In Situ Synthesis of Silicon–Carbon Composites and Application as Lithium-Ion Battery Anode Materials


Abstract
:1. Introduction
2. Materials and Methods
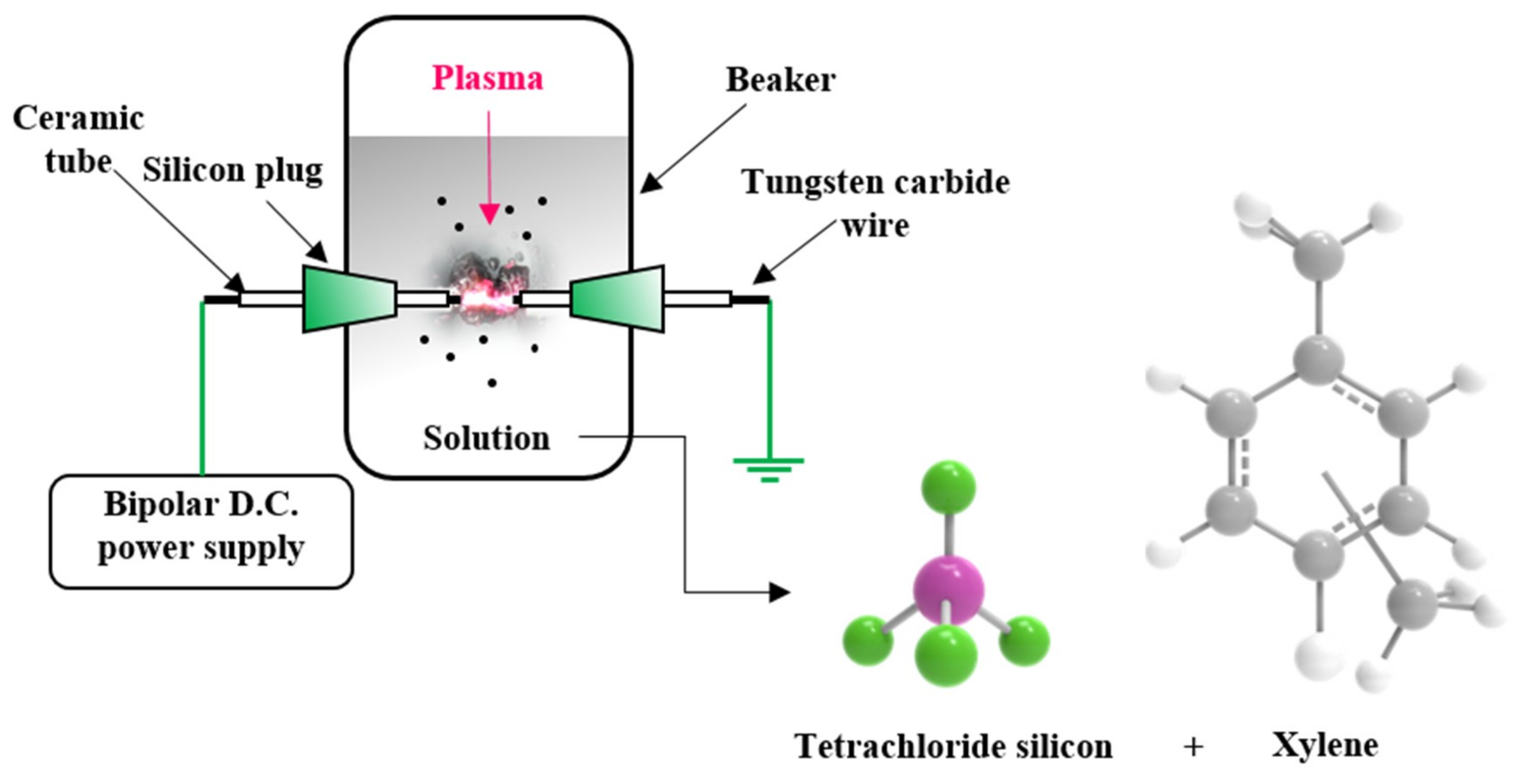
2.1. Synthesis of Si–C Composite
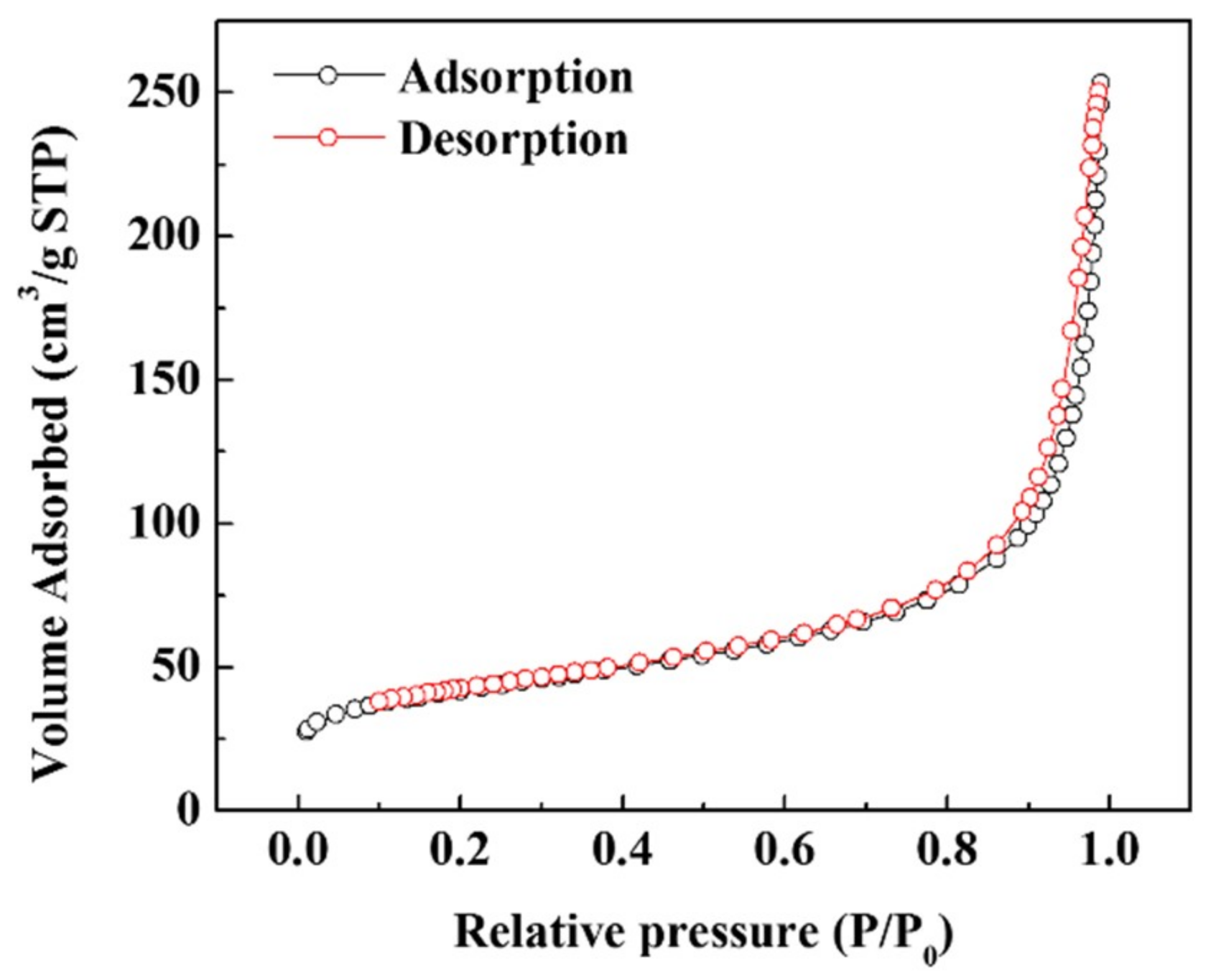
2.2. Material Characterization
2.3. Electrode Manufacturing and Electrochemical Test
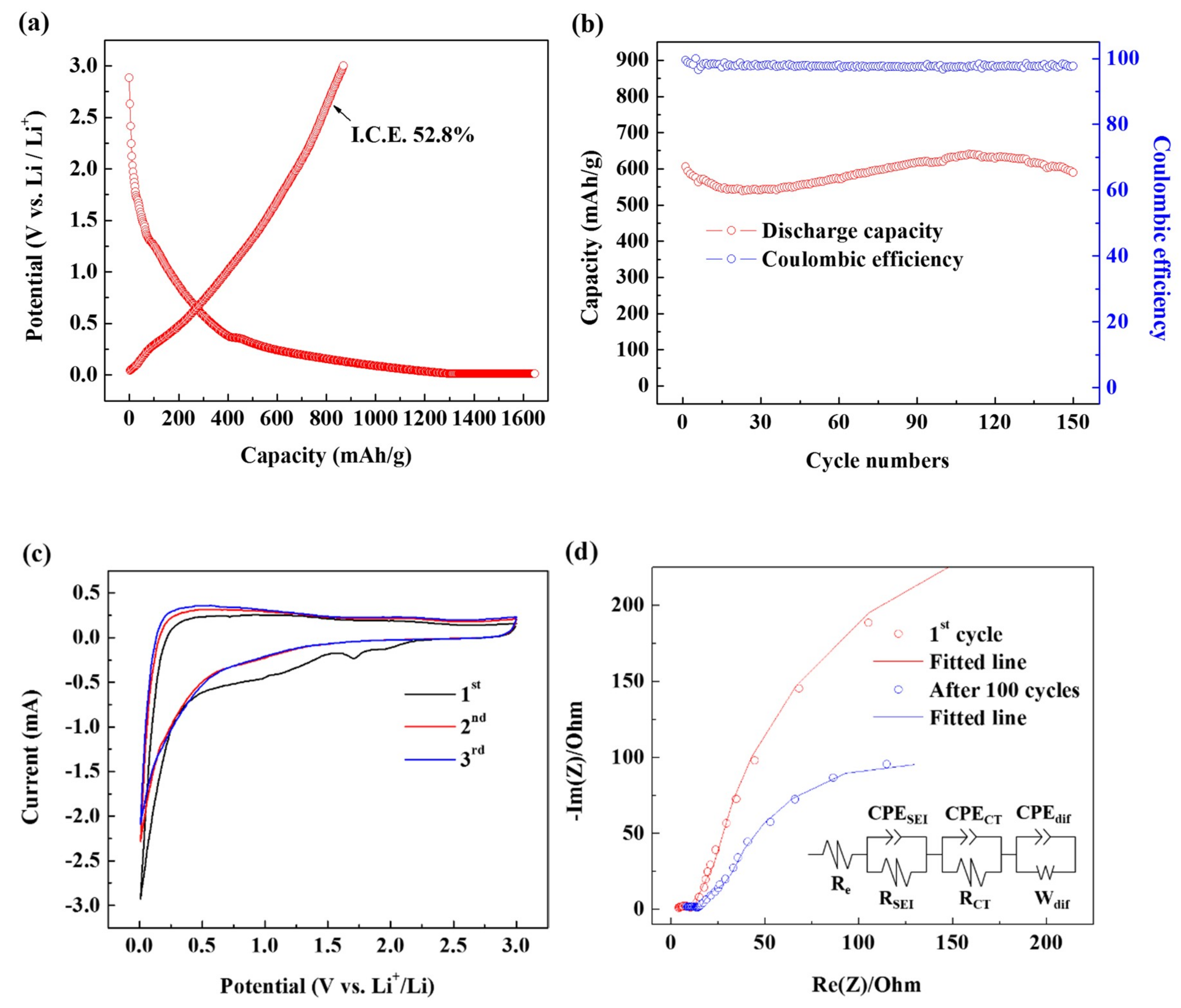
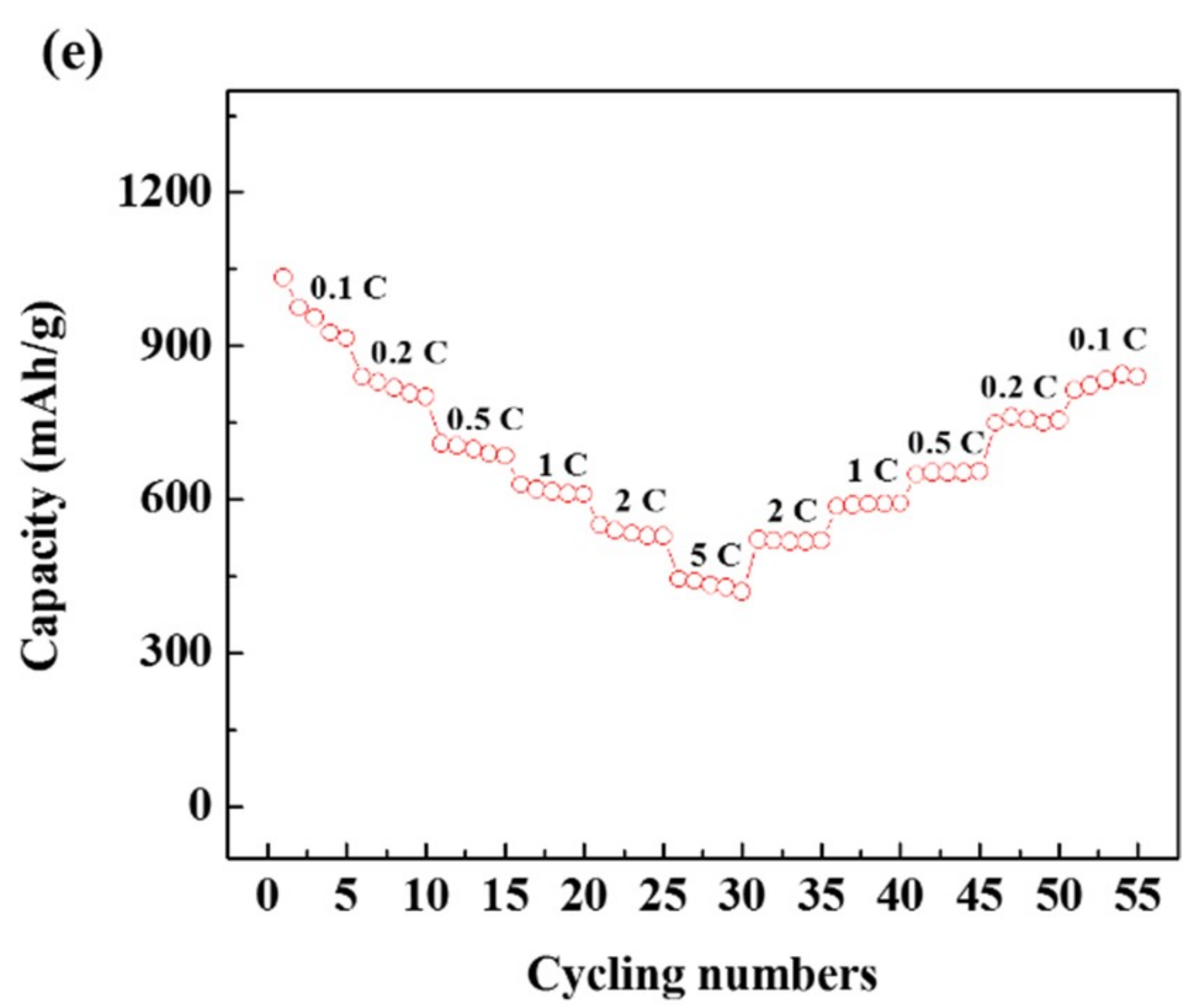
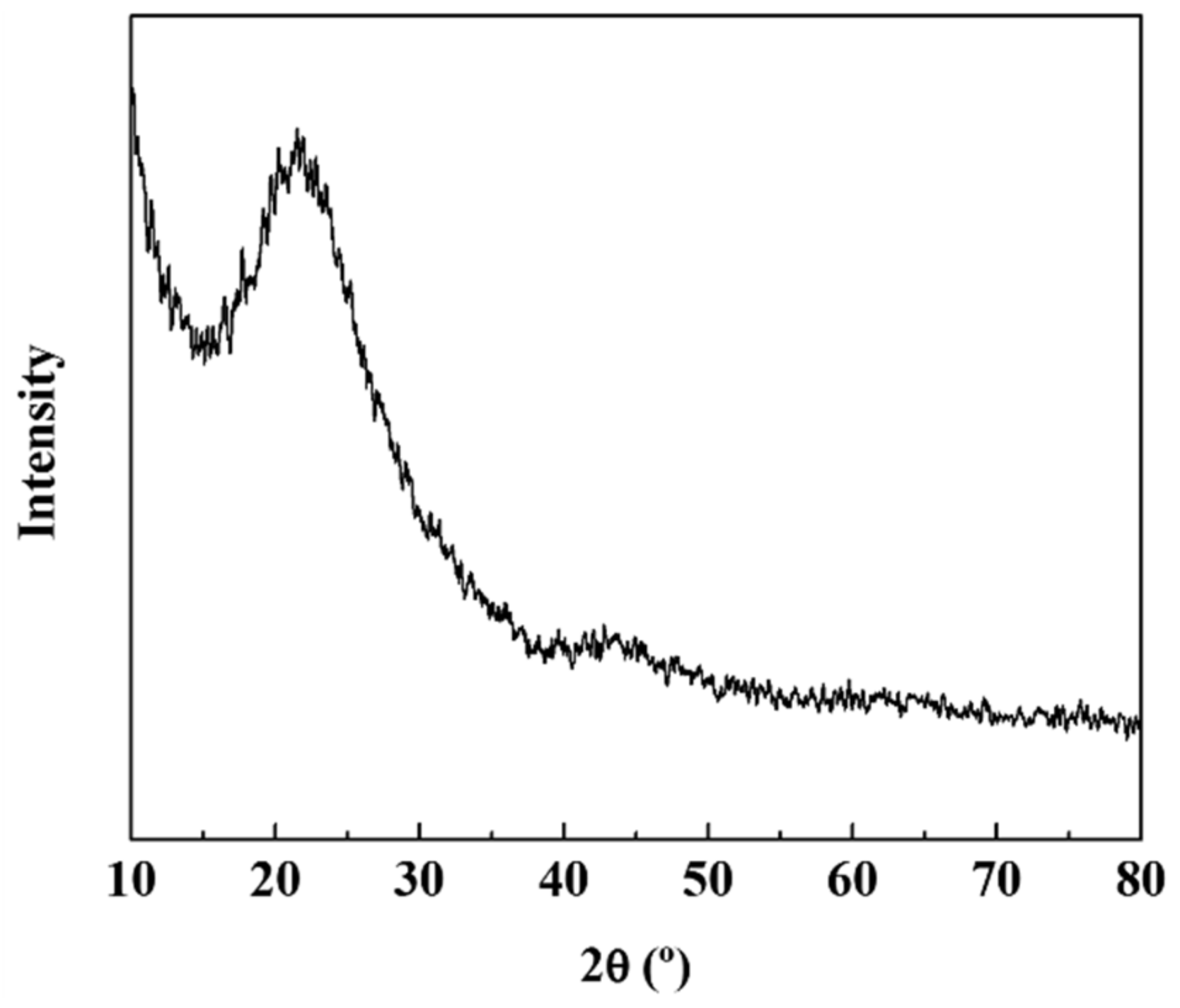
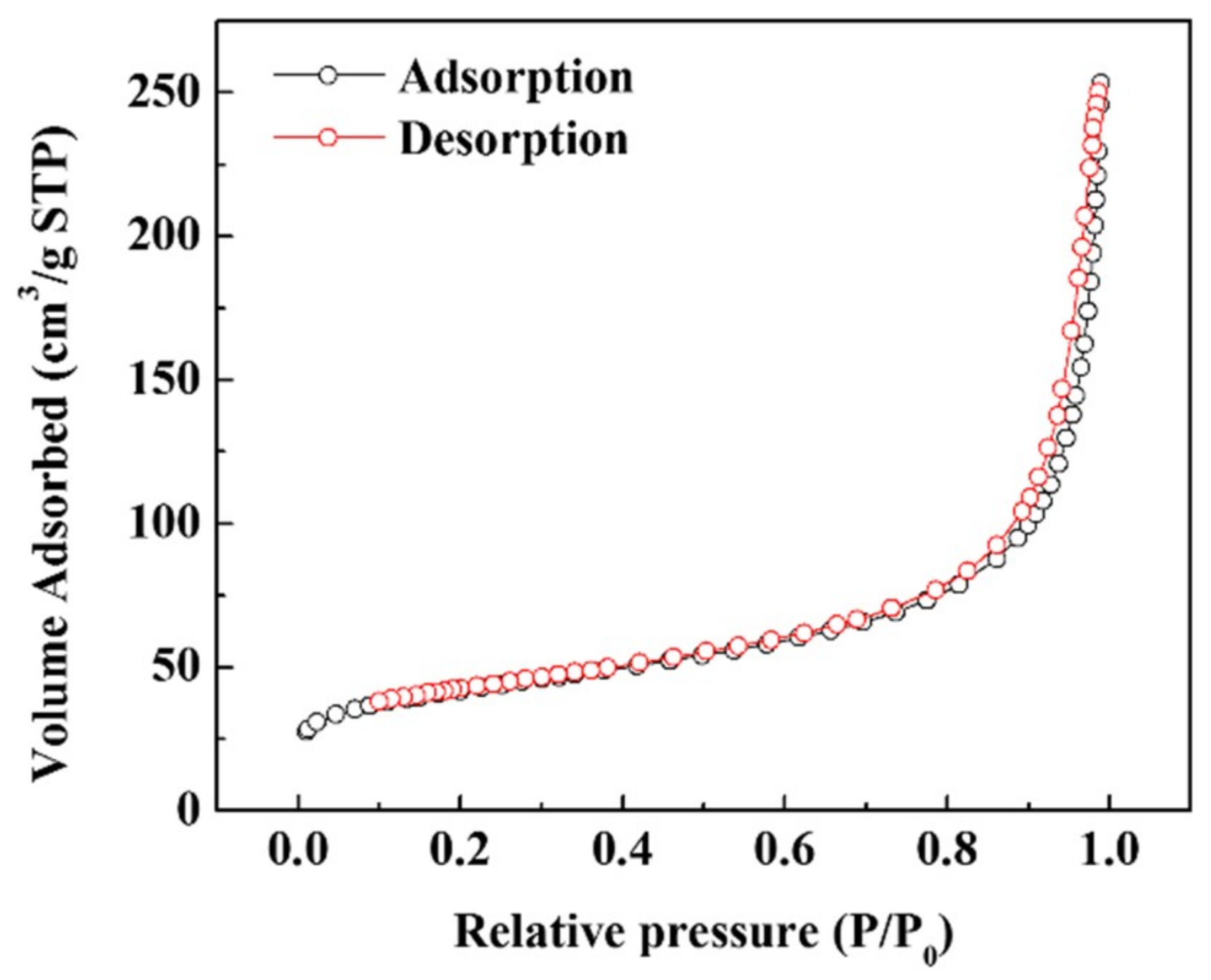
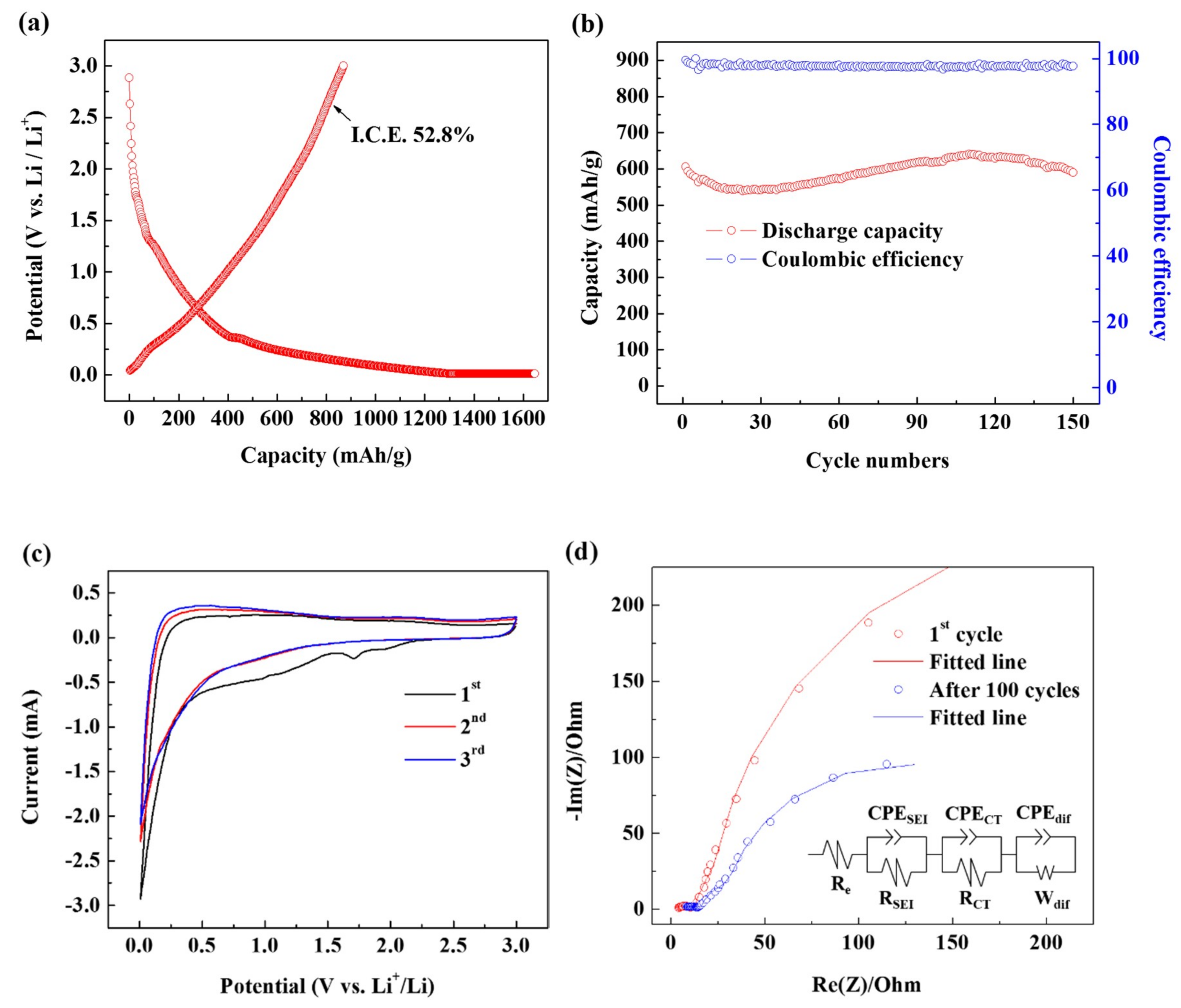
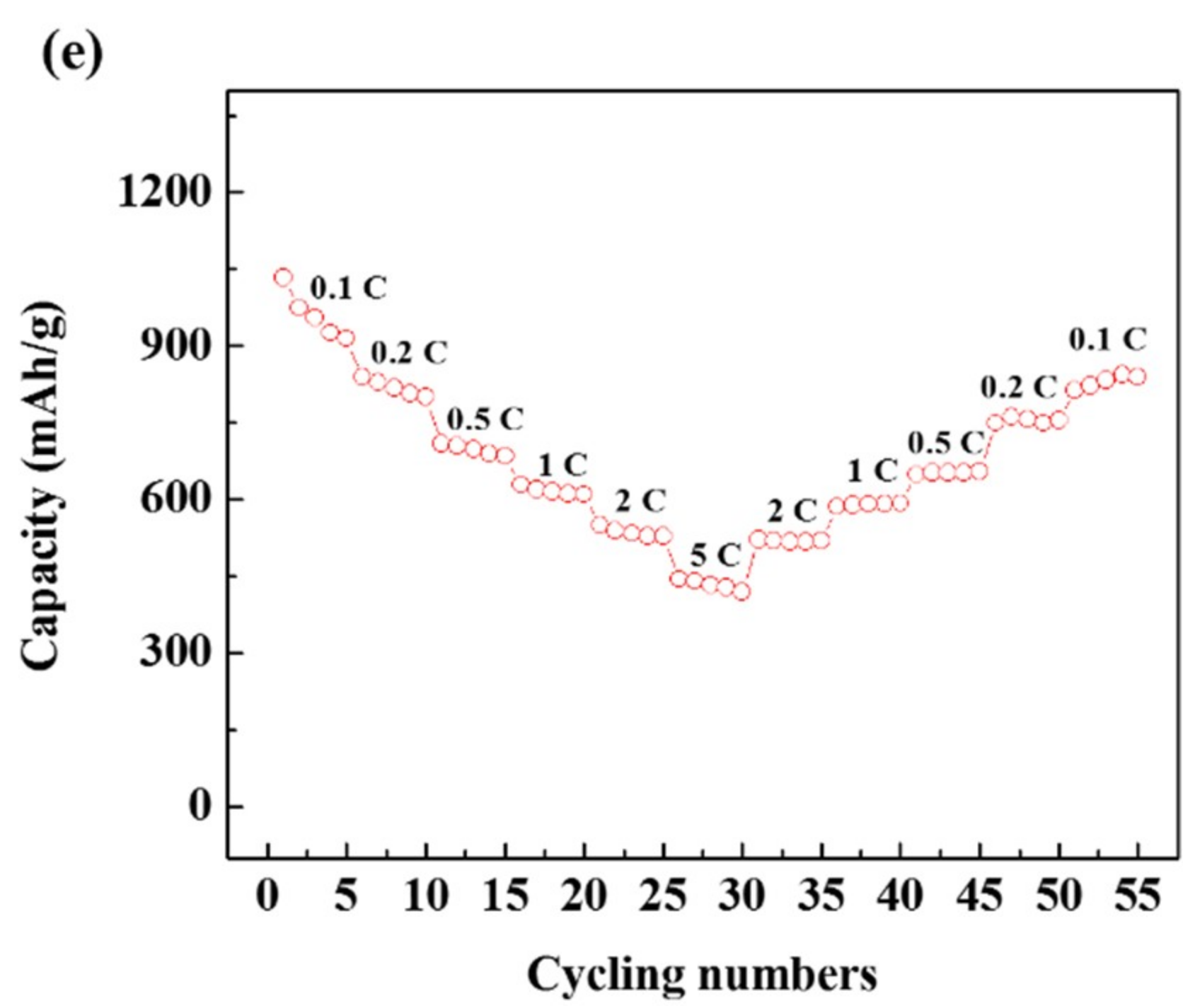
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nijhawan, S.; McMurry, P.H.; Swihart, M.T.; Suh, S.-M.; Girshick, S.L.; Campbell, S.A.; Brockmann, J.E. An experimental and numerical study of particle nucleation and growth during low-pressure thermal decomposition of silane. J. Aerosol Sci. 2003, 34, 691–711. [Google Scholar] [CrossRef]
- Li, Z.; Ruckenstein, E. Water-soluble poly (acrylic acid) grafted luminescent silicon nanoparticles and their use as fluorescent biological staining labels. Nano Lett. 2004, 4, 1463–1467. [Google Scholar] [CrossRef]
- Antoniadis, H. Silicon ink high efficiency solar cells. In Proceedings of the 2009 34th IEEE Photovoltaic Specialists Conference (PVSC), Philadelphia, PA, USA, 7–12 June 2009; pp. 650–654. [Google Scholar]
- Gupta, A.; Wiggers, H. Surface chemistry and photoluminescence property of functionalized silicon nanoparticles. Phys. E Low-Dimens. Syst. Nanostruct. 2009, 41, 1010–1014. [Google Scholar] [CrossRef]
- He, Y.; Piper, D.M.; Gu, M.; Travis, J.J.; George, S.M.; Lee, S.-H.; Genc, A.; Pullan, L.; Liu, J.; Mao, S.X.; et al. In situ transmission electron microscopy probing of native oxide and artificial layers on silicon nanoparticles for lithium ion batteries. ACS Nano 2014, 8, 11816–11823. [Google Scholar] [CrossRef]
- Erogbogbo, F.; Lin, T.; Tucciarone, P.M.L.; Joie, K.M.; Lai, L.; Patki, G.D.; Prasad, P.N.; Swihart, M.T. On-demand hydrogen generation using nanosilicon: Splitting water without light, heat, or electricity. Nano Lett. 2013, 13, 451–456. [Google Scholar] [CrossRef]
- Yu, T.; Wang, F.; Xu, Y.; Ma, L.; Pi, X.; Yang, D. Graphene Coupled with Silicon Quantum Dots for High-Performance Bulk-Silicon-Based Schottky-Junction Photodetectors. Adv. Mater. 2016, 28, 4912–4919. [Google Scholar] [CrossRef]
- Iqbal, M.; Purkait, T.K.; Goss, G.G.; Bolton, J.R.; Gamal El-Din, M.; Veinot, J.G. Application of engineered Si nanoparticles in light-induced advanced oxidation remediation of a water-borne model contaminant. ACS Nano 2016, 10, 5405–5412. [Google Scholar] [CrossRef]
- Rodriguez, I.; Shi, L.; Lu, X.; Korgel, B.A.; Alvarez-Puebla, R.; Meseguer, F. Silicon nanoparticles as Raman scattering enhancers. Nanoscale 2014, 6, 5666–5670. [Google Scholar] [CrossRef]
- Reynolds, W.N. Physical Properties of Graphite; Elsevier Pubishing Co. Ltd.: New York, NY, USA, 1968. [Google Scholar]
- Song, X.; Kinoshita, K.; Tran, T. Microstructural characterization of lithiated graphite. J. Electrochem. Soc. 1996, 143, L120–L123. [Google Scholar] [CrossRef]
- Endo, M.; Kim, C.; Nishimura, K.; Fujino, T.; Miyashita, K. Recent development of carbon materials for Li ion batteries. Carbon 2000, 38, 183–197. [Google Scholar] [CrossRef]
- Tamai, H.; Matsuoka, S.; Ishihara, M.; Yasuda, H. New carbon materials from pitch containing organotin compounds for anode of lithium ion batteries. Carbon 2001, 39, 1515–1523. [Google Scholar] [CrossRef]
- Erk, C.; Brezesinski, T.; Sommer, H.; Schneider, R.; Janek, J. Toward silicon anodes for next-generation lithium ion batteries: A comparative performance study of various polymer binders and silicon nanopowders. ACS Appl. Mater. Interfaces 2013, 5, 7299–7307. [Google Scholar] [CrossRef]
- Li, H.; Huang, X.; Chen, L.; Zhou, G.; Zhang, Z.; Yu, D.; Mo, Y.J.; Pei, N. The crystal structural evolution of nano-Si anode caused by lithium insertion and extraction at room temperature. Solid State Ion. 2000, 135, 181–191. [Google Scholar] [CrossRef]
- Park, M.-H.; Kim, M.G.; Joo, J.; Kim, K.; Kim, J.; Ahn, S.; Cui, Y.; Jaephil Cho, J. Silicon nanotube battery anodes. Nano Lett. 2009, 9, 3844–3847. [Google Scholar] [CrossRef]
- Szczech, J.R.; Jin, S. Nanostructured silicon for high capacity lithium battery anodes. Energy Environ. Sci. 2011, 4, 56–72. [Google Scholar] [CrossRef]
- Thackeray, M.M.; Wolverton, C.; Isaacs, E.D. Electrical energy storage for transportation—Approaching the limits of, and going beyond, lithium-ion batteries. Energy Environ. Sci. 2012, 5, 7854–7863. [Google Scholar] [CrossRef]
- Boukamp, B.; Lesh, G.; Huggins, R. All-solid lithium electrodes with mixed-conductor matrix. J. Electrochem. Soc. 1981, 128, 725–729. [Google Scholar] [CrossRef]
- Kasavajjula, U.; Wang, C.; Appleby, A.J. Nano-and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J. Power Sources 2007, 163, 1003–1039. [Google Scholar] [CrossRef]
- He, Y.; Yu, X.; Wang, Y.; Li, H.; Huang, X. Alumina-coated patterned amorphous silicon as the anode for a lithium-ion battery with high Coulombic efficiency. Adv. Mater. 2011, 23, 4938–4941. [Google Scholar] [CrossRef]
- El-Demellawi, J.K.; Holt, C.R.; Abou-Hamad, E.; Al-Talla, Z.A.; Saih, Y.; Chaieb, S. Room-temperature reactivity of silicon nanocrystals with solvents: The case of ketone and hydrogen production from secondary alcohols: Catalysis? ACS Appl. Mater. Interfaces 2015, 7, 13794–13800. [Google Scholar] [CrossRef]
- Joo, J.; Liu, X.; Kotamraju, V.R.; Ruoslahti, E.; Nam, Y.; Sailor, M.J. Gated luminescence imaging of silicon nanoparticles. ACS Nano 2015, 9, 6233–6241. [Google Scholar] [CrossRef]
- Sugimoto, H.; Fujii, M.; Imakita, K.; Hayashi, S.; Akamatsu, K. All-inorganic near-infrared luminescent colloidal silicon nanocrystals: High dispersibility in polar liquid by phosphorus and boron codoping. J. Phys. Chem. C 2012, 116, 17969–17974. [Google Scholar] [CrossRef]
- Hori, Y.; Kano, S.; Sugimoto, H.; Imakita, K.; Fujii, M. Size-dependence of acceptor and donor levels of boron and phosphorus codoped colloidal silicon nanocrystals. Nano Lett. 2016, 16, 2615–2620. [Google Scholar] [CrossRef]
- Kramer, N.J.; Schramke, K.S.; Kortshagen, U.R. Plasmonic properties of silicon nanocrystals doped with boron and phosphorus. Nano Lett. 2015, 15, 5597–5603. [Google Scholar] [CrossRef]
- Miller, J.B.; Dandu, N.; Velizhanin, K.A.; Anthony, R.J.; Kortshagen, U.R.; Kroll, D.M.; Kilina, S.; Hobbie, E.K. Enhanced luminescent stability through particle interactions in silicon nanocrystal aggregates. ACS Nano 2015, 9, 9772–9782. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Kim, D.-H.; Kim, S.-H.; Lee, E.-K.; Park, S.-K.; Lee, J.-W.; Yun, Y.-S.; Choi, S.-Y.; Kang, J. Nano Hard Carbon Anodes for Sodium-Ion Batteries. Nanomaterials 2019, 9, 793. [Google Scholar] [CrossRef]
- Kang, J.; Kim, D.-Y.; Chae, S.-A.; Saito, N.; Choi, S.-Y.; Kim, K.-H. Maximization of sodium storage capacity of pure carbon material used in sodium-ion batteries. J. Mater. Chem. A 2019. [Google Scholar] [CrossRef]
- Morishita, T.; Ueno, T.; Panomsuwan, G.; Hieda, J.; Yoshida, A.; Bratescu, M.A.; Saito, N. Fastest formation routes of nanocarbons in solution plasma processes. Sci. Rep. 2016, 6, 36880. [Google Scholar] [CrossRef]
- Kang, J.; Li, O.L.; Saito, N. Hierarchical meso–macro structure porous carbon black as electrode materials in Li–air battery. J. Power Sources 2014, 261, 156–161. [Google Scholar] [CrossRef]
- Kang, J.; Li, O.L.; Saito, N. Synthesis of structure-controlled carbon nano spheres by solution plasma process. Carbon 2013, 60, 292–298. [Google Scholar] [CrossRef]
- MacDonald, J.; Johnson, W. Impedance Spectroscopy; Macdonald, J.R., Ed.; John Wiley and Sons: New York, NY, USA, 1987. [Google Scholar]
- Mulder, W.; Sluyters, J.; Pajkossy, T.; Nyikos, L. Tafel current at fractal electrodes: Connection with admittance spectra. J. Electroanal. Chem. Interfacial Electrochem. 1990, 285, 103–115. [Google Scholar] [CrossRef]
- Schiller, C.; Strunz, W. The evaluation of experimental dielectric data of barrier coatings by means of different models. Electrochim. Acta 2001, 46, 3619–3625. [Google Scholar] [CrossRef]
- Kim, C.-H.; Pyun, S.-I.; Kim, J.-H. An investigation of the capacitance dispersion on the fractal carbon electrode with edge and basal orientations. Electrochim. Acta 2003, 48, 3455–3463. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Re (Ω) | RSEI (Ω) | RCT (Ω) | |
|---|---|---|---|
| 1st cycle | 1.761 | 11.01 | 12.28 |
| After 100 cycles | 3.689 | 14.07 | 15.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.-Y.; Kim, H.-V.; Kang, J. In Situ Synthesis of Silicon–Carbon Composites and Application as Lithium-Ion Battery Anode Materials. Materials 2019, 12, 2871. https://doi.org/10.3390/ma12182871
Kim D-Y, Kim H-V, Kang J. In Situ Synthesis of Silicon–Carbon Composites and Application as Lithium-Ion Battery Anode Materials. Materials. 2019; 12(18):2871. https://doi.org/10.3390/ma12182871
Chicago/Turabian StyleKim, Dae-Yeong, Han-Vin Kim, and Jun Kang. 2019. "In Situ Synthesis of Silicon–Carbon Composites and Application as Lithium-Ion Battery Anode Materials" Materials 12, no. 18: 2871. https://doi.org/10.3390/ma12182871
APA StyleKim, D.-Y., Kim, H.-V., & Kang, J. (2019). In Situ Synthesis of Silicon–Carbon Composites and Application as Lithium-Ion Battery Anode Materials. Materials, 12(18), 2871. https://doi.org/10.3390/ma12182871