Ru-Doped Wells–Dawson Polyoxometalate as Efficient Catalyst for Glycerol Hydrogenolysis to Propanediols



Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
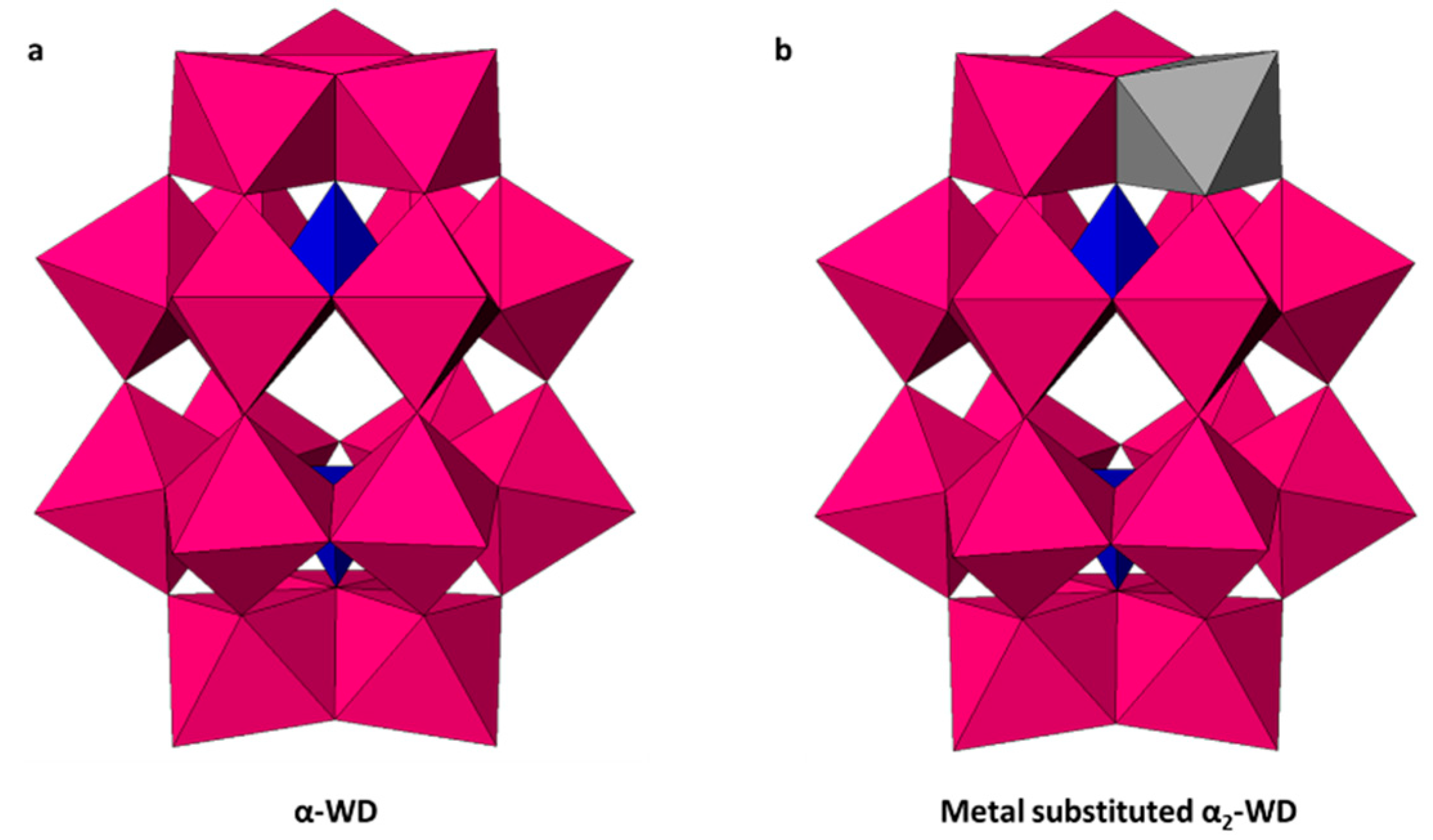
2.2. Synthesis of WD-POMs
2.3. Characterization of WD-POMs
2.4. Catalytic Hydrogenolysis of Glycerol with WD-POMs
2.5. Product Analysis
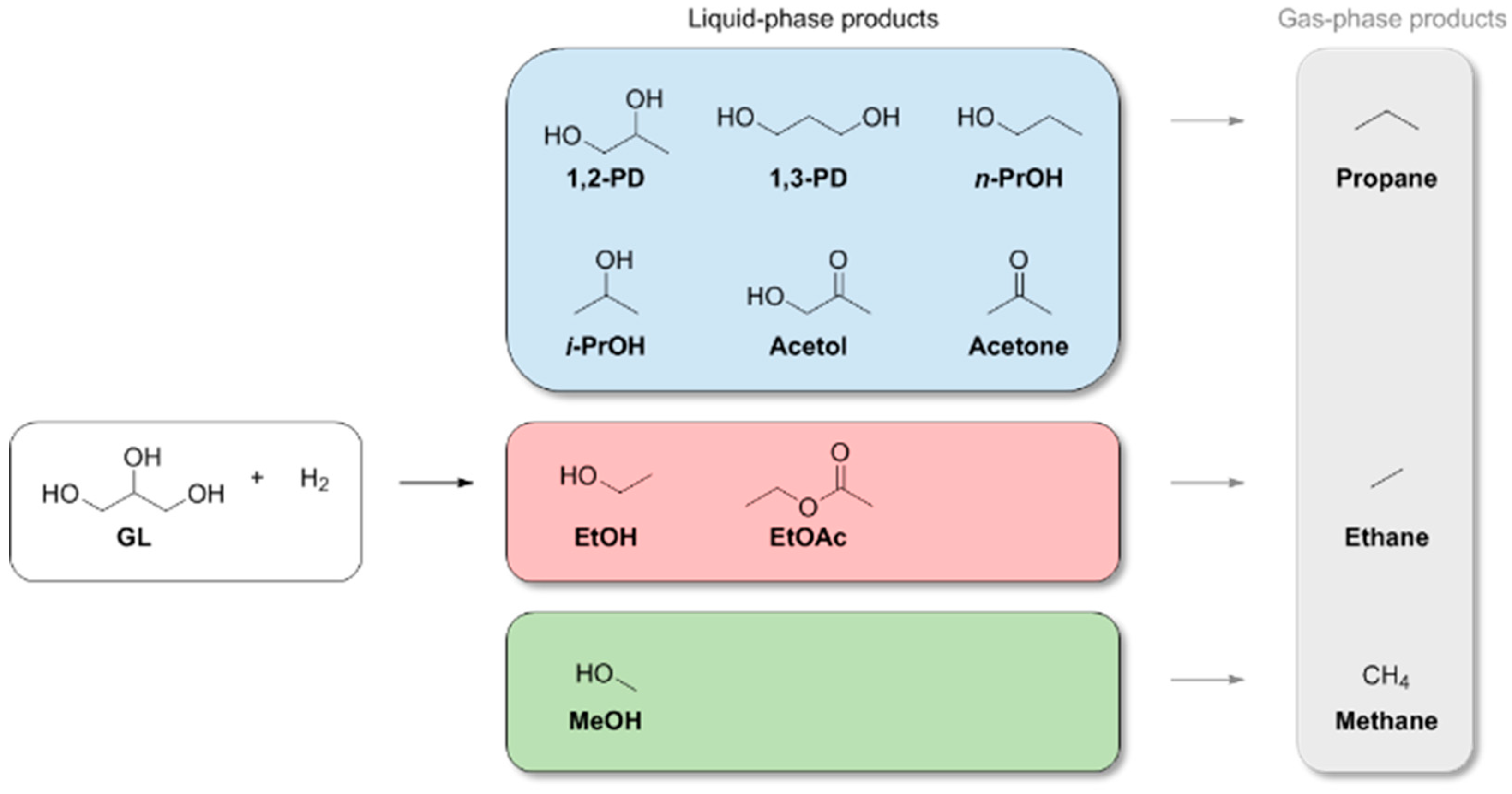
3. Results and Discussion
3.1. Catalytic Hydrogenolysis of Glycerol
3.1.1. Catalyst Screening
3.1.2. Catalysis with Ru-WD
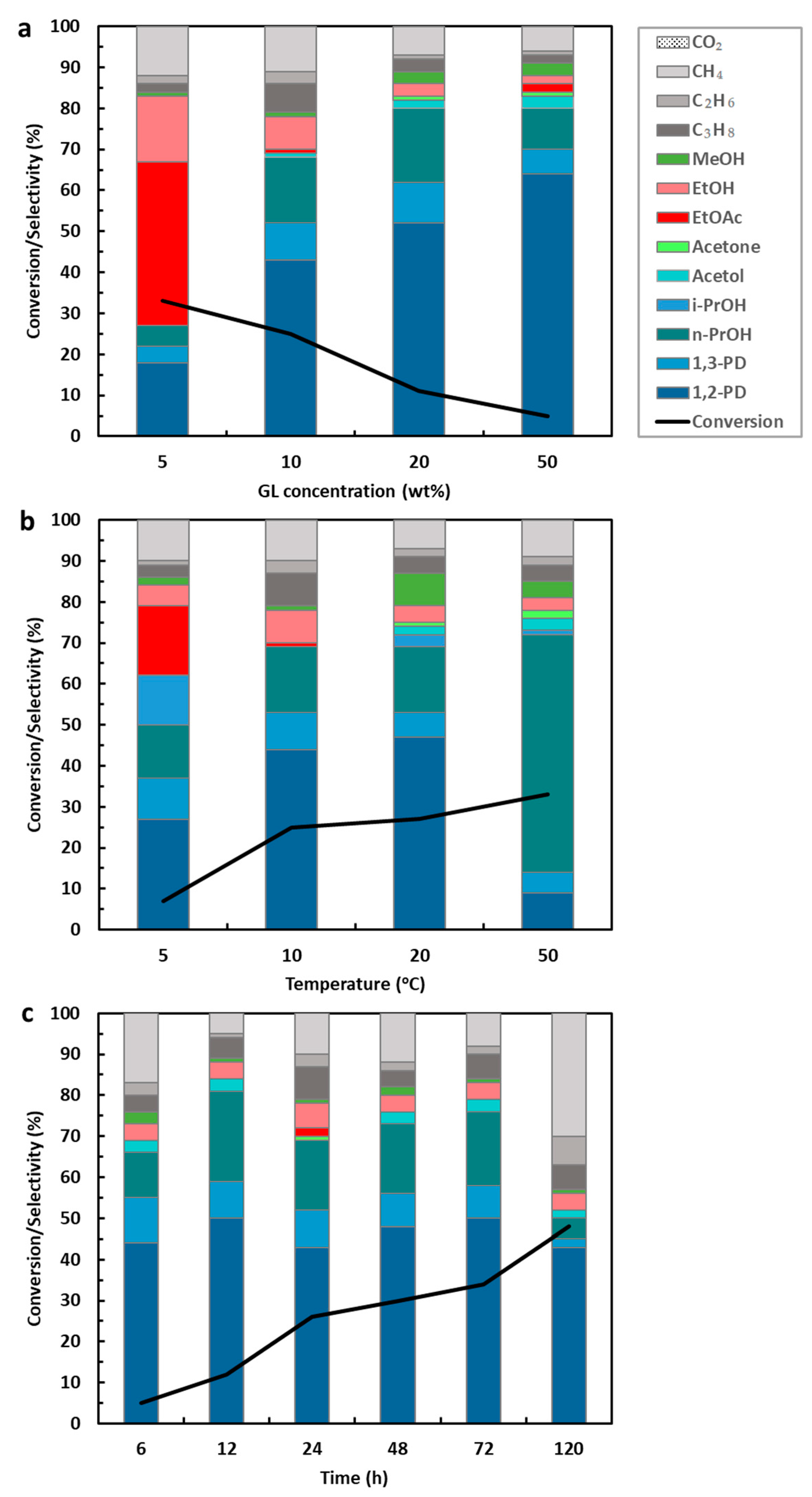
Effect of Glycerol Concentration, Temperature, and Reaction Time
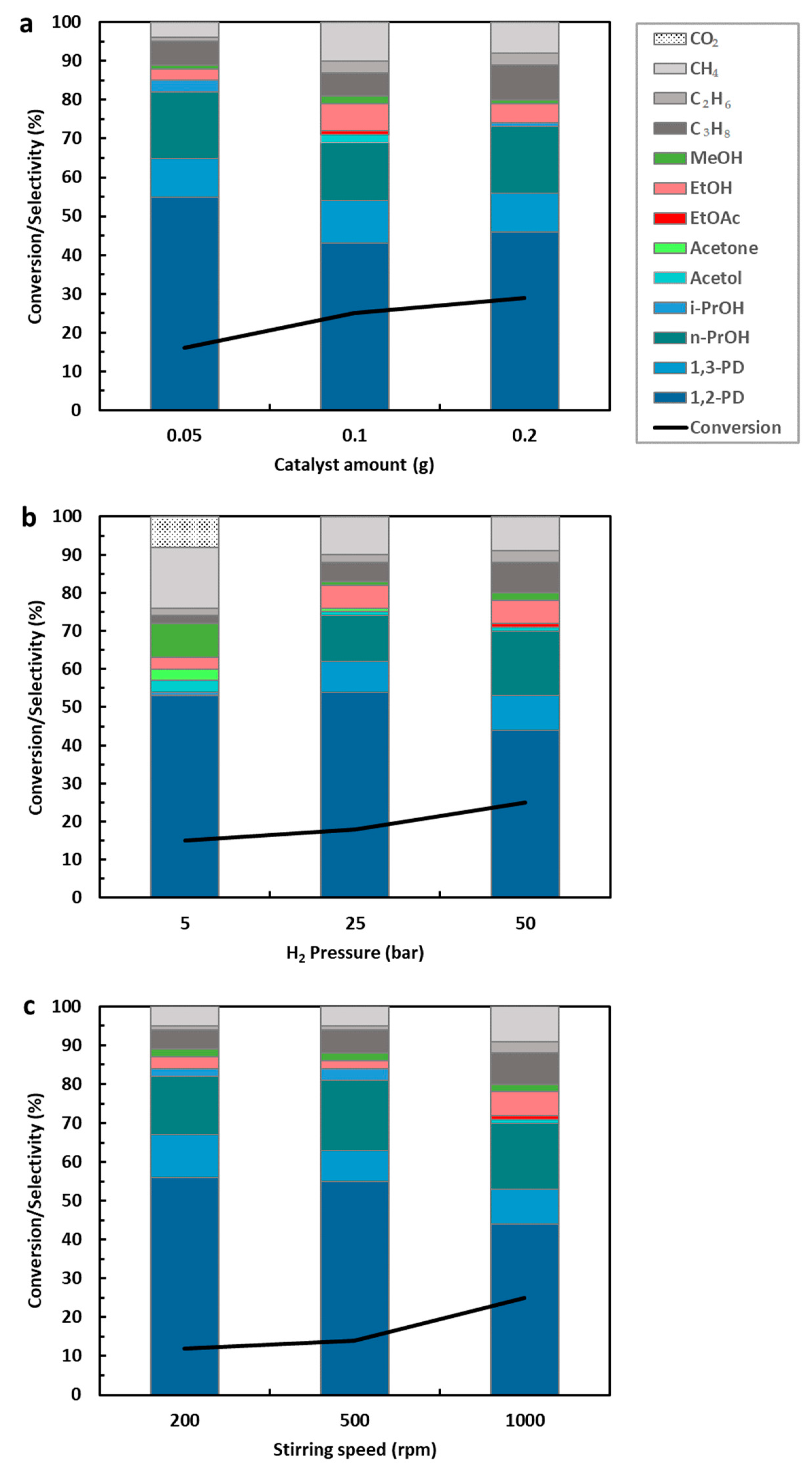
Effect of Catalyst Concentration, Hydrogen Pressure, and Stirring Speed
Catalyst Reuse
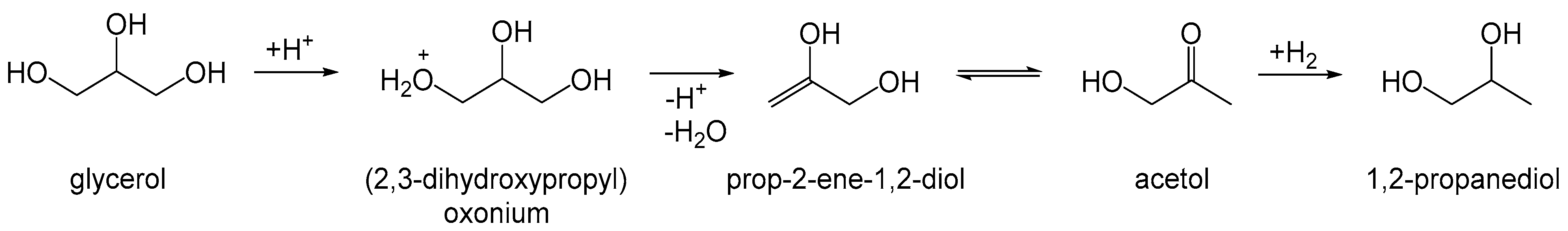
3.1.3. Characterization of WD-POMs
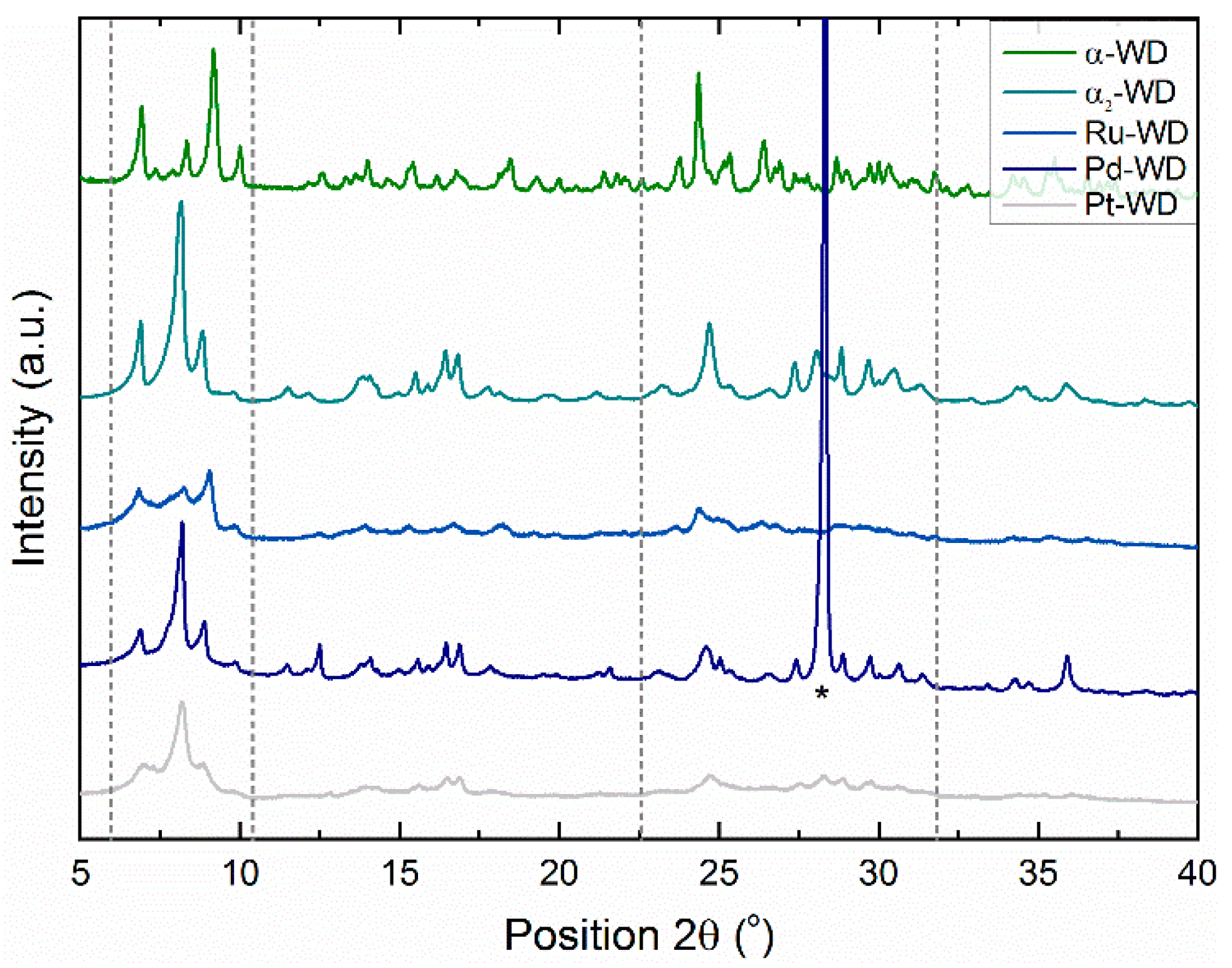
Structural Analysis by 31P-NMR, ATR-FTIR, and XRD
Elemental Analysis by XRF and ICP
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Fernando, S.; Adhikari, S.; Chandrapal, C.; Murali, N. Biorefineries: Current Status, Challenges, and Future Direction. Energy Fuels 2006, 20, 1727–1737. [Google Scholar] [CrossRef]
- Biddy, M.J.; Scarlata, C.; Kinchin, C. Chemicals from Biomass: A Market Assessment of Bioproducts with Near-Term Potential; National Renewable Energy Laboratory: Golden, CO, USA, 2016.
- International Energy Agency (IEA). World Energy Outlook 2013; International Energy Agency: Paris, France, 2013. [Google Scholar]
- Pagliaro, M.; Rossi, M. The Future of Glycerol: New Usages for a Versatile Raw Material; The Royal Society of Chemistry Publishing: Cambridge, UK, 2008. [Google Scholar]
- ten Dam, J.; Hanefeld, U. Renewable Chemicals: Dehydroxylation of Glycerol and Polyols. ChemSusChem 2011, 4, 1017–1034. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Tomishige, K. Heterogeneous catalysis of the glycerol hydrogenolysis. Catal. Sci. Technol. 2011, 1, 179–190. [Google Scholar] [CrossRef]
- Corma, A.; Iborra, S.; Velty, A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; della Pina, C. From glycerol to value-added products. Angew. Chem. Int. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Julkapli, N.M.; Yehye, W.A. Catalytic conversion of biodiesel derived raw glyceril to value added products. Renew. Sustain. Energy Rev. 2015, 41, 113–127. [Google Scholar] [CrossRef]
- Schlaf, M. Selective deoxygenation of sugar polyols to α,ω-diols and other oxygen content reduced materials—a new challenge to homogeneous ionic hydrogenation and hydrogenolysis catalysis. Dalton Trans. 2006, 39, 4645–4653. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yamada, Y.; Sato, S.; Ueda, W. Glycerol hydrogenolysis into useful C3 chemicals. Appl. Catal. B Environ. 2016, 193, 75–92. [Google Scholar] [CrossRef]
- Dasari, M.A.; Kiatsimkul, P.-P.; Sutterlin, W.R.; Suppes, G.J. Low-pressure hydrogenolysis of glycerol to propylene glycol. Appl. Catal. A Gen. 2005, 281, 225–231. [Google Scholar] [CrossRef]
- Huang, Z.W.; Cui, F.; Kang, H.X.; Chen, J.; Xia, C.G. Characterization and catalytic properties of the CuO/SiO2 catalysts prepared by precipitation-gel method in the hydrogenolysis of glycerol to 1,2-propanediol: Effect of residual sodium. Appl. Catal. A Gen. 2009, 366, 288. [Google Scholar] [CrossRef]
- Chopade, S.P.; Miller, D.J.; Jackson, J.E.; Werpy, T.A.; John, J.; Frye, G.; Zacher, A.H. Catalysts and process for hydrogenolysis of sugar alcohols to polyols. US6291725B1, 18 September 2000. [Google Scholar]
- Misono, M. Heterogeneous catalysis by heteropoly compounds of molybdenum and tungsten. Catal. Rev. 1987, 29, 269–321. [Google Scholar] [CrossRef]
- Maksimov, G.M.; Kozhevnikov, I.V. Heteropolyacids as catalysts for synthesis of methyl tert-butyl ether. React. Kinet. Catal. Lett. 1989, 39, 317–322. [Google Scholar] [CrossRef]
- Wang, C.; Bu, X.; Ma, J.; Liu, C.; Chou, K.; Wang, X.; Li, Q. Wells–Dawson type Cs5.5H0.5P2W18O62 based Co/Al2O3 as binfunctional catalysts for direct production of clean-gasoline fuel through Fischer–Tropsch synthesis. Catal. Today 2016, 274, 82–87. [Google Scholar] [CrossRef]
- Mansuy, D.; Bartoli, J.F.; Battioni, P.; Lyon, D.K.; Finke, R.G. Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. 2. Catalysis of olefin epoxidation and aliphatic and aromatic hydroxylations starting from.alpha.2-P2W17O61(Mn+.cntdot.Br)(n-11) (Mn+ = Mn3+,Fe3+,Co2+,Ni2+,Cu2+), including quantitative comparisons to metalloporphyrin catalysts. J. Am. Chem. Soc. 1991, 113, 7222–7226. [Google Scholar]
- Khenkin, A.M.; Neumann, R. Mixed-Addenda Vanadium-Substituted Polyfluorooxometalates: Synthesis, Characterization, and Catalytic Aerobic Oxidation. Inorg. Chem. 2000, 39, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Ben-Daniel, R.; Khenkin, A.M.; Neumann, R. The nickel subsituted quasi Wells-Dawson type polyfluoroxometalate [NiII(H2O)H2F6NaW17O55]9−, as a Uniquely Active Nickel-Based Catalyst for the Activation of Hydrogen Peroxide and the Epoxidation of Alkenes and Alkenols. Chem. A Eur. J. 2000, 6, 3722–3728. [Google Scholar] [CrossRef]
- Lyon, D.K.; Finke, R.G. Polyoxoanions as soluble metal oxide analogs. 6. Catalytic activity and initial kinetic and mechanistic studies of polyoxoanion-supported, atomically dispersed iridium(I), (1,5-COD)Ir.cntdot.P2W15Nb3O628-. Inorg. Chem. 1990, 29, 1787–1789. [Google Scholar] [CrossRef]
- Briand, L.E.; Baronetti, G.T.; Thomas, H.J. The state of the art on Wells–Dawson heteropoly-compounds A review of their properties and applications. Appl. Catal. A Gen. 2003, 256, 37–50. [Google Scholar] [CrossRef]
- Lyon, D.K.; Miller, W.K.; Novet, T.; Domaille, P.J.; Evitt, E.; Johnson, D.C.; Finke, R.G. Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. 1. The synthesis and characterization of aqueous-soluble potassium salts of.alpha.2-P2W17O61(Mn+.cntdot.OH2)(n-10) and organic solvent soluble tetra-n-butylammonium salts of.alpha.2-P2W17O61(Mn+.cntdot.Br)(n-11) (M = Mn3+,Fe3+,Co2+,Ni2+,Cu2+). J. Am. Chem. Soc. 1991, 113, 7209–7221. [Google Scholar]
- Maris, E.P.; Davis, R.J. Hydrogenolysis of glycerol over carbon-supported Ru and Pt. J. Catal. 2007, 249, 328–337. [Google Scholar] [CrossRef]
- Iqbal, S.; Kondrat, S.A.; Jones, D.R.; Schoenmakers, D.C.; Edwards, J.K.; Lu, L.; Yeo, B.R.; Wells, P.P.; Gibson, E.K.; Morgan, D.J.; et al. Ruthenium Nanoparticles Supported on Carbon: An Active Catalyst for the Hydrogenation of Lactic Acid to 1,2-Propanediol. ACS Catal. 2015, 5, 5047–5059. [Google Scholar] [CrossRef]
- Kandasamy, S.; Samudrala, S.P.; Bhattacharya, S. Vapour Phase Hydrogenolysis of Glycerol over NaY-Zeolite Supported Ru Catalysts for Targeted Selectivity towards 1,2-Propanediol. In Proceedings of the 2018 2nd International Conference on Green Energy and Applications (ICGEA), Singapore, 24–26 March 2018; pp. 14–18. [Google Scholar]
- Mbomekalle, I.M.; Lu, Y.W.; Keita, B.; Nadjo, L. Simple, high yield and reagent-saving synthesis of pure α-K6P2W18O62·14H2O. Inorg. Chem. Commun. 2004, 7, 86–90. [Google Scholar] [CrossRef]
- Han, X.; Xu, L.; Li, F.; Jiang, N. A New Series of Nanoporous Ionic Crystals Based on Polyoxometalates—Synthesis, Crystal Structures, and Adsorption Properties. Eur. J. Inorg. Chem. 2011, 29, 4564–4570. [Google Scholar] [CrossRef]
- Randall, W.J.; Lyon, D.K.; Domaille, P.J.; Finke, R.G.; Khenkin, A.M.; Hill, A.C. Transition Metal Complexes of the Lacunary Heteropolytungstate, [P2W17O61]10−. Inorg. Synth. 1998, 32, 242–268. [Google Scholar]
- Contant, R. Inorganic Synthesis; Ginsberg, A.P., Ed.; Inorganic Syntheses Inc.: Hoboken, NJ, USA, 1990; Volume 27, p. 104. [Google Scholar]
- Abbessi, M.; Contant, R.; Thouvenot, R.; Herveé, G. Dawson type heteropolyanions. 1. Multinuclear (phosphorus-31, vanadium-51, tungsten-183) NMR structural investigations of octadeca(molybdotungstovanado)diphosphates.alpha.-1,2,3-[P2MM’2W15O62]n− (M, M’ = Mo, V, W): syntheses of new related compounds. Inorg. Chem. 1991, 30, 1695–1702. [Google Scholar] [CrossRef]
- Contant, R.; Abbessi, M.; Canny, J.; Belhouari, A.; Keita, B.; Nadjo, L. Iron-Substituted Dawson-Type Tungstodiphosphates: Synthesis, Characterization, and Single or Multiple Initial Electronation Due to the Substituent Nature or Position. J. Inorg. Chem. 1997, 36, 4961–4967. [Google Scholar] [CrossRef]
- Sakai, Y.; Shinohara, A.; Hayashi, K.; Nomiya, K. Synthesis and Characterization of Two Novel, Mono-Lacunary Dawson Polyoxometalate-Based, Water-Soluble Organometallic Ruthenium(II) Complexes: Molecular Structure of [{(C6H6)Ru(H2O)}(α2-P2W17O61)]8–. Eur. J. Inorg. Chem. 2006, 2006, 163–171. [Google Scholar] [CrossRef]
- Vanhaecht, S.; Absillis, G.; Parac-Vogt, T.N. Hydrolysis of DNA model substrates catalyzed by metal-substituted Wells–Dawson polyoxometalates. Dalton Trans. 2012, 41, 10028. [Google Scholar] [CrossRef]
- Contant, R.; Abbessi, M.; Thouvenot, R.; Hervé, G. Dawson Type Heteropolyanions. 3. Syntheses and 31P, 51V, and 183W NMR Structural Investigation of Octadeca(molybdo−tungsto−vanado)diphosphates Related to the [H2P2W12O48]12− Anion. Inorg. Chem. 2004, 43, 3597–3604. [Google Scholar] [CrossRef]
- Dawson, B. The structure of the 9(18)-heteropoly anion in potassium 9(18)-tungstophosphate, K6(P2W18O62)·14H2O. Acta Crystallogr. 1953, 6, 113–126. [Google Scholar] [CrossRef]
- Matsumoto, K.Y.; Sasaki, Y.J. Crystal structure of α-P2W18O626– anion. Chem. Soc. Chem. Commun. 1975, 16, 691–692. [Google Scholar] [CrossRef]
- Liu, H.; Yue, B.; Sun, W.; Chen, Z.; Jin, S.; Deng, J.; Xie, G.; Shao, Q.; Wu, L. Synthesis and characterization of noble-metal-substituted Dawson-type polyoxometalates. Transit. Met. Chem. 1997, 22, 321–325. [Google Scholar] [CrossRef]
- Jorris, T.L.; Kozik, M.; Casan-Pastor, N.; Domaille, P.J.; Finke, R.G.; Miller, W.K.; Baker, L.C.W. Effects of paramagnetic and diamagnetic transition-metal monosubstitutions on tungsten-183 and phosphorus-31 NMR spectra for Keggin and Wells-Dawson heteropolytungstate derivatives. Correlations and corrections. Tungsten-183 NMR two-dimensional INADEQUATE studies of.alpha.-[(D2O)ZnO4Xn+W11O34](10-n)- wherein Xn+ = Si4+ and P5+. J. Am. Chem. Soc. 1987, 109, 7402–7408. [Google Scholar]
- Stapleton, A.J.; Sloan, M.E.; Napper, N.J.; Burns, R.C. Transition metal-substituted Dawson anions as chemo- and regio-selective oxygen transfer catalysts for H2O2 in the epoxidation of allylic alcohols. Dalton Trans. 2009, 9603–9615. [Google Scholar] [CrossRef] [PubMed]
- Izarova, N.V.; Banerjee, A.; Kortz, U. Noble metals in polyoxometalate chemistry: palladium-containing derivatives of the monolacunary Keggin and Wells-Dawson tungstophosphates. Inorg. Chem. 2011, 50, 10379–10386. [Google Scholar] [CrossRef] [PubMed]
- Comuzzi, C.; Dolcetti, G.; Trovarelli, A.; Cavani, F.; Trifirò, F.; Llorca, J.; Finke, R.G. The solid-state rearrangement of the Wells-Dawson K6P2W18O62·10H2O to a stable Keggin-type heteropolyanion phase: a catalyst for the selective oxidation of isobutane to isobutene. Catal. Lett. 1996, 36, 75–79. [Google Scholar] [CrossRef]
- Ueda, T.; Nishimoto, Y.; Saito, R.; Ohnishi, M.; Nambu, J. Vanadium(V)-Substitution Reactions of Wells–Dawson-Type Polyoxometalates: From [X2M18O62]6− (X = P, As; M = Mo, W) to [X2VM17O62]7−. Inorganics 2015, 3, 355. [Google Scholar] [CrossRef]
- Contant, R.; Thouvenot, R. A reinvestigation of isomerism in the Dawson structure: syntheses and 183W NMR structural characterization of three new polyoxotungstates [X2W18O62]6− (X=PV, AsV). Inorg. Chim. Acta 1993, 212, 41–50. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Conversion (%) b | pH c | Product Selectivity in Phases (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Liquid b | Gas d | ||||||||||
| 1,2-PD | 1,3-PD | n-PrOH | EtOAc | Acetone | Acetol | CH4 | |||||
| 1 | Ru-WD | 26 | 5.6 | 42 | 9 | 17 | 1 | 0 | 1 | 10 | |
| 2 | RuCl3 | 17 | 3.1 | 17 | 4 | 12 | 2 | 0 | 0 | 44 | |
| 3 | Ru/C e | 17 | 4.1 | 31 | 1 | 2 | 0 | 0 | 0 | 44 | |
| 4 | Pd-WD | 1 | 5.9 | 60 | 0 | 14 | 0 | 0 | 7 | 1 | |
| 5 | PdCl2 | 3 | 2.7 | 16 | 19 | 59 | 0 | 0 | 0 | 0 | |
| 6 | Pd/C e | 2 | 3.4 | 74 | 0 | 6 | 0 | 0 | 0 | 0 | |
| 7 | Pt-WD | 3 | 6.0 | 63 | 15 | 8 | 0 | 2 | 1 | 0 | |
| 8 | H2PtCl6 | 7 | 2.5 | 13 | 18 | 57 | 0 | 0 | 0 | 6 | |
| 9 | Pt/C e | 6 | 4.1 | 78 | 6 | 6 | 0 | 0 | 0 | 0 | |
| 10 | α-WD | 1 | 6.4 | 8 | 0 | 5 | 39 | 8 | 14 | 1 | |
| 11 | α2-WD | 0 | 6.3 | 18 | 0 | 0 | 0 | 18 | 35 | 1 | |
| 12 | none | 0 | 7.0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Entry | Conversion (%) b | Converted GL (g) | Product Selectivity in Phases (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Liquid b | Gas c | |||||||||||||
| 1,2-PD | 1,3-PD | n-PrOH | EtOAc | Acetone | Acetol | EtOH | MeOH | CH4 | C2H6 | C3H8 | ||||
| 1 | 26 | 0.26 | 42 | 9 | 17 | 1 | 0 | 1 | 7 | 1 | 10 | 3 | 7 | |
| 2 d | 14 | 0.24 | 49 | 11 | 20 | 2 | 0 | 0 | 6 | 1 | 4 | 1 | 4 | |
| 3 e | 10 | 0.25 | 54 | 7 | 14 | 0 | 0 | 0 | 8 | 3 | 6 | 2 | 5 | |
| Material | Abbreviation | Color | Water Content (mol%) a |
|---|---|---|---|
| α-K6P2W18O62 | α-WD | Yellow | 8 |
| α2-K10P2W17O61 | α2-WD | White | 16 |
| α2-K7P2W17O61Ru | Ru-WD | Black | 9 |
| α2-K8P2W17O61Pd | Pd-WD | Brown | 8 |
| α2-K8P2W17O61Pt | Pt-WD | Light brown | 11 |
| Material | W/P Ratio (mol/mol) | K/P Ratio (mol/mol) | M/P Ratio (mol/mol) b | |||||
|---|---|---|---|---|---|---|---|---|
| XRF | ICP | Theo. | XRF | ICP | Theo. | ICP | Theo. | |
| α-WD | 9.1 | 8.8 | 9.0 | 3.0 | 2.3 | 3.0 | 0.0 | 0.0 |
| α2-WD | 8.4 | 8.1 | 8.5 | 5.1 | 4.4 | 5.0 | 0.0 | 0.0 |
| Ru-WD c | 8.6 | 8.1 | 8.5 | 4.1 | 5.2 | 3.5 | 0.7 | 0.5 |
| Pd-WD d | 9.2 | 8.1 | 8.5 | 5.2 | 3.9 | 4.0 | 0.4 | 0.5 |
| Pt-WD d | 9.9 | 8.3 | 8.5 | 5.2 | 8.0 | 4.0 | 0.3 | 0.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modvig, A.; Kumpidet, C.; Riisager, A.; Albert, J. Ru-Doped Wells–Dawson Polyoxometalate as Efficient Catalyst for Glycerol Hydrogenolysis to Propanediols. Materials 2019, 12, 2175. https://doi.org/10.3390/ma12132175
Modvig A, Kumpidet C, Riisager A, Albert J. Ru-Doped Wells–Dawson Polyoxometalate as Efficient Catalyst for Glycerol Hydrogenolysis to Propanediols. Materials. 2019; 12(13):2175. https://doi.org/10.3390/ma12132175
Chicago/Turabian StyleModvig, Amalie, Chiraphat Kumpidet, Anders Riisager, and Jakob Albert. 2019. "Ru-Doped Wells–Dawson Polyoxometalate as Efficient Catalyst for Glycerol Hydrogenolysis to Propanediols" Materials 12, no. 13: 2175. https://doi.org/10.3390/ma12132175
APA StyleModvig, A., Kumpidet, C., Riisager, A., & Albert, J. (2019). Ru-Doped Wells–Dawson Polyoxometalate as Efficient Catalyst for Glycerol Hydrogenolysis to Propanediols. Materials, 12(13), 2175. https://doi.org/10.3390/ma12132175