ethynylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)cyclobut-3-ene-1,2-dione
4.4. General Procedure for Michael Addition of Pentane-2,4-dione (2) to Trans-β-nitrostyrene (1).
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nag, A.; Gergelitsová, I.; Veselý, J.; Adly, F.G.; Ghanem, A.; Moriuchi, T.; Ohmura, S.D.; Hirao, T.; McCluskey, A.; Simone, M.I.; et al. Asymmetric Synthesis of Drugs and Natural Products, 1st ed.; CRC Press: Boca Raton, FL, USA, 2019; pp. 1–485. [Google Scholar]
- Houk, K.N.; List, B. Asymmetric organocatalysis. Acc. Chem. Res. 2004, 37, 487. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Zhan, G.; Du, W.; Chen, Y.C. Switchable divergent asymmetric synthesis via organocatalysis. Chem. Soc. Rev. 2017, 46, 1675–1692. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L. Biocatalysis in Drug Development-Highlights of the Recent Patent Literature. Org. Proc. Res. Dev. 2018, 22, 1063–1080. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. (Camb.) 2008, 22, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.P.; Jacobsen, E.N. Privileged chiral catalysts. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Yeboah, E.M.O.; Yeboah, S.O.; Singh, G.S. Recent applications of Cinchona alkaloids and their derivatives as catalysts in metal-free asymmetric synthesis. Tetrahedron 2011, 67, 1725–1762. [Google Scholar] [CrossRef]
- Kato, K.; Zhang, M.R.; Suzuki, K. Synthesis of (R,S)-[4-C-11]baclofen via Michael addition of nitromethane labeled with short-lived C-11. Bioorg. Med. Chem. Lett. 2009, 19, 6222–6224. [Google Scholar] [CrossRef]
- Hajzer, V.; Fisera, R.; Latika, A.; Durmis, J.; Kollar, J.; Frecer, V.; Tucekova, Z.; Miertus, S.; Kostolansky, F.; Vareckova, E.; et al. Stereoisomers of oseltamivir—Synthesis, in silico prediction and biological evaluation. Org. Biomol. Chem. 2017, 15, 1828–1841. [Google Scholar] [CrossRef]
- Jung, J.C.; Park, O.S. Efficient asymmetric synthesis of prostaglandin E-1. Z Naturforsch B 2007, 62, 556–560. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.N.; Takemoto, Y. Enantio- and diastereoselective Michael reaction of 1,3-dicarbonyl compounds to nitroolefins catalyzed by a bifunctional thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Hamza, A.; Schubert, G.; Soos, T.; Papai, I. Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: Competing routes to C-C bond formation. J. Am. Chem. Soc. 2006, 128, 13151–13160. [Google Scholar] [CrossRef] [PubMed]
- Kotai, B.; Kardos, G.; Hamza, A.; Farkas, V.; Papai, I.; Soos, T. On the Mechanism of Bifunctional Squaramide- Catalyzed Organocatalytic Michael Addition: A Protonated Catalyst as an Oxyanion Hole. Chem. Eur. J. 2014, 20, 5631–5639. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Mika, L.T.; Csampai, A.; Holczbauer, T.; Kardos, G.; Soos, T. Mechanistic investigations of a bifunctional squaramide organocatalyst in asymmetric Michael reaction and observation of stereoselective retro-Michael reaction. RSC Adv. 2015, 5, 95079–95086. [Google Scholar] [CrossRef]
- Grayson, M.N. Mechanism and Origins of Stereoselectivity in the Cinchona Thiourea- and Squaramide-Catalyzed Asymmetric Michael Addition of Nitroalkanes to Enones. J. Org. Chem. 2017, 82, 4396–4401. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Benaglia, M.; Coccia, F.; Cozzi, F.; Puglisi, A. Solid Supported 9-Amino-9-deoxy-epi-quinine as Efficient Organocatalyst for Stereoselective Reactions in Batch and Under Continuous Flow Conditions. Adv. Synth. Catal. 2015, 357, 377–383. [Google Scholar] [CrossRef]
- Kacprzak, K.M.; Maier, N.M.; Lindner, W. Highly efficient immobilization of Cinchona alkaloid derivatives to silica gel via click chemistry. Tetrahedron Lett. 2006, 47, 8721–8726. [Google Scholar] [CrossRef]
- Lee, J.W.; Mayer-Gall, T.; Opwis, K.; Song, C.E.; Gutmann, J.S.; List, B. Organotextile Catalysis. Science 2013, 341, 1225–1229. [Google Scholar] [CrossRef]
- Parvez, M.M.; Haraguchi, N.; Itsuno, S. Synthesis of Cinchona Alkaloid-Derived Chiral Polymers by Mizoroki-Heck Polymerization and Their Application to Asymmetric Catalysis. Macromolecules 2014, 47, 1922–1928. [Google Scholar] [CrossRef]
- Kohout, M.; Wernisch, S.; Tuma, J.; Hettegger, H.; Picha, J.; Lindner, W. Effect of different immobilization strategies on chiral recognition properties of Cinchona-based anion exchangers. J. Sep. Sci. 2018, 41, 1355–1364. [Google Scholar] [CrossRef]
- Fredriksen, K.A.; Kristensen, T.E.; Hansen, T. Combined bead polymerization and Cinchona organocatalyst immobilization by thiol-ene addition. Beilstein J. Org. Chem. 2012, 8, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Song, C.E.; Blaser, H.-U.; Ager, D.J.; Deshmukh, R.R.; Ryu, D.H.; Shin, U.S.; Lee, J.E.; Yang, J.W.; Park, H.-G.; Jeong, B.-S.; et al. Cinchona Alkaloids in Synthesis and Catalysis, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 1–506. [Google Scholar]
- Kumpuga, B.T.; Itsuno, S. Synthesis of Crosslinked Chiral Polysiloxanes of Cinchona Alkaloid Derivatives and their Applications in Asymmetric Catalysis. Asian J. Org. Chem. 2019, 8, 251–256. [Google Scholar] [CrossRef]
- Etzenbach-Effers, K.; Berkessel, A. Noncovalent Organocatalysis Based on Hydrogen Bonding: Elucidation of Reaction Paths by Computational Methods. Top. Curr. Chem. 2009, 291, 1–27. [Google Scholar]
- Knowles, R.R.; Jacobsen, E.N. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Didaskalou, C.; Kupai, J.; Cseri, L.; Barabas, J.; Vass, E.; Holtzl, T.; Szekely, G. Membrane-Grafted Asymmetric Organocatalyst for an Integrated Synthesis-Separation Platform. ACS Catal. 2018, 8, 7430–7438. [Google Scholar] [CrossRef]
- Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. Asymmetric synthesis with cinchona-decorated cyclodextrin in a continuous-flow membrane reactor. J. Catal. 2019, 371, 255–261. [Google Scholar] [CrossRef]
- Nagy, S.; Dargo, G.; Kisszekelyi, P.; Feher, Z.; Simon, A.; Barabas, J.; Holtzl, T.; Matravolgyi, B.; Karpati, L.; Drahos, L.; et al. New enantiopure binaphthyl-cinchona thiosquaramides: Synthesis and application for enantioselective organocatalysis. New J. Chem. 2019, 43, 5948–5959. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csampai, A.; Soos, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.G.; Lippert, K.M.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Li, X.; Cheng, J.P. Equilibrium acidities of cinchona alkaloid organocatalysts bearing 6′-hydrogen bonding donors in DMSO. Org. Chem. Front. 2016, 3, 170–176. [Google Scholar] [CrossRef]
- Ni, X.; Li, X.; Wang, Z.; Cheng, J.P. Squaramide Equilibrium Acidities in DMSO. Org. Lett. 2014, 16, 1786–1789. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.S.; Yang, C.; Li, X.; Cheng, J.P. Computational Study on the pK(a) Shifts in Proline Induced by Hydrogen-Bond-Donating Cocatalysts. J. Org. Chem. 2014, 79, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, V.E.; Yuen, K.K.Y.; Smith, D.G.; Ho, J.M.; Qin, L.; Turner, P.; Jolliffe, K.A. Deltamides and Croconamides: Expanding the Range of Dual H-bond Donors for Selective Anion Recognition. Chem. Eur. J. 2018, 24, 1140–1150. [Google Scholar] [CrossRef]
- Ho, J.M.; Zwicker, V.E.; Yuen, K.K.Y.; Jolliffe, K.A. Quantum Chemical Prediction of Equilibrium Acidities of Ureas, Deltamides, Squaramides, and Croconamides. J. Org. Chem. 2017, 82, 10732–10736. [Google Scholar] [CrossRef] [PubMed]
- Busschaert, N.; Elmes, R.B.P.; Czech, D.D.; Wu, X.; Kirby, I.L.; Peck, E.M.; Hendzel, K.D.; Shaw, S.K.; Chan, B.; Smith, B.D.; et al. Thiosquaramides: pH switchable anion transporters. Chem. Sci. 2014, 5, 3617–3626. [Google Scholar] [CrossRef]
- Lu, T.X.; Wheeler, S.E. Origin of the Superior Performance of (Thio)Squaramides over (Thio)Ureas in Organocatalysis. Chem. Eur. J. 2013, 19, 15141–15147. [Google Scholar] [CrossRef]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of Chiral, Bifunctional Thiosquaramides: Enantioselective Michael Additions of Barbituric Acids to Nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef]
- Rombola, M.; Rawal, V.H. Dicyclopentyl Dithiosquarate as an Intermediate for the Synthesis of Thiosquaramides. Org. Lett. 2018, 20, 514–517. [Google Scholar] [CrossRef]
- Braje, W.M.; Frackenpohl, J.; Schrake, O.; Wartchow, R.; Beil, W.; Hoffmann, H.M.R. Synthesis of 10,11-didehydro Cinchona alkaloids and key derivatives. Helv. Chim. Acta 2000, 83, 777–792. [Google Scholar] [CrossRef]
- McCooey, S.H.; Connon, S.J. Readily accessible 9-epi-amino cinchona alkaloid derivatives promote efficient, highly enantioselective additions of aldehydes and ketones to nitroolefins. Org. Lett. 2007, 9, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Vakulya, B.; Varga, S.; Soos, T. Epi-cinchona based thiourea organocatalyst family as an efficient asymmetric Michael addition promoter: Enantioselective conjugate addition of nitroalkanes to chalcones and alpha,beta-unsaturated N-acylpyrroles. J. Org. Chem. 2008, 73, 3475–3480. [Google Scholar] [CrossRef] [PubMed]
- del Pozo, S.; Vera, S.; Oiarbide, M.; Palomo, C. Catalytic Asymmetric Synthesis of Quaternary Barbituric Acids. J. Am. Chem. Soc. 2017, 139, 15308–15311. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.Y.; Some, S.; Lee, J.H.; Kim, J.Y.; Song, M.J.; Lee, S.; Zhang, Y.J.; Song, C.E. Organocatalytic Enantioselective Michael-Addition of Malonic Acid Half-Thioesters to beta-Nitroolefins: From Mimicry of Polyketide Synthases to Scalable Synthesis of gamma-Amino Acids. Adv. Synth. Catal. 2011, 353, 3196–3202. [Google Scholar] [CrossRef]
- Grosseheilmann, J.; Kragl, U. Simple and Effective Catalyst Separation by New CO2-Induced Switchable Organocatalysts. ChemSusChem 2017, 10, 2685–2691. [Google Scholar] [CrossRef]
- Madarasz, A.; Dosa, Z.; Varga, S.; Soos, T.; Csampai, A.; Papai, I. Thiourea Derivatives as Bronsted Acid Organocatalysts. ACS Catal. 2016, 6, 4379–4387. [Google Scholar] [CrossRef]
- Hammar, P.; Marcelli, T.; Hiemstra, H.; Himo, F. Density functional theory study of the Cinchona thiourea-catalyzed henry reaction: Mechanism and enantioselectivity. Adv. Synth. Catal. 2007, 349, 2537–2548. [Google Scholar] [CrossRef]
- Quinonero, D.; Frontera, A.; Suner, G.A.; Morey, J.; Costa, A.; Ballester, P.; Deya, P.M. Squaramide as a binding unit in molecular recognition. Chem. Phys. Lett. 2000, 326, 247–254. [Google Scholar] [CrossRef]
- Quinonero, D.; Prohens, R.; Garau, C.; Frontera, A.; Ballester, P.; Costa, A.; Deya, P.M. A theoretical study of aromaticity in squaramide complexes with anions. Chem. Phys. Lett. 2002, 351, 115–120. [Google Scholar] [CrossRef]
- Allen, R.I.; Box, K.J.; Comer, J.E.; Peake, C.; Tam, K.Y. Multiwavelength spectrophotometric determination of acid dissociation constants of ionizable drugs. J. Pharm. Biomed. Anal. 1998, 17, 699–712. [Google Scholar] [CrossRef]
- Tam, K.Y.; Takacs-Novak, K. Multi-wavelength spectrophotometric determination of acid dissociation constants: A validation study. Anal. Chim. Acta 2001, 434, 157–167. [Google Scholar] [CrossRef]
- Avdeef, A.; Comer, J.E.A.; Thomson, S.J. Ph-Metric Log. 3. Glass-Electrode Calibration in Methanol Water, Applied to Pka Determination of Water-Insoluble Substances. Anal. Chem. 1993, 65, 42–49. [Google Scholar] [CrossRef]
- TakacsNovak, K.; Box, K.J.; Avdeef, A. Potentiometric pK(a) determination of water-insoluble compounds: Validation study in methanol/water mixtures. Int. J. Pharm. 1997, 151, 235–248. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Shao, Y.H.; Gan, Z.T.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.T.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. A universal approach to solvation modeling. Acc. Chem. Res. 2008, 41, 760–768. [Google Scholar] [CrossRef]
- Chipman, D.M.; Dupuis, M. Implementation of solvent reaction fields for electronic structure. Theor. Chem. Acc. 2002, 107, 90–102. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Generalized Born Solvation Model SM12. J. Chem. Theory Comput. 2013, 9, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.G.; Dixon, D.A. Absolute hydration free energy of the proton from first-principles electronic structure calculations. J. Phys. Chem. A 2001, 105, 11534–11540. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, V.S.; LLC Schrodinger: New York, NY, USA, 2010.
- Palacio, C.; Connon, S.J. A New Class of Urea-Substituted Cinchona Alkaloids Promote Highly Enantioselective Nitroaldol reactions of Trifluoromethylketones. Org. Lett. 2011, 13, 1298–1301. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalystb | pKa | Entry | Catalystb | pKa |
|---|---|---|---|---|---|
| 1 | HQ | 9.10 | 7 | HQ-SQ | 7.86 |
| 2 | Q | 8.52 | 8 | Q-SQ | 6.95 |
| 3 | DQ | 7.40 | 9 | DQ-SQ | 6.13 |
| 4 | HQ-N | 9.71 | 10 | HQ-TU | 8.61 |
| 5 | Q-N | 9.10 | 11 | Q-TU | 8.06 |
| 6 | DQ-N | 8.10 | 12 | DQ-TU | 7.02 |
| Entry | Catalyst | pKa, NH1 | pKa, NH2 |
|---|---|---|---|
| 1 | HQ-SQ | 9.57 | 11.73 |
| 2 | Q-SQ | 9.29 | >12# |
| 3 | DQ-SQ | 9.20 | >12# |
| 4 | HQ-TU | 10.87 | 12 # |
| 5 | Q-TU | 10.84 | 12 # |
| 6 | DQ-TU | 10.78 | 12 # |
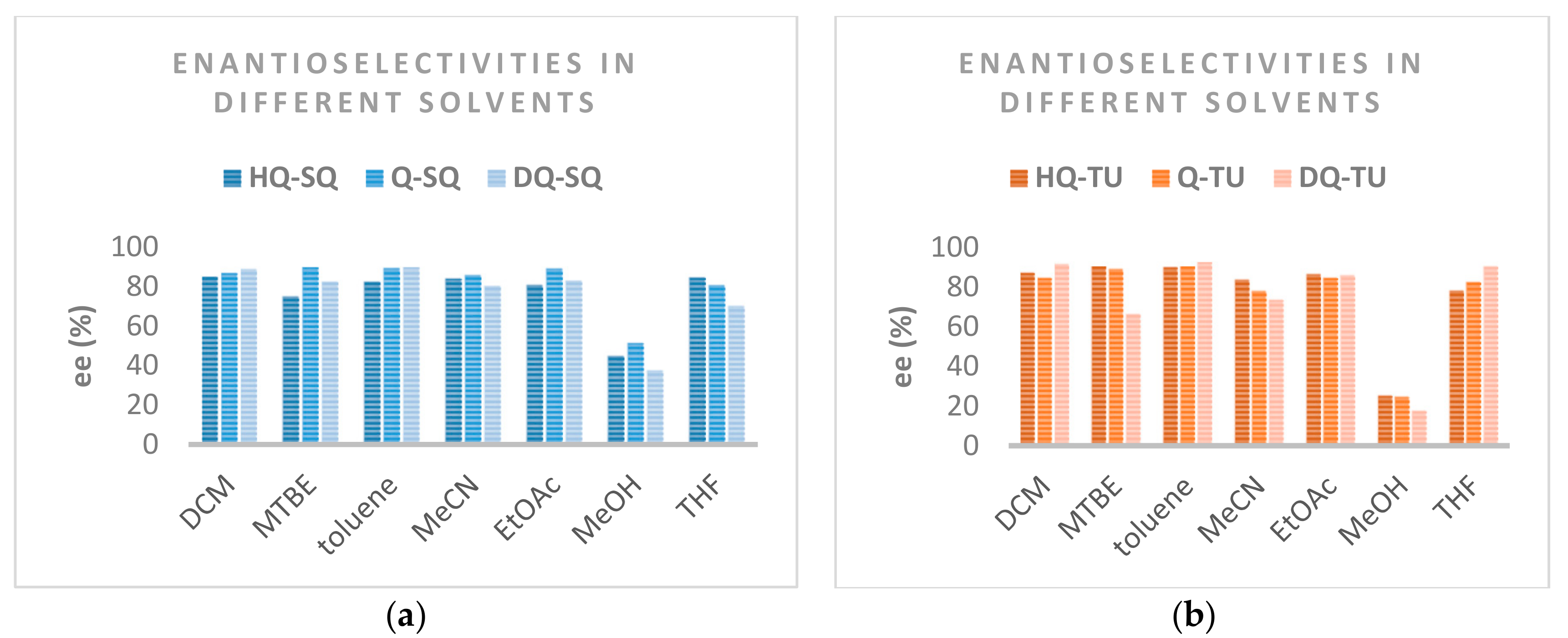
| Entry | Catalyst | Solvent | Yield [%]b | ee [%]c |
|---|---|---|---|---|
| 1 | HQ-SQ | DCM | 99 | 85 |
| 2 | HQ-SQ | MTBE | 100 | 75 |
| 3 | HQ-SQ | toluene | 100 | 82 |
| 4 | HQ-SQ | MeCN | 97 | 84 |
| 5 | HQ-SQ | EtOAc | 99 | 81 |
| 6 | HQ-SQ | MeOH | 77 | 45 |
| 7 | HQ-SQ | THF | 99 | 85 |
| 8 | Q-SQ | DCM | 98 | 87 |
| 9 | Q-SQ | MTBE | 99 | 90 |
| 10 | Q-SQ | toluene | 99 | 89 |
| 11 | Q-SQ | MeCN | 99 | 86 |
| 12 | Q-SQ | EtOAc | 100 | 89 |
| 13 | Q-SQ | MeOH | 76 | 51 |
| 14 | Q-SQ | THF | 99 | 81 |
| 15 | DQ-SQ | DCM | 99 | 89 |
| 16 | DQ-SQ | MTBE | 76 | 82 |
| 17 | DQ-SQ | toluene | 87 | 90 |
| 18 | DQ-SQ | MeCN | 88 | 80 |
| 19 | DQ-SQ | EtOAc | 89 | 83 |
| 20 | DQ-SQ | MeOH | 76 | 38 |
| 21 | DQ-SQ | THF | 95 | 70 |
| 22 | HQ-TU | DCM | 93 | 87 |
| 23 | HQ-TU | MTBE | 99 | 91 |
| 24 | HQ- TU | toluene | 95 | 90 |
| 25 | HQ- TU | MeCN | 94 | 84 |
| 26 | HQ- TU | EtOAc | 99 | 86 |
| 27 | HQ- TU | MeOH | 76 | 25 |
| 28 | HQ- TU | THF | 97 | 78 |
| 29 | Q- TU | DCM | 100 | 85 |
| 30 | Q- TU | MTBE | 99 | 89 |
| 31 | Q- TU | toluene | 100 | 91 |
| 32 | Q- TU | MeCN | 99 | 78 |
| 33 | Q- TU | EtOAc | 100 | 85 |
| 34 | Q- TU | MeOH | 82 | 25 |
| 35 | Q- TU | THF | 99 | 82 |
| 36 | DQ- TU | DCM | 98 | 91 |
| 37 | DQ- TU | MTBE | 97 | 66 |
| 38 | DQ- TU | toluene | 99 | 93 |
| 39 | DQ- TU | MeCN | 85 | 73 |
| 40 | DQ- TU | EtOAc | 80 | 86 |
| 41 | DQ- TU | MeOH | 76 | 17 |
| 42 | DQ- TU | THF | 98 | 90 |
| Compound | Neutral | Deprotonated (anion) | Protonated (cation) | |||||
|---|---|---|---|---|---|---|---|---|
| i | ii | iii | iv | v | vi | vii | viii | |
| HQ-TU | 0 | 32 | 59 | 64 | 0 (150) | 9 (159) | 0 (−44) | 62 (−14) |
| Q-TU | 0 | 32 | 63 | 65 | 0 (148) | 10 (157) | 0 (−46) | 69 (−9) |
| DQ-TU | 0 | 40 | 66 | 70 | 0 (144) | 12 (157) | 0 (−40) | 64 (−9) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, S.; Fehér, Z.; Dargó, G.; Barabás, J.; Garádi, Z.; Mátravölgyi, B.; Kisszékelyi, P.; Dargó, G.; Huszthy, P.; Höltzl, T.; et al. Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring. Materials 2019, 12, 3034. https://doi.org/10.3390/ma12183034
Nagy S, Fehér Z, Dargó G, Barabás J, Garádi Z, Mátravölgyi B, Kisszékelyi P, Dargó G, Huszthy P, Höltzl T, et al. Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring. Materials. 2019; 12(18):3034. https://doi.org/10.3390/ma12183034
Chicago/Turabian StyleNagy, Sándor, Zsuzsanna Fehér, Gergő Dargó, Júlia Barabás, Zsófia Garádi, Béla Mátravölgyi, Péter Kisszékelyi, Gyula Dargó, Péter Huszthy, Tibor Höltzl, and et al. 2019. "Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring" Materials 12, no. 18: 3034. https://doi.org/10.3390/ma12183034
APA StyleNagy, S., Fehér, Z., Dargó, G., Barabás, J., Garádi, Z., Mátravölgyi, B., Kisszékelyi, P., Dargó, G., Huszthy, P., Höltzl, T., Balogh, G. T., & Kupai, J. (2019). Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring. Materials, 12(18), 3034. https://doi.org/10.3390/ma12183034